
Zinc Supplementation Enhances the Pro-Death Function of UPR in Lymphoma Cells Exposed to Radiation

 ,
,  , , and
, , and
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Cultures and Treatments
2.2. Cell Viability
2.3. Apoptosis Analysis
2.4. siRNA Interference
2.5. Western Blot Analysis
2.6. Antibodies
2.7. Densitometric Analysis
3. Results
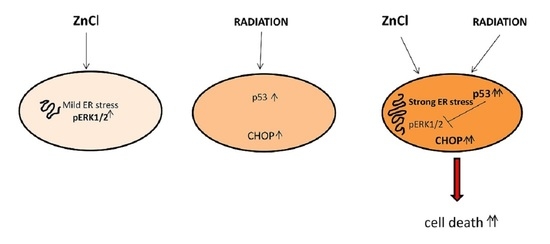
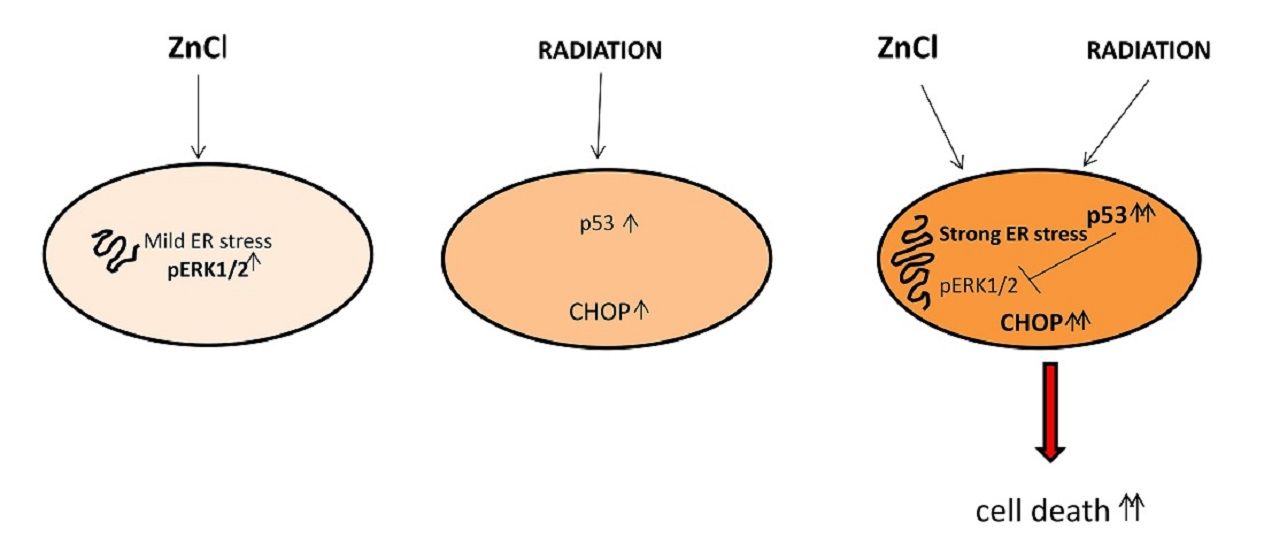
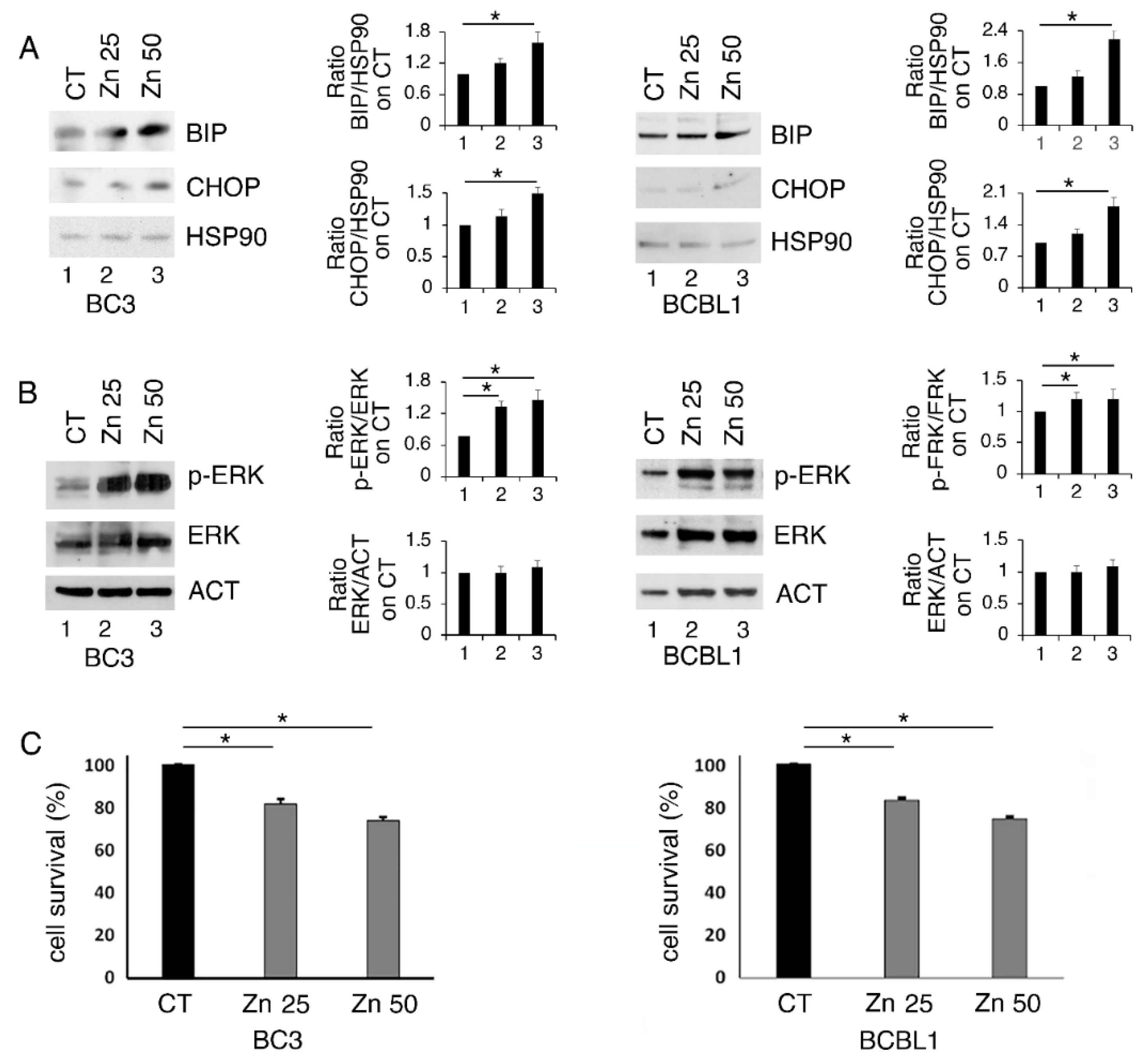
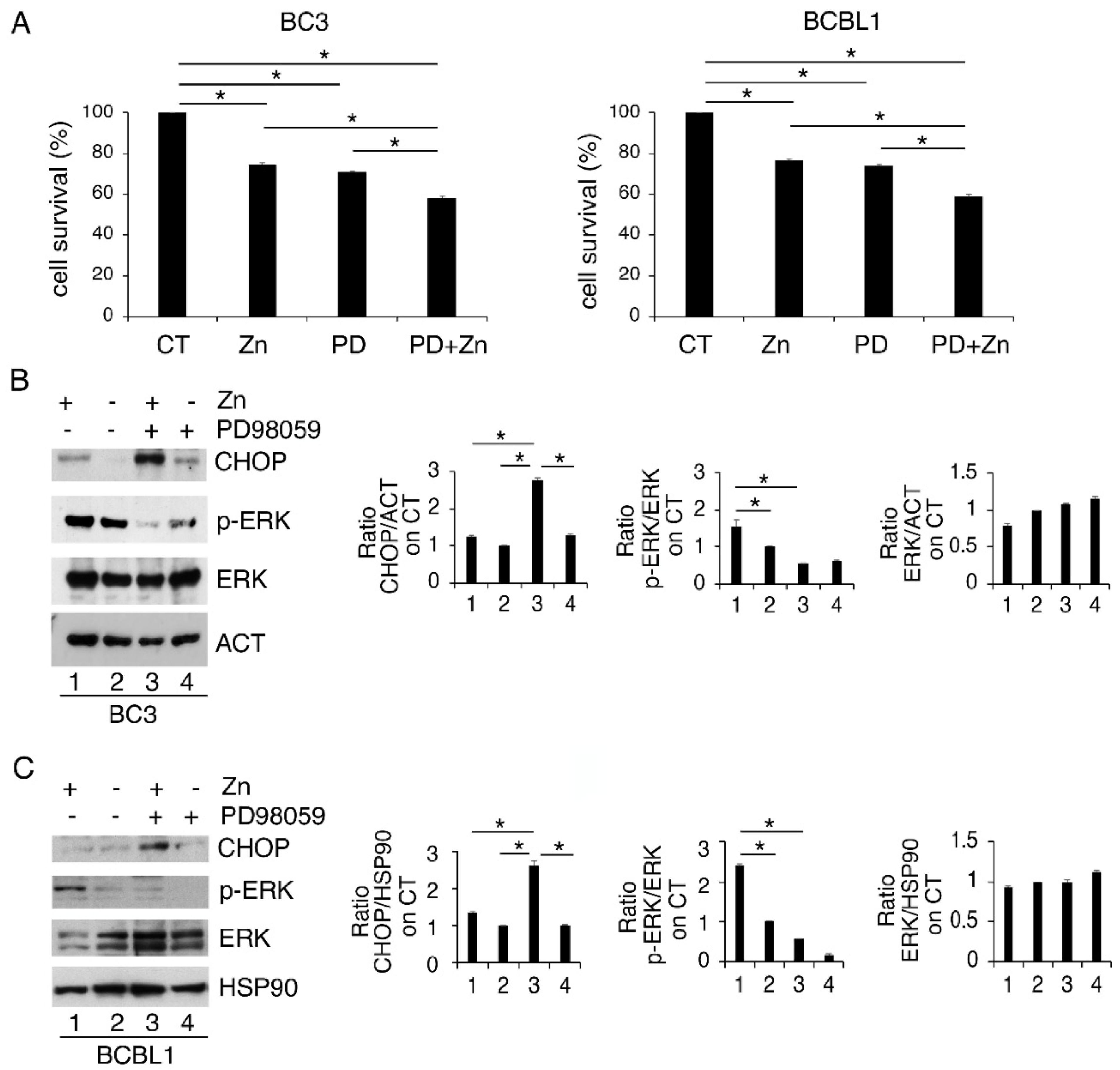
3.1. Zinc Triggers Mild ER Stress, Increases ERK1/2 Phosphorylation and Slightly Affects Cell Survival in PEL Cells
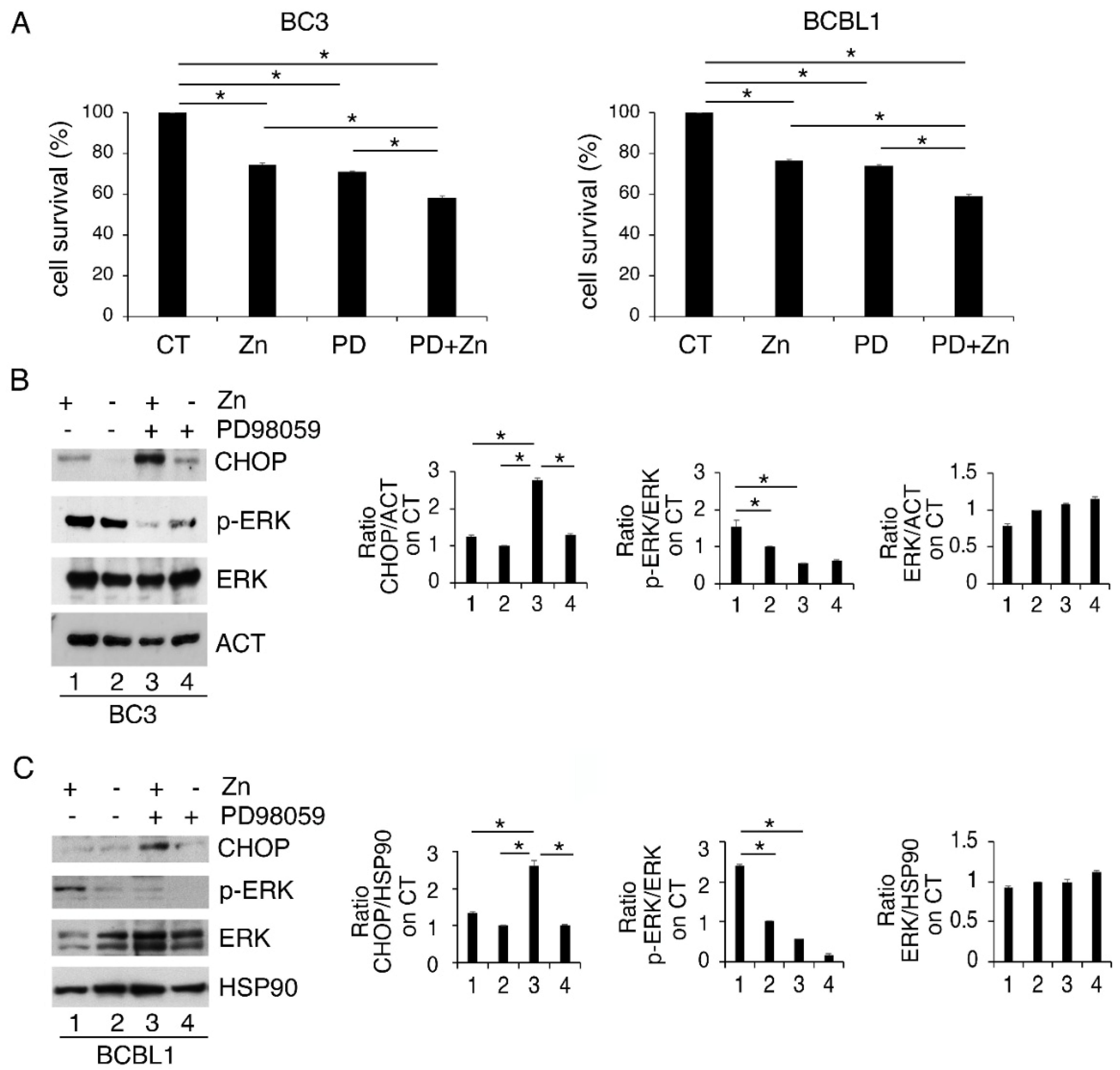
3.2. ERK1/2 Inhibition Upregulates CHOP and Reduces the Survival of Zinc-Treated PEL Cells
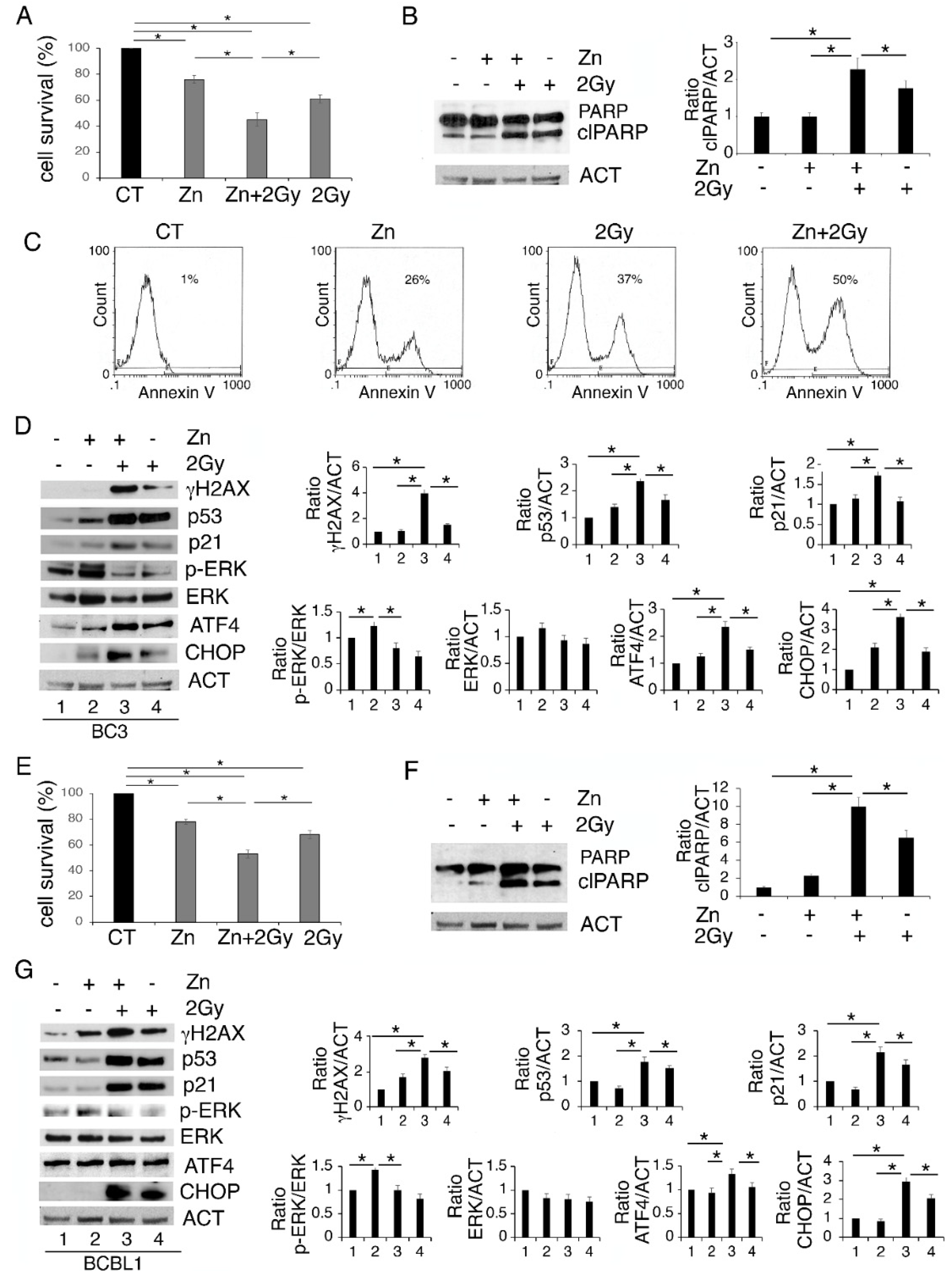
3.3. Zinc in Combination with Low-Dose Radiation Enhances DNA Damage and p53 Activation, Reduces ERK1/2 Phosphorylation, Upregulates ATF4/CHOP Axis and Further Impairs PEL Cell Survival
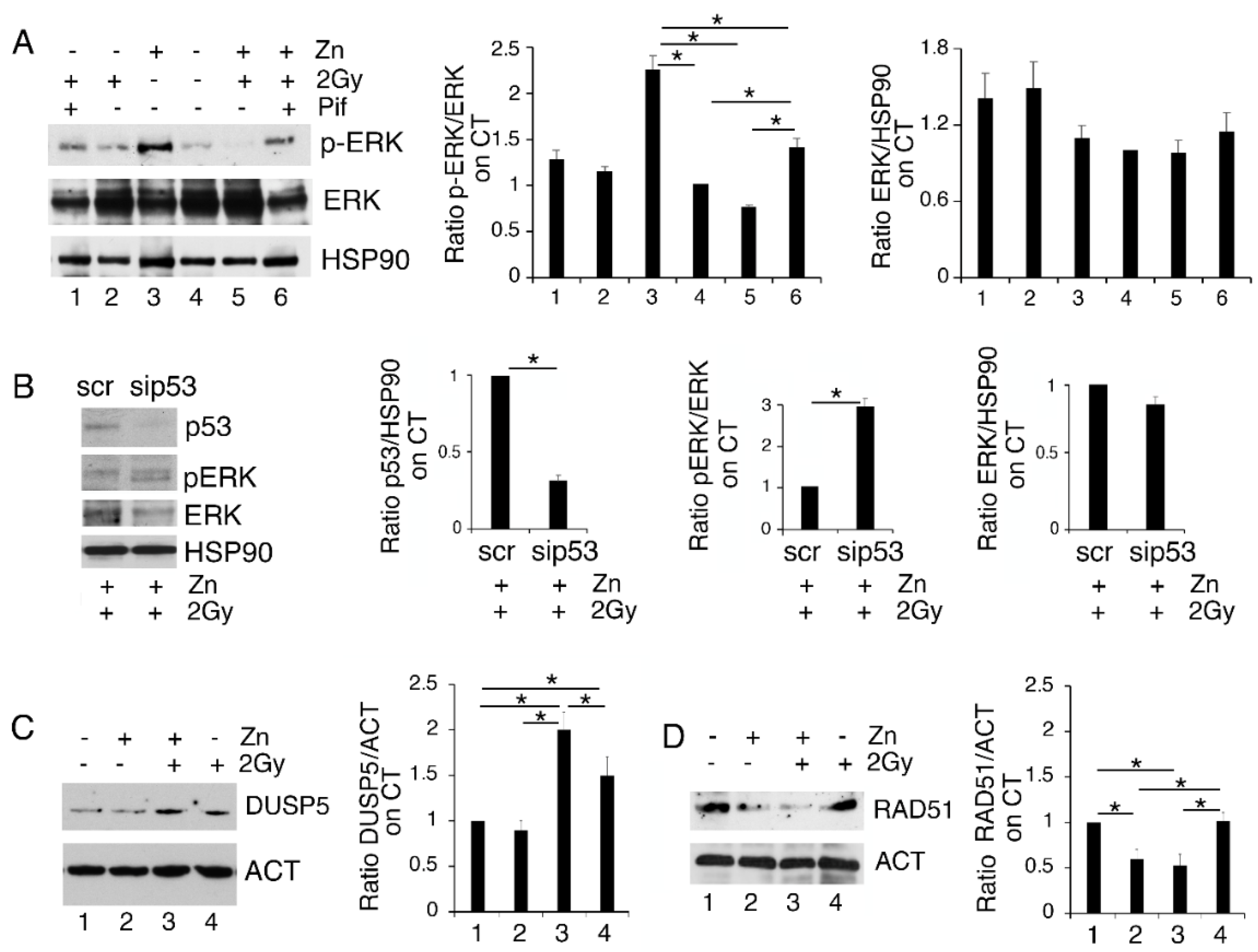
3.4. p53 Activation Contributes to ERK1/2 Dephosphorylation in PEL Cells Undergoing Zinc/Radiation Treatment
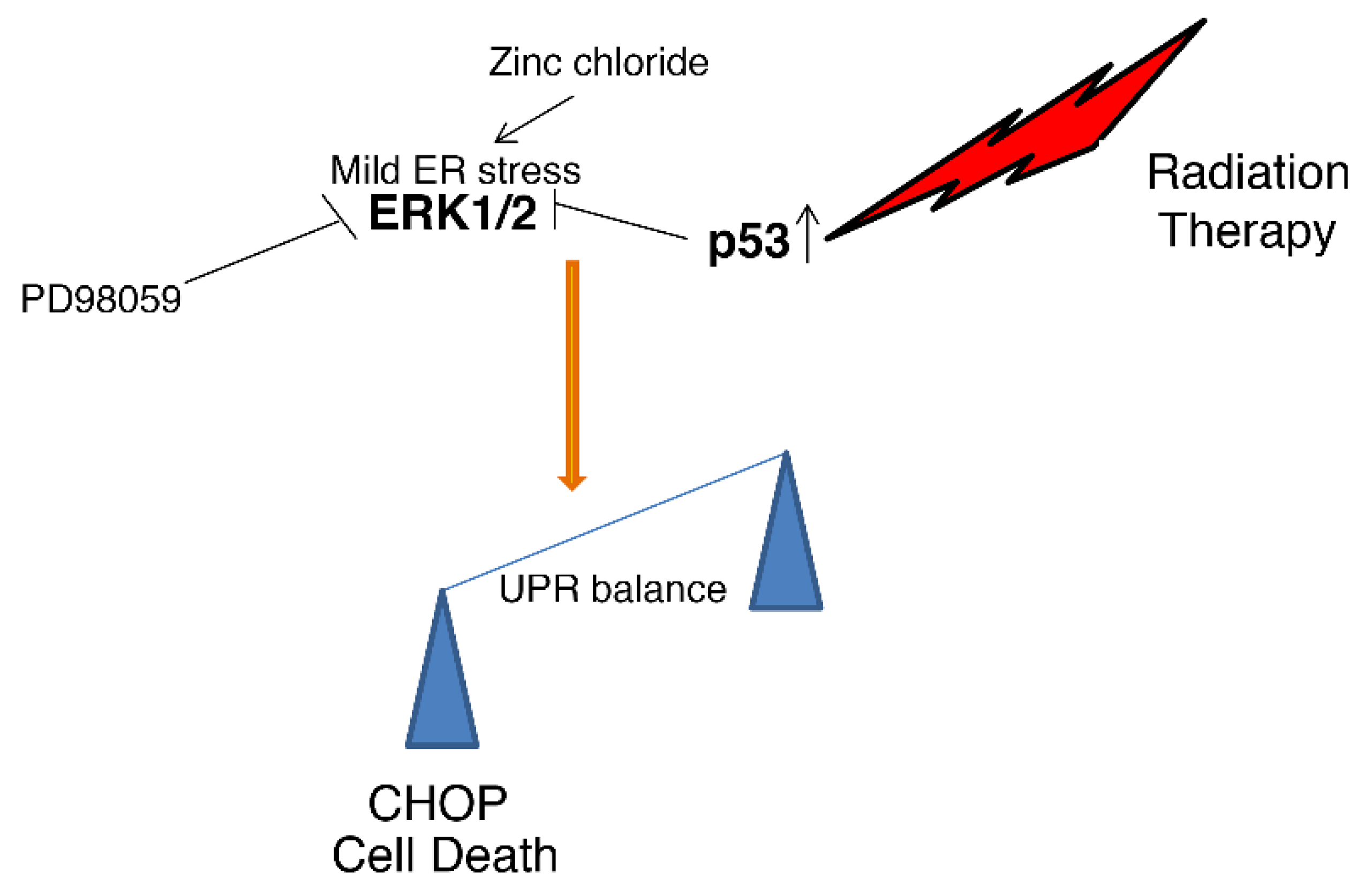
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Puca, R.; Nardinocchi, L.; Porru, M.; Simon, A.J.; Rechavi, G.; Leonetti, C.; Givol, D.; D’Orazi, G. Restoring p53 active conformation by zinc increases the response of mutant p53 tumor cells to anticancer drugs. Cell Cycle 2011, 10, 1679–1689. [Google Scholar] [CrossRef]
- Margalit, O.; Simon, A.J.; Yakubov, E.; Puca, R.; Yosepovich, A.; Avivi, C.; Jacob-Hirsch, J.; Gelernter, I.; Harmelin, A.; Barshack, I.; et al. Zinc supplementation augments in vivo antitumor effect of chemotherapy by restoring p53 function. Int. J. Cancer 2012, 131, E562–E568. [Google Scholar] [CrossRef] [PubMed]
- Cirone, M.; Garufi, A.; Di Renzo, L.; Granato, M.; Faggioni, A.; D’Orazi, G. Zinc supplementation is required for the cytotoxic and immunogenic effects of chemotherapy in chemoresistant p53-functionally deficient cells. Oncoimmunology 2013, 2, e26198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garufi, A.; Ubertini, V.; Mancini, F.; D’Orazi, V.; Baldari, S.; Moretti, F.; Bossi, G.; D’Orazi, G. The beneficial effect of Zinc(II) on low-dose chemotherapeutic sensitivity involves p53 activation in wild-type p53-carrying colorectal cancer cells. J. Exp. Clin. Cancer Res. 2015, 34, 87. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2018, 9, 3083. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Quiroz, M.; Blondel, A.; Sagredo, A.; Hetz, C.; Chevet, E.; Pedeux, R. When Endoplasmic Reticulum Proteostasis Meets the DNA Damage Response. Trends Cell Biol. 2020, 30, 881–891. [Google Scholar] [CrossRef]
- Zhan, Q.; Carrier, F.; Fornace, A.J., Jr. Induction of cellular p53 activity by DNA-damaging agents and growth arrest. Mol. Cell. Biol. 1993, 13, 4242–4250. [Google Scholar] [CrossRef]
- Lin, W.C.; Chuang, Y.C.; Chang, Y.S.; Lai, M.D.; Teng, Y.N.; Su, I.J.; Wang, C.C.; Lee, K.H.; Hung, J.H. Endoplasmic reticulum stress stimulates p53 expression through NF-kappaB activation. PLoS ONE 2012, 7, e39120. [Google Scholar] [CrossRef] [Green Version]
- Fusee, L.T.S.; Marin, M.; Fahraeus, R.; Lopez, I. Alternative Mechanisms of p53 Action During the Unfolded Protein Response. Cancers 2020, 12, 401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Tang, X.Y.; Liu, H.; Jiang, Z.W. Inhibition of mitogen-activated protein kinase signaling pathway sensitizes breast cancer cells to endoplasmic reticulum stress-induced apoptosis. Oncol. Rep. 2016, 35, 2113–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darling, N.J.; Cook, S.J. The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim. Biophys. Acta 2014, 1843, 2150–2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sklar, M.D. The ras oncogenes increase the intrinsic resistance of NIH 3T3 cells to ionizing radiation. Science 1988, 239, 645–647. [Google Scholar] [CrossRef]
- Munshi, A.; Ramesh, R. Mitogen-activated protein kinases and their role in radiation response. Genes Cancer 2013, 4, 401–408. [Google Scholar] [CrossRef]
- El-Deiry, W.S.; Harper, J.W.; O’Connor, P.M.; Velculescu, V.E.; Canman, C.E.; Jackman, J.; Pietenpol, J.A.; Burrell, M.; Hill, D.E.; Wang, Y.; et al. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 1994, 54, 1169–1174. [Google Scholar]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [Green Version]
- Santarelli, R.; Pompili, C.; Gilardini Montani, M.S.; Romeo, M.A.; Gonnella, R.; D’Orazi, G.; Cirone, M. Lovastatin reduces PEL cell survival by phosphorylating ERK1/2 that blocks the autophagic flux and engages a cross-talk with p53 to activate p21. IUBMB Life 2021, 73, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Lei, T.; Guo, P.; Yu, J.; Xu, Q.; Luo, Y.; Ke, R.; Huang, D. Mechanisms shaping the role of ERK1/2 in cellular senescence (Review). Mol. Med. Rep. 2019, 19, 759–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persons, D.L.; Yazlovitskaya, E.M.; Pelling, J.C. Effect of extracellular signal-regulated kinase on p53 accumulation in response to cisplatin. J. Biol. Chem. 2000, 275, 35778–35785. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.S. The functional interactions between the p53 and MAPK signaling pathways. Cancer Biol. Ther. 2004, 3, 156–161. [Google Scholar] [CrossRef] [Green Version]
- Ueda, K.; Arakawa, H.; Nakamura, Y. Dual-specificity phosphatase 5 (DUSP5) as a direct transcriptional target of tumor suppressor p53. Oncogene 2003, 22, 5586–5591. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Lee, S.H.; Yoon, M.H.; Park, B.J. A new p53 target gene, RKIP, is essential for DNA damage-induced cellular senescence and suppression of ERK activation. Neoplasia 2013, 15, 727–737. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.B.; Rahemtullah, A.; Hochberg, E. Primary effusion lymphoma. Oncologist 2007, 12, 569–576. [Google Scholar] [CrossRef] [Green Version]
- Gonnella, R.; Gilardini Montani, M.S.; Guttieri, L.; Romeo, M.A.; Santarelli, R.; Cirone, M. IRE1 Alpha/XBP1 Axis Sustains Primary Effusion Lymphoma Cell Survival by Promoting Cytokine Release and STAT3 Activation. Biomedicines 2021, 9, 118. [Google Scholar] [CrossRef]
- Arena, A.; Gilardini Montani, M.S.; Romeo, M.A.; Benedetti, R.; Gaeta, A.; Cirone, M. DNA damage triggers an interplay between wtp53 and c-Myc affecting lymphoma cell proliferation and Kaposi sarcoma herpesvirus replication. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1869, 119168. [Google Scholar] [CrossRef]
- Azriel-Tamir, H.; Sharir, H.; Schwartz, B.; Hershfinkel, M. Extracellular zinc triggers ERK-dependent activation of Na+/H+ exchange in colonocytes mediated by the zinc-sensing receptor. J. Biol. Chem. 2004, 279, 51804–51816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.K.; Chakraborty, P.K.; Roussa, E.; Wolff, N.A.; Thevenod, F. ERK1/2-dependent bestrophin-3 expression prevents ER-stress-induced cell death in renal epithelial cells by reducing CHOP. Biochim. Biophys. Acta 2012, 1823, 1864–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolland, H.; Ma, T.S.; Ramlee, S.; Ramadan, K.; Hammond, E.M. Links between the unfolded protein response and the DNA damage response in hypoxia: A systematic review. Biochem. Soc. Trans. 2021, 49, 1251–1263. [Google Scholar] [CrossRef] [PubMed]
- Arias-Lopez, C.; Lazaro-Trueba, I.; Kerr, P.; Lord, C.J.; Dexter, T.; Iravani, M.; Ashworth, A.; Silva, A. p53 modulates homologous recombination by transcriptional regulation of the RAD51 gene. EMBO Rep. 2006, 7, 219–224. [Google Scholar] [CrossRef] [Green Version]
- Ko, J.C.; Su, Y.J.; Lin, S.T.; Jhan, J.Y.; Ciou, S.C.; Cheng, C.M.; Lin, Y.W. Suppression of ERCC1 and Rad51 expression through ERK1/2 inactivation is essential in emodin-mediated cytotoxicity in human non-small cell lung cancer cells. Biochem. Pharmacol. 2010, 79, 655–664. [Google Scholar] [CrossRef]
- Yamamori, T.; Meike, S.; Nagane, M.; Yasui, H.; Inanami, O. ER stress suppresses DNA double-strand break repair and sensitizes tumor cells to ionizing radiation by stimulating proteasomal degradation of Rad51. FEBS Lett. 2013, 587, 3348–3353. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.J.; Chen, S.; Wu, P.; Hu, C.S.; Thorne, R.F.; Luo, C.M.; Hersey, P.; Zhang, X.D. Inhibition of MEK blocks GRP78 up-regulation and enhances apoptosis induced by ER stress in gastric cancer cells. Cancer Lett. 2009, 274, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Yan, J.; Tang, D. Extracellular signal-regulated kinases modulate DNA damage response—A contributing factor to using MEK inhibitors in cancer therapy. Curr. Med. Chem. 2011, 18, 5476–5482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dufey, E.; Bravo-San Pedro, J.M.; Eggers, C.; Gonzalez-Quiroz, M.; Urra, H.; Sagredo, A.I.; Sepulveda, D.; Pihan, P.; Carreras-Sureda, A.; Hazari, Y.; et al. Genotoxic stress triggers the activation of IRE1alpha-dependent RNA decay to modulate the DNA damage response. Nat. Commun. 2020, 11, 2401. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonnella, R.; Guttieri, L.; Gilardini Montani, M.S.; Santarelli, R.; Bassetti, E.; D’Orazi, G.; Cirone, M. Zinc Supplementation Enhances the Pro-Death Function of UPR in Lymphoma Cells Exposed to Radiation. Biology 2022, 11, 132. https://doi.org/10.3390/biology11010132
Gonnella R, Guttieri L, Gilardini Montani MS, Santarelli R, Bassetti E, D’Orazi G, Cirone M. Zinc Supplementation Enhances the Pro-Death Function of UPR in Lymphoma Cells Exposed to Radiation. Biology. 2022; 11(1):132. https://doi.org/10.3390/biology11010132
Chicago/Turabian StyleGonnella, Roberta, Luisa Guttieri, Maria Saveria Gilardini Montani, Roberta Santarelli, Erica Bassetti, Gabriella D’Orazi, and Mara Cirone. 2022. "Zinc Supplementation Enhances the Pro-Death Function of UPR in Lymphoma Cells Exposed to Radiation" Biology 11, no. 1: 132. https://doi.org/10.3390/biology11010132
APA StyleGonnella, R., Guttieri, L., Gilardini Montani, M. S., Santarelli, R., Bassetti, E., D’Orazi, G., & Cirone, M. (2022). Zinc Supplementation Enhances the Pro-Death Function of UPR in Lymphoma Cells Exposed to Radiation. Biology, 11(1), 132. https://doi.org/10.3390/biology11010132