
Keystone Taxa and Predictive Functional Analysis of Sphagnum palustre Tank Microbiomes in Erxianyan Peatland, Central China
 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
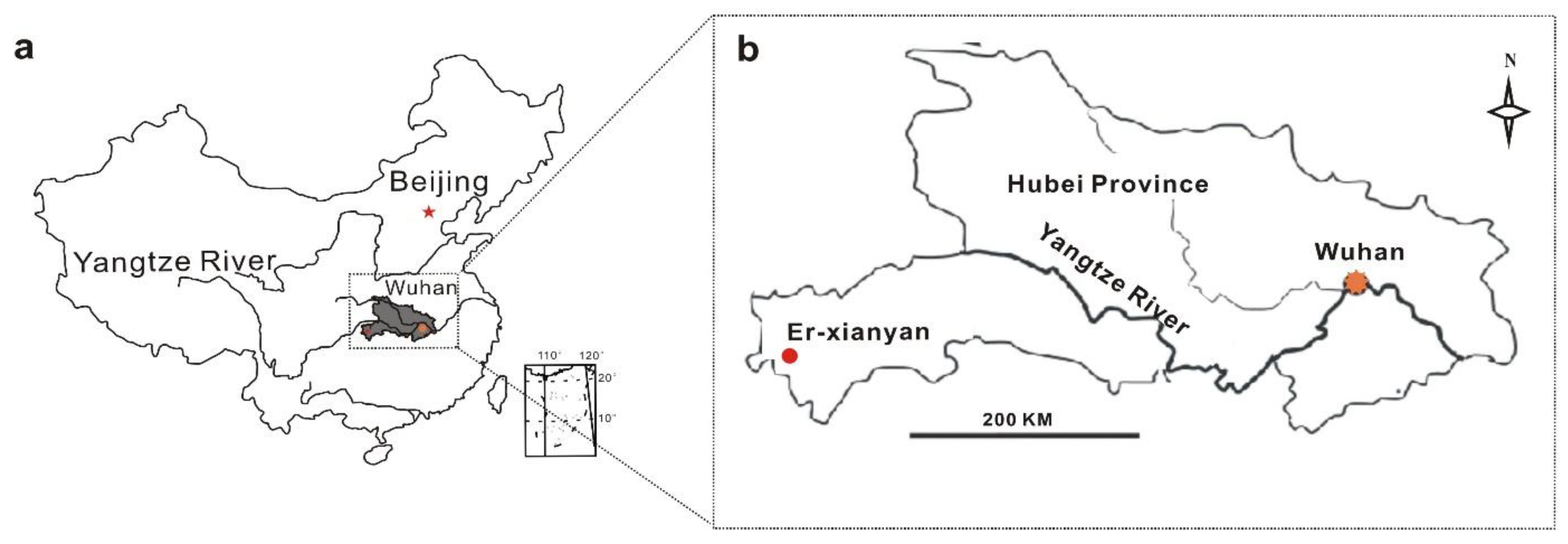
2.1. Study Site Description and Sphagnum Collection
2.2. Genomic DNA Extraction and High-Throughput Sequencing
2.3. Bioinformatics Analysis
3. Results
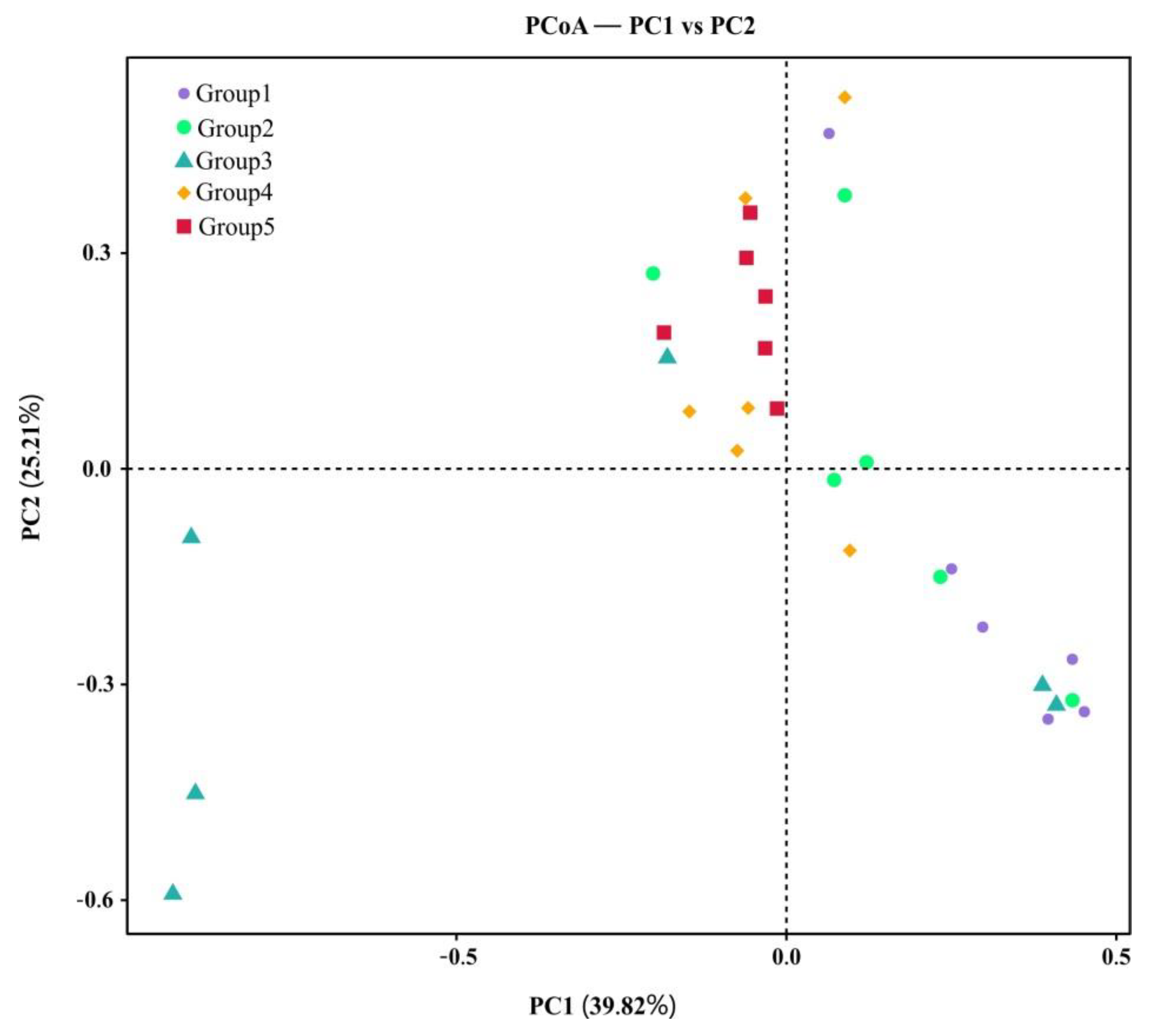
3.1. Community Composition and Biodiversity Assessment
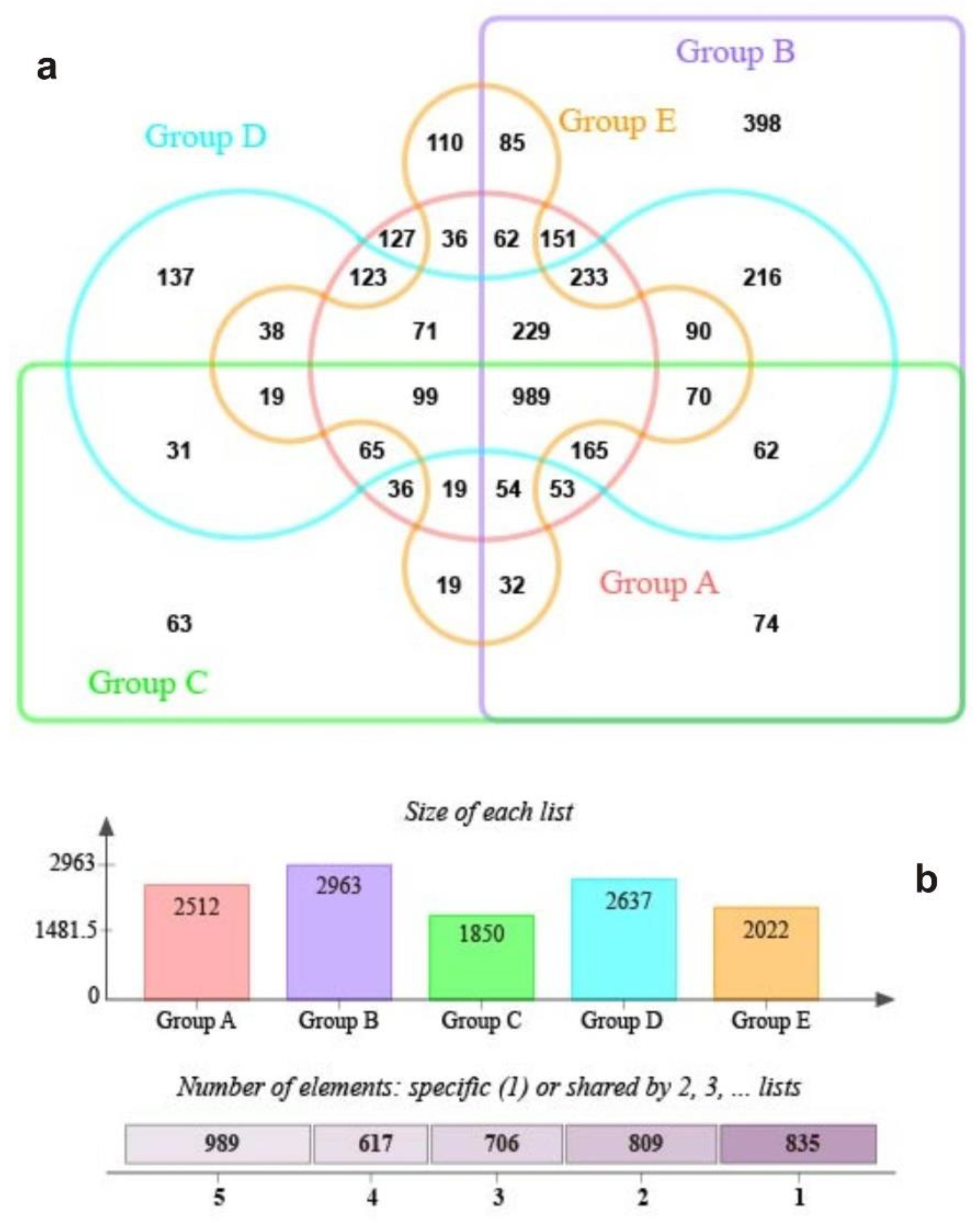
3.2. The S.-palustre-Associated Core Microbiome
3.3. Functional Prediction of Microbial Communities
4. Discussion
4.1. Proteobacteria Dominant in S.-palustre-Associated Microbiomes
4.2. The S.-palustre-Associated Core Microbiome
4.3. Methodological Limitations and Future Aspects
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vitt, D.H.; House, M. Bryophytes as key indicators of ecosystem function and structure of northern peatlands. Bryophyt. Divers. Evol. 2021, 43, 253–264. [Google Scholar] [CrossRef]
- Ma, X.Y.; Xu, H.; Cao, Z.Y.; Shu, L.; Zhu, R.L. Will climate change cause the global peatland to expand or contract? Evidence from the habitat shift pattern of Sphagnum mosses. Glob. Chang. Biol. 2022, 1–14. [Google Scholar] [CrossRef]
- Dise, N.B. Peatland response to global change. Science 2009, 326, 810–811. [Google Scholar] [CrossRef] [PubMed]
- Limpens, J.; Berendse, F.; Blodau, C.; Canadell, J.G.; Freeman, C.; Holden, J.; Roulet, N.; Rydin, H.; Schaepman-Strub, G. Peatlands and the carbon cycle: From local processes to global implications—A synthesis. Biogeosciences 2008, 5, 1475–1491. [Google Scholar] [CrossRef]
- Shaw, A.J.; Schmutz, J.; Devos, N.; Shu, S.; Carrell, A.A.; Weston, D.J. The sphagnum genome project: A new model for ecological and evolutionary genomics. Adv. Bot. Res. 2016, 78, 1–18. [Google Scholar]
- Shcherbakov, A.V.; Bragina, A.V.; Kuzmina, E.Y.; Berg, C.; Muntyan, A.N.; Makarova, N.M.; Malfanova, N.V.; Cardinale, M.; Berg, G.; Chebotar, V.K.; et al. Endophytic bacteria of Sphagnum mosses as promising objects of agricultural microbiology. Microbiology 2013, 82, 306–315. [Google Scholar] [CrossRef]
- Shcherbakov, A.V.; Kuzmina, E.Y.; Lapshina, E.D.; Shcherbakova, E.N.; Chebotar, V.K. Taxonomic diversity of bacterial populations inhabiting gametophytes of Sphagnum mosses from different geographic regions of Russia. Agron. Res. 2015, 13, 192–201. [Google Scholar]
- Carrell, A.A.; Kolton, M.; Glass, J.B.; Pelletier, D.A.; Warren, M.J.; Kostka, J.E.; Iversen, C.M.; Hanson, P.J.; Weston, D.J. Experimental warming alters the community composition, diversity, and N2 fixation activity of peat moss (Sphagnum fallax) microbiomes. Glob. Chang. Biol. 2019, 25, 2993–3004. [Google Scholar] [CrossRef]
- Weston, D.J.; Timm, C.M.; Walker, A.P.; Gu, L.; Muchero, W.; Schmutz, J.; Shaw, A.J.; Tuskan, G.A.; Warren, J.M.; Wullschleger, S.D. Sphagnum physiology in the context of changing climate: Emergent influences of genomics, modelling and host–microbiome interactions on understanding ecosystem function. Plant Cell Environ. 2015, 38, 1737–1751. [Google Scholar] [CrossRef]
- Kostka, J.E.; Weston, D.J.; Glass, J.B.; Lilleskov, E.A.; Shaw, A.J.; Turetsky, M.R. The Sphagnum microbiome: New insights from an ancient plant lineage. New Phytol. 2016, 211, 57–64. [Google Scholar] [CrossRef]
- Basińska, A.M.; Reczuga, M.K.; Gąbka, M.; Stróżecki, M.; Łuców, D.; Samson, M.; Urbaniak, M.; Leśny, J.; Chojnicki, B.H.; Gilbert, D.; et al. Experimental warming and precipitation reduction affect the biomass of microbial communities in a Sphagnum peatland. Ecol. Indic. 2020, 112, 106059. [Google Scholar] [CrossRef]
- Carrell, A.A.; Lawrence, T.J.; Cabugao, K.G.M.; Carper, D.L.; Pelletier, D.A.; Lee, J.H.; Jawdy, S.S.; Grimwood, J.; Schmutz, J.; Hanson, P.J.; et al. Habitat-adapted microbial communities mediate Sphagnum peatmoss resilience to warming. New Phytol. 2022, 234, 2111–2125. [Google Scholar] [CrossRef] [PubMed]
- Afzal, M.; Khan, Q.M.; Sessitsch, A. Endophytic bacteria: Prospects and applications for the phytoremediation of organic pollutants. Chemosphere 2014, 117, 232–242. [Google Scholar] [CrossRef]
- Raymond, J.A. Dependence on epiphytic bacteria for freezing protection in an Antarctic moss, Bryum argenteum. Environ. Microbiol. Rep. 2016, 8, 14–19. [Google Scholar] [CrossRef]
- Opelt, K.; Berg, C.; Schonmann, S.; Eberl, L.; Berg, G. High specificity but contrasting biodiversity of Sphagnum-associated bacterial and plant communities in bog ecosystems independent of the geographical region. ISME J. 2007, 1, 502–516. [Google Scholar] [CrossRef]
- Opelt, K.; Chobot, V.; Hadacek, F.; Schonmann, S.; Eberl, L.; Berg, G. Investigations of the structure and function of bacterial communities associated with Sphagnum mosses. Environ. Microbiol. 2007, 9, 2795–2809. [Google Scholar] [CrossRef]
- Opelt, K.; Berg, G. Diversity and antagonistic potential of bacteria associated with bryophytes from nutrient-poor habitats of the Baltic Sea Coast. Appl. Environ. Microbiol. 2004, 70, 6569–6579. [Google Scholar] [CrossRef]
- Tang, J.Y.; Ma, J.; Li, X.D.; Li, Y.H. Illumina sequencing-based community analysis of bacteria associated with different bryophytes collected from Tibet, China. BMC Microbiol. 2016, 16, 276. [Google Scholar] [CrossRef]
- Tian, W.; Xiang, X.; Ma, L.; Evers, S.; Wang, R.; Qiu, X.; Wang, H. Rare species shift the structure of bacterial communities across sphagnum compartments in a subalpine peatland. Front. Microbiol. 2020, 10, 3138. [Google Scholar] [CrossRef]
- Stéphane, C.; Abdul, S.; Hanna, F.; Angela, S. A review on the plant microbiome: Ecology, functions, and emerging trends in microbial application. J. Adv. Res. 2019, 19, 29–37. [Google Scholar]
- Risely, A. Applying the core microbiome to understand host–microbe systems. J. Anim. Ecol. 2020, 89, 1549–1558. [Google Scholar] [CrossRef]
- Neu, A.T.; Allen, E.E.; Roy, K. Defining and quantifying the core microbiome: Challenges and prospects. Proc. Natl. Acad. Sci. USA 2021, 118, e2104429118. [Google Scholar] [CrossRef]
- Vandenkoornhuyse, P.; Quaiser, A.; Duhamel, M.; Van, A.L.; Dufresne, A. The importance of the microbiome of the plant holobiont. New Phytol. 2015, 206, 1196–1206. [Google Scholar] [CrossRef]
- Lemanceau, P.; Blouin, M.; Muller, D.; Moënne-Loccoz, Y. Let the core microbiota be functional. Trends Plant Sci. 2017, 7, 583–595. [Google Scholar] [CrossRef]
- Pan, J.; Jeffrey, P.; Edwards, M.A.; Amy, P.; Mark, I.A. Impact of water chemistry, pipe material and stagnation on the building plumbing microbiome. PLoS ONE 2015, 10, e0141087. [Google Scholar]
- Zarraonaindia, I.; Owens, S.M.; Weisenhorn, P.; West, K.; Hampton-Marcell, J.; Lax, S.; Bokulich, N.A.; Mills, D.A.; Martin, G.; Taghavi, S.; et al. The soil microbiome influences grapevine-associated microbiota. mBio 2015, 6, e02527-14. [Google Scholar] [CrossRef]
- Chen, H.; Wu, H.; Yan, B.; Zhao, H.; Liu, F.; Zhang, H.; Sheng, Q.; Miao, F.; Liang, Z. Core microbiome of medicinal plant salvia miltiorrhiza seed: A rich reservoir of beneficial microbes for secondary metabolism? Int. J. Mol. Sci. 2018, 19, 672. [Google Scholar] [CrossRef]
- Chen, S.Y.; Li, J.J.; Lin, J.; Bao, K.X.; Fan, J.Q.; Zhang, R.Q.; He, W. High-throughput sequencing fungal community structures in aging tobacco strips from different growing areas and stalk positions. Tob. Sci. Technol. 2018, 51, 12–19. [Google Scholar]
- Kaul, S.; Sharma, T.; Dhar, M.K. “Omics” tools for better understanding the plant–endophyte interactions. Front. Plant Sci. 2016, 7, 955. [Google Scholar] [CrossRef]
- Qin, Y.; Mitchell, E.A.; Lamentowicz, M.; Payne, R.J.; Lara, E.; Gu, Y.; Huang, X.; Wang, H. Ecology of testate amoebae in peatlands of central China and development of a transfer function for paleohydrological reconstruction. J. Paleolimnol. 2013, 50, 319–330. [Google Scholar] [CrossRef]
- Qin, Y.; Payne, R.J.; Gu, Y.; Huang, X.; Wang, H. Ecology of testate amoebae in Dajiuhu peatland of Shennongjia Mountains, China, in relation to hydrology. Front. Earth Sci. 2012, 6, 57–65. [Google Scholar] [CrossRef]
- Michaelis, D. The Sphagnum Species of the World; Schweizerbart, Science Publishers: Stuttgart, Germany, 2019. [Google Scholar]
- Castrillo, G.; Teixeira, P.J.P.L.; Paredes, S.H.; Law, T.F.; De Lorenzo, L.; Feltcher, M.E.; Finkel, O.M.; Breakfield, N.W.; Mieczkowski, P.; Jones, C.D.; et al. Root microbiota drive direct integration of phosphate stress and immunity. Nature 2017, 543, 513–518. [Google Scholar] [CrossRef]
- Louca, S.; Parfrey, L.W.; Doebeli, M. Decoupling function and taxonomy in the global ocean microbiome. Science 2016, 353, 1272–1277. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahe, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, L.; Gu, Y.; Coleman-Derr, D. MetaCoMET: A web platform for discovery and visualization of the core microbiome. Bioinformatics 2016, 32, 3469–3470. [Google Scholar] [CrossRef]
- Kip, N.; Fritz, C.; Langelaan, E.S.; Pan, Y.; Bodrossy, L.; Pancotto, V.; Jetten, M.S.M.; Smolders, A.J.P.; Op den Camp, H.J.M. Methanotrophic activity and diversity in different Sphagnum magellanicum dominated habitats in the southernmost peat bogs of Patagonia. Biogeosciences 2011, 9, 47–55. [Google Scholar] [CrossRef]
- Xiang, X.; Wang, H.; Tian, W.; Wang, R.; Gong, L.; Xu, Y. Composition and function of bacterial communities of bryophytes and their underlying sediments in the Dajiuhu Peatland, central China. J. Earth Sci. 2020. Available online: https://kns.cnki.net/kcms/detail/42.1788.P.20201222.1817.004.html (accessed on 11 July 2021).
- Danilova, O.V.; Belova, S.E.; Gagarinova, I.V.; Dedysh, S.N. Microbial community composition and methanotroph diversity of a subarctic wetland in Russia. Microbiology 2016, 85, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Leff, J.W.; Jones, S.E.; Prober, S.M.; Barberán, A.; Borer, E.T.; Firn, J.L.; Harpole, W.S.; Hobbie, S.E.; Hofmockel, K.S.; Knops, J.M.; et al. Consistent responses of soil microbial communities to elevated nutrient inputs in grasslands across the globe. Proc. Natl. Acad. Sci. USA 2015, 112, 10967–10972. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Wang, H.; Gong, L.; Liu, Q. Vertical variations and associated ecological function of bacterial communities from Sphagnum to underlying sediments in Dajiuhu Peatland. Sci. China-Earth Sci. 2014, 57, 1013–1020. [Google Scholar] [CrossRef]
- Mishra, S.; Lee, W.A.; Hooijer, A.; Reuben, S.; Sudiana, I.M.; Idris, A.; Swarup, S. Microbial and metabolic profiling reveal strong influence of water table and land-use patterns on classification of degraded tropical peatlands. Biogeosciences 2014, 11, 14009–14042. [Google Scholar] [CrossRef]
- Toju, H.; Peay, K.G.; Yamamichi, M.; Narisawa, K.; Hiruma, K.; Naito, K.; Fukuda, S.; Ushio, M.; Nakaoka, S.; Onoda, Y.; et al. Core microbiomes for sustainable agroecosystems. Nat. Plants 2018, 4, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yu, L.; Zhang, J.; Zhang, X.; Xue, Y.; Liu, J.; Xiao, Z. Characterization of the core microbiome in tobacco leaves during aging. Microbiol. Open 2020, 9, e984. [Google Scholar] [CrossRef] [PubMed]
- Rui, J.; Li, J.; Zhang, S.; Xuefeng, Y.; Wang, Y.; Li, X. The core populations and co-occurrence patterns of prokaryotic communities in household biogas digesters. Biotechnol. Biofuels 2015, 8, 158. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-M.; Lu, Z.-M.; Shi, J.-S.; Xu, Z. Exploring flavour-producing core microbiota in multispecies solid-state fermentation of traditional Chinese vinegar. Sci. Rep. 2016, 6, 26818. [Google Scholar] [CrossRef]
- Tian, W.; Wang, H.M.; Xiang, X.; Wang, R.C.; Xu, Y. Structural variations of bacterial community driven by Sphagnum microhabitat differentiation in a subalpine peatland. Front. Microbiol. 2019, 10, 1661. [Google Scholar] [CrossRef] [PubMed]
- Kulichevskaya, I.S.; Ivanova, A.O.; Baulina, O.I.; Bodelier, P.L.; Damste, J.S.S.; Dedysh, S.N. Singulisphaera acidiphila gen. nov., sp. nov., a non-filamentous, Isosphaera-like planctomycete from acidic northern wetlands. Int. J. Syst. Evol. Microbiol. 2008, 58, 1186–1193. [Google Scholar] [CrossRef]
- Alica, C.; Jií, B.; Eva, K.; Zuzana, U.; Tomá, P. Spatial heterogeneity of belowground microbial communities linked to peatland microhabitats with different plant dominants. FEMS Microbiol. Ecol. 2019, 95, fiz130. [Google Scholar]
- Ma, B.; Cai, Y.; Bork, E.W.; Chang, S.X. Defoliation intensity and elevated precipitation effects on microbiome and interactome depend on site type in northern mixed-grass prairie. Soil Biol. Biochem. 2018, 122, 163–172. [Google Scholar] [CrossRef]
- Peng, M.; Jia, H.; Wang, Q. The effect of land use on bacterial communities in saline–alkali soil. Curr. Microbiol. 2017, 74, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Zinke, L.A.; Kiel, R.B.; James, M.M.; Wheat, C.G.; Orcutt, B.N.; Amend, J.P. Sediment microbial communities influenced by cool hydrothermal fluid migration. Front. Microbiol. 2018, 9, 1249. [Google Scholar] [CrossRef]
- Takaichi, S.; Maoka, T.; Takasaki, K.; Hanada, S. Carotenoids of Gemmatimonas aurantiaca (Gemmatimonadetes): Identification of a novel carotenoid, deoxyoscillol 2-rhamnoside, and proposed biosynthetic pathway of oscillol 2,2-dirhamnoside. Microbiology 2010, 156, 757–763. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total Tags | Taxon Tags | OTUs | Observed Species | Shannon | Simpson | Chao1 | ACE | Goods Coverage (%) |
|---|---|---|---|---|---|---|---|---|---|
| E0W01 | 70,028 | 64,973 | 1015 | 809 | 3.34 | 0.77 | 994.25 | 1000.77 | 99.60 |
| E0W03 | 70,144 | 65,305 | 499 | 386 | 2.76 | 0.77 | 503.61 | 549.77 | 99.70 |
| E0W1A | 70,260 | 64,355 | 753 | 615 | 3.93 | 0.82 | 726.78 | 769.59 | 99.70 |
| E0W3A | 70,137 | 65,126 | 735 | 597 | 4.91 | 0.92 | 697.72 | 717.74 | 99.70 |
| E0W1B | 75,644 | 71,054 | 926 | 770 | 2.78 | 0.57 | 917.00 | 998.60 | 99.60 |
| E0W3B | 70,168 | 65,832 | 1466 | 1261 | 4.16 | 0.76 | 1415.29 | 1460.88 | 99.50 |
| E2W01 | 70,086 | 58,693 | 2152 | 2028 | 9.19 | 1.00 | 2092.96 | 2108.92 | 99.70 |
| E2W03 | 70,098 | 59,469 | 1109 | 914 | 4.25 | 0.82 | 1059.44 | 1106.76 | 99.60 |
| E2W1A | 70,197 | 66,422 | 615 | 497 | 2.40 | 0.57 | 613.04 | 653.66 | 99.70 |
| E2W3A | 70,736 | 66,471 | 779 | 624 | 2.95 | 0.69 | 768.01 | 782.86 | 99.70 |
| E2W1B | 62,775 | 55,600 | 1770 | 1610 | 5.59 | 0.91 | 1644.79 | 1703.36 | 99.70 |
| E2W3B | 68,365 | 63,327 | 748 | 601 | 4.42 | 0.91 | 722.26 | 753.98 | 99.70 |
| E4W01 | 70,121 | 65,995 | 857 | 681 | 2.54 | 0.64 | 852.01 | 893.87 | 99.60 |
| E4W03 | 70,039 | 64,692 | 911 | 762 | 5.00 | 0.91 | 907.76 | 955.94 | 99.60 |
| E4W1A | 70,135 | 30,225 | 1021 | 854 | 4.23 | 0.81 | 975.72 | 1015.33 | 99.60 |
| E4W3A | 75,810 | 38,562 | 775 | 644 | 4.11 | 0.86 | 791.02 | 831.05 | 99.70 |
| E4W1B | 70,114 | 32,545 | 927 | 773 | 4.25 | 0.81 | 920.86 | 952.33 | 99.60 |
| E4W3B | 70,141 | 65,377 | 714 | 576 | 3.22 | 0.77 | 728.80 | 754.16 | 99.70 |
| E10W01 | 70,168 | 63,701 | 1995 | 1809 | 7.77 | 0.99 | 1996.37 | 2004.69 | 99.40 |
| E10W03 | 70,113 | 66,461 | 1030 | 852 | 2.98 | 0.57 | 1007.37 | 1035.15 | 99.60 |
| E10W1A | 70,138 | 64,424 | 1057 | 901 | 6.40 | 0.98 | 1049.68 | 1081.34 | 99.60 |
| E10W3A | 70,131 | 64,665 | 1072 | 927 | 6.38 | 0.96 | 1046.72 | 1068.99 | 99.70 |
| E10W1B | 73,679 | 69,889 | 851 | 701 | 4.55 | 0.84 | 805.33 | 851.42 | 99.70 |
| E10W3B | 70,130 | 64,983 | 1136 | 1004 | 6.61 | 0.97 | 1157.46 | 1166.40 | 99.60 |
| E18W01 | 70,162 | 65,309 | 1349 | 1169 | 5.74 | 0.91 | 1323.50 | 1367.93 | 99.50 |
| E18W03 | 70,195 | 65,172 | 1833 | 1633 | 6.20 | 0.92 | 1804.13 | 1840.30 | 99.40 |
| E18W1A | 70,053 | 65,817 | 940 | 795 | 5.13 | 0.86 | 952.96 | 984.06 | 99.60 |
| E18W3A | 70,081 | 65,481 | 1114 | 921 | 5.31 | 0.92 | 1116.03 | 1167.87 | 99.50 |
| E18W1B | 70,213 | 64,520 | 1413 | 1227 | 7.00 | 0.97 | 1397.65 | 1435.94 | 99.50 |
| E18W3B | 70,134 | 66,921 | 704 | 602 | 5.09 | 0.89 | 714.13 | 735.06 | 99.70 |
| Total | 2,110,195 | 1,851,366 | 32,266 | 27,543 | |||||
| Mean | 70,340 | 61,712 | 1076 | 918 | 4.77 | 0.84 | 1056.75 | 1091.62 | 99.62 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Man, B.; Xiang, X.; Zhang, J.; Cheng, G.; Zhang, C.; Luo, Y.; Qin, Y. Keystone Taxa and Predictive Functional Analysis of Sphagnum palustre Tank Microbiomes in Erxianyan Peatland, Central China. Biology 2022, 11, 1436. https://doi.org/10.3390/biology11101436
Man B, Xiang X, Zhang J, Cheng G, Zhang C, Luo Y, Qin Y. Keystone Taxa and Predictive Functional Analysis of Sphagnum palustre Tank Microbiomes in Erxianyan Peatland, Central China. Biology. 2022; 11(10):1436. https://doi.org/10.3390/biology11101436
Chicago/Turabian StyleMan, Baiying, Xing Xiang, Junzhong Zhang, Gang Cheng, Chao Zhang, Yang Luo, and Yangmin Qin. 2022. "Keystone Taxa and Predictive Functional Analysis of Sphagnum palustre Tank Microbiomes in Erxianyan Peatland, Central China" Biology 11, no. 10: 1436. https://doi.org/10.3390/biology11101436
APA StyleMan, B., Xiang, X., Zhang, J., Cheng, G., Zhang, C., Luo, Y., & Qin, Y. (2022). Keystone Taxa and Predictive Functional Analysis of Sphagnum palustre Tank Microbiomes in Erxianyan Peatland, Central China. Biology, 11(10), 1436. https://doi.org/10.3390/biology11101436