
Transcriptome Analysis of Two Near-Isogenic Lines with Different NUE under Normal Nitrogen Conditions in Wheat


 ,
, 
 ,
,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Experimental Design
2.2. Dry Matter and Amount of Nitrogen
2.3. Net Photosynthetic Rate (Pn)
2.4. GS and GOGAT Activity Measurement
2.5. RNA Extraction and Illumina Sequencing
2.6. Transcriptome Sequencing Analysis
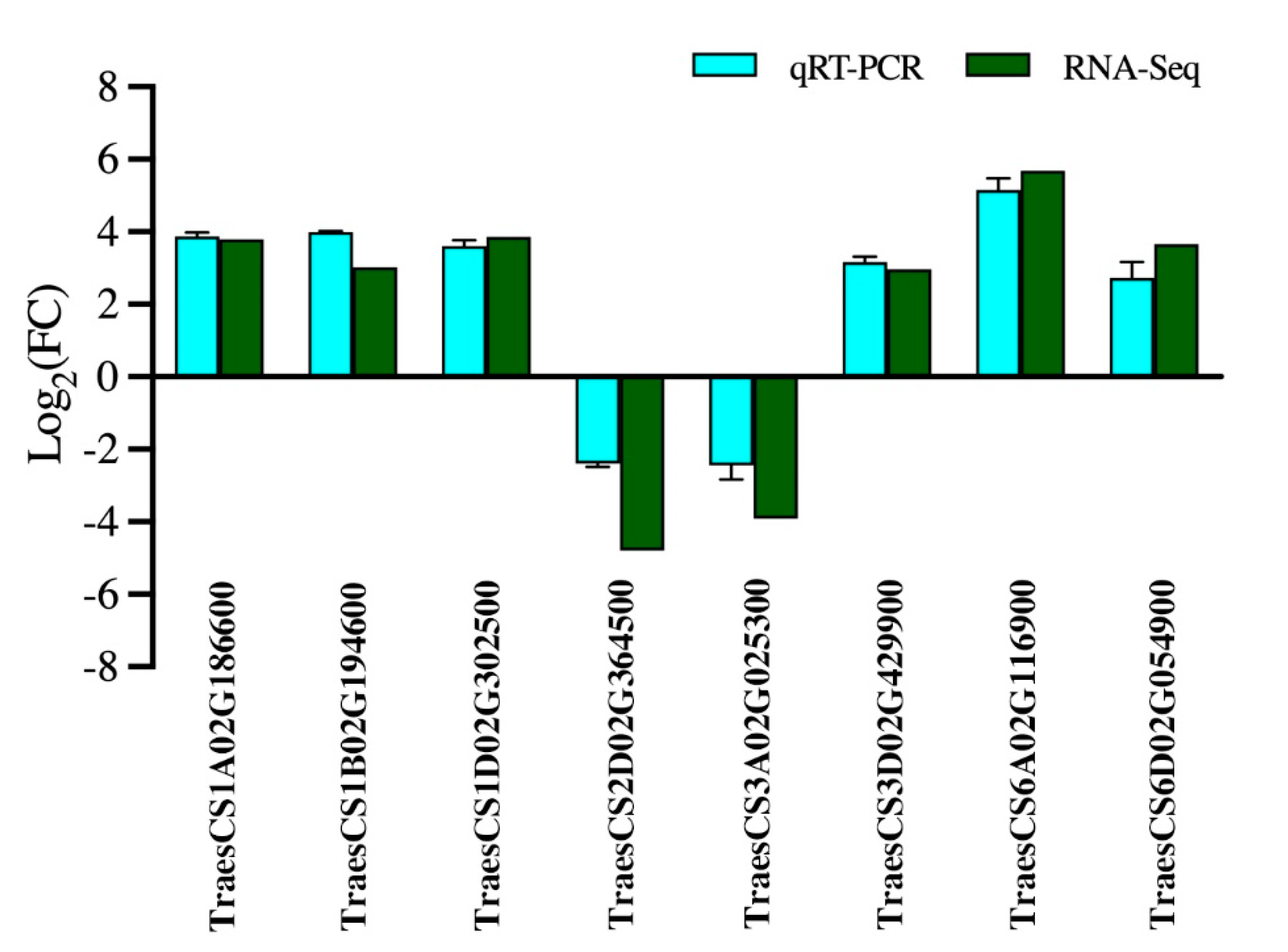
2.7. Quantitative Real-Time PCR (qRT-PCR) Analysis
2.8. Statistical Analysis
3. Results
3.1. Effect of Normal Nitrogen on Agronomic Traits of Two Wheat NILs
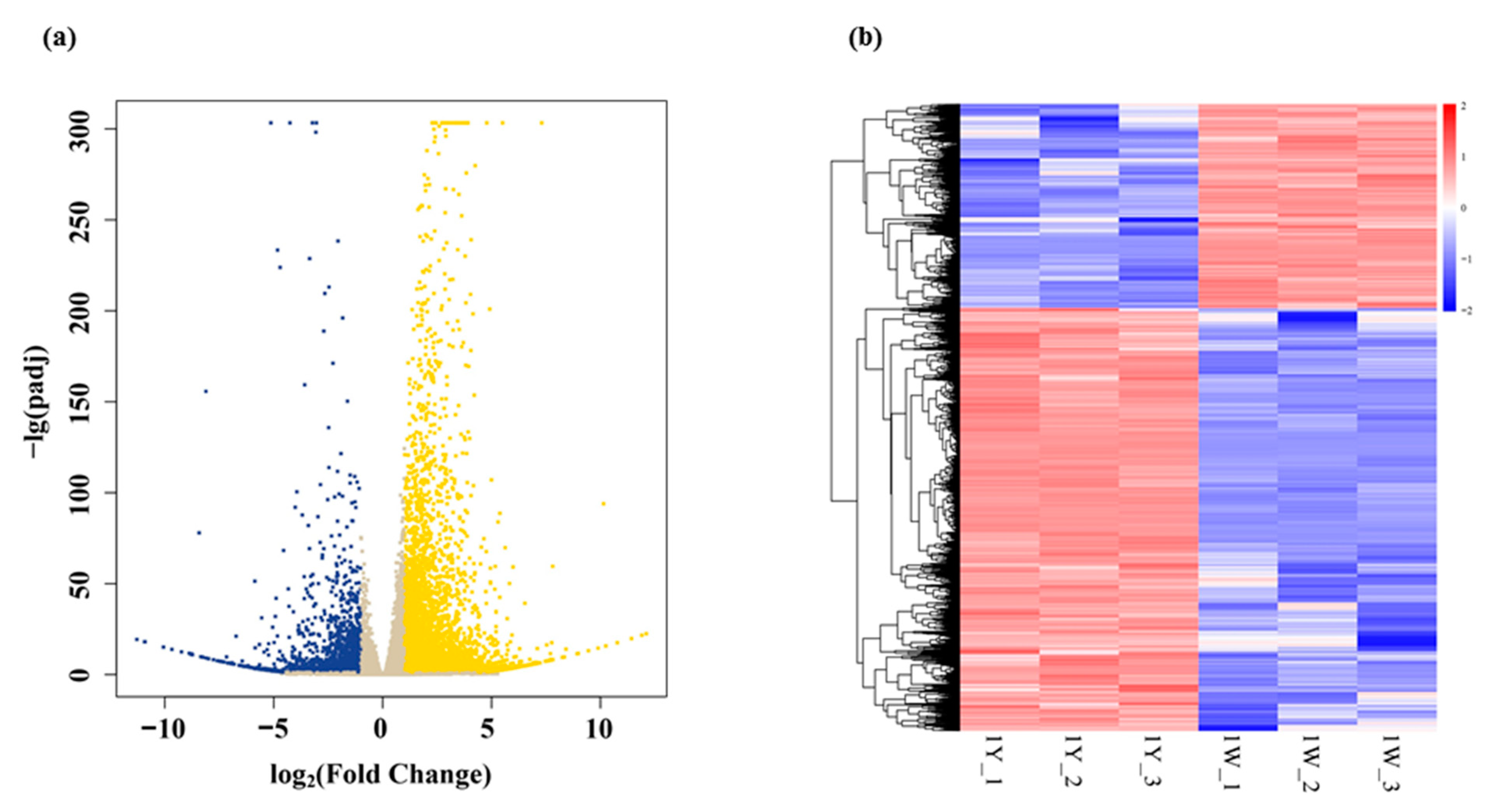
3.2. Transcriptome Sequencing Results
3.3. Analysis of Differentially Expressed Genes (DEGs)
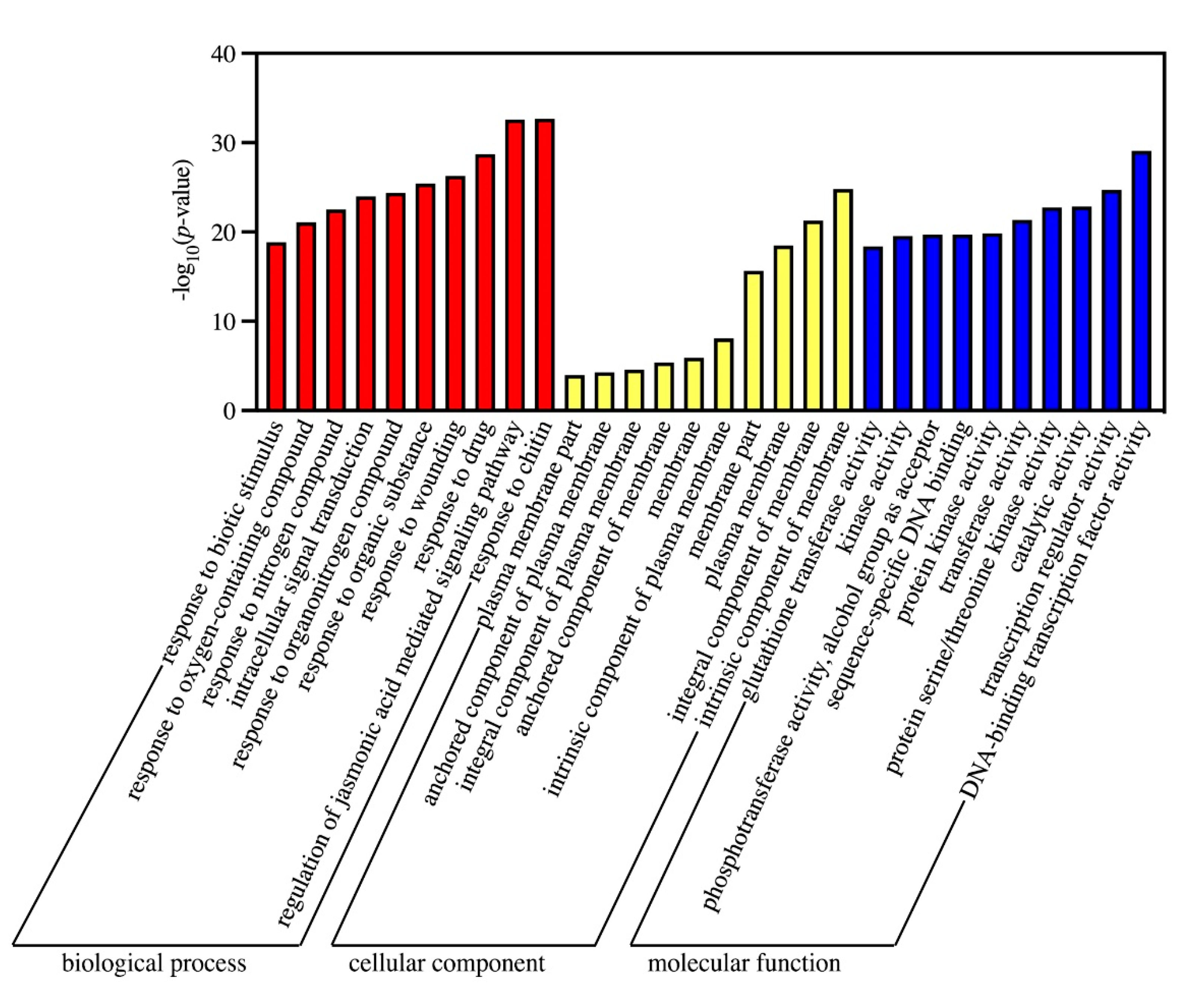
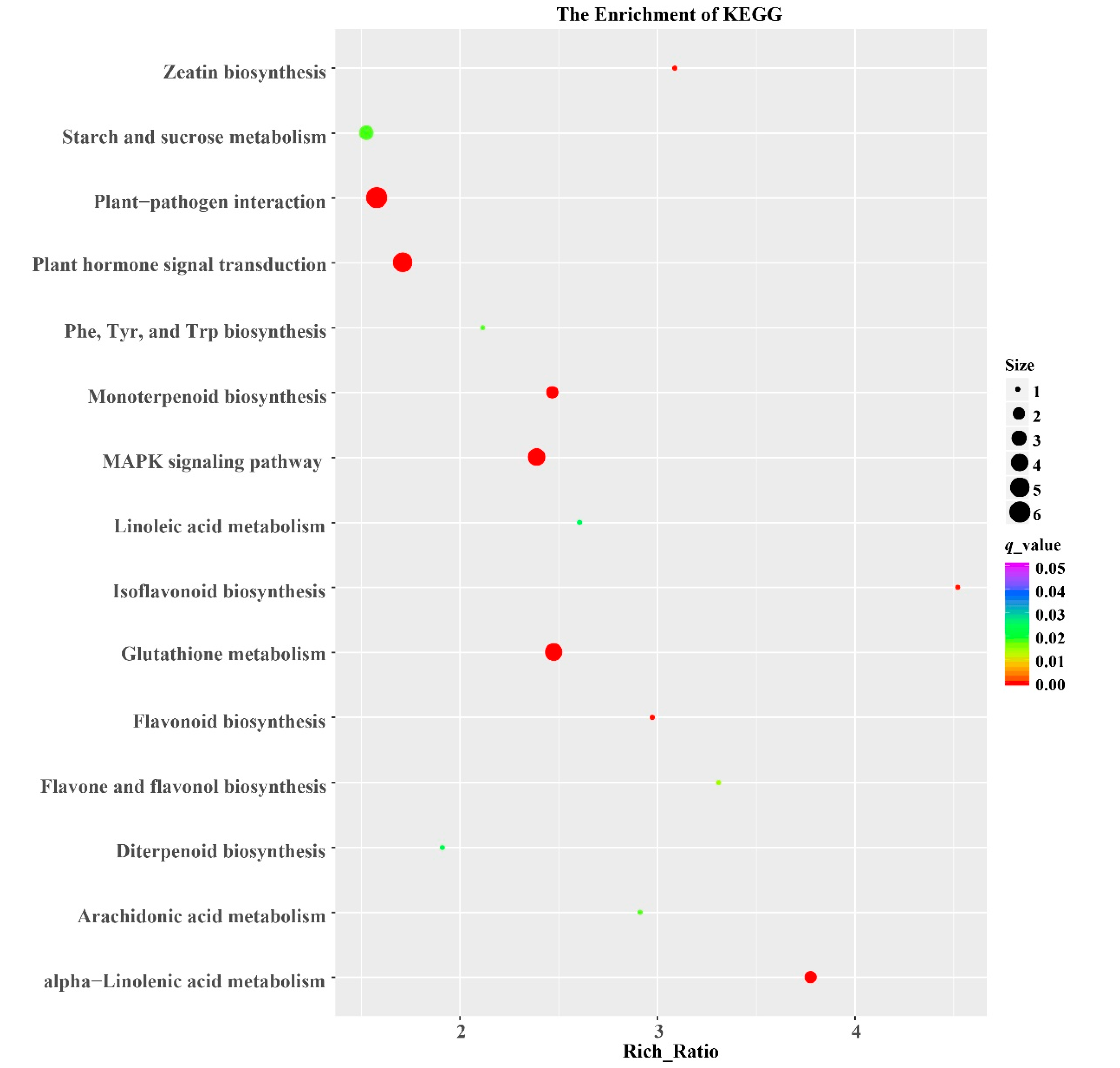
3.4. GO and KEGG Analysis of DEGs
3.5. Expression Analysis of Genes Associated with Nitrogen Metabolism
3.6. Expression Profiling of Genes Related to Carbon Metabolism
3.7. Regulation of Differential Gene Expression by TFs
3.8. Expression of GS, GOGAT, and GDH Gene Families in Wheat Seedlings
4. Discussion
4.1. Nitrogen Metabolism
4.2. Carbon Metabolism
4.3. Transcription Factors (TFs)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weber, K.; Burow, M. Nitrogen-essential macronutrient and signal controlling flowering time. Physiol. Plant 2018, 162, 251–260. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Fan, X.; Miller, A.J. Plant nitrogen assimilation and use efficiency. Annu. Rev. Plant Biol. 2012, 63, 153–182. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; de Vries, W.; Liu, X.; Hao, T.; Zeng, M.; Shen, J.; Zhang, F. Enhanced acidification in Chinese croplands as derived from element budgets in the period 1980–2010. Sci. Total Environ. 2018, 618, 1497–1505. [Google Scholar] [CrossRef] [Green Version]
- Duan, J.; Shao, Y.; He, L.; Li, X.; Hou, G.; Li, S.; Feng, W.; Zhu, Y.; Wang, Y.; Xie, Y. Optimizing nitrogen management to achieve high yield, high nitrogen efficiency and low nitrogen emission in winter wheat. Sci. Total Environ. 2019, 697, 134088. [Google Scholar] [CrossRef]
- Good, A.G.; Shrawat, A.K.; Muench, D.G. Can less yield more? Is reducing nutrient input into the environment compatible with maintaining crop production? Trends Plant Sci. 2004, 9, 597–605. [Google Scholar] [CrossRef]
- Hawkesford, M.J.; Riche, A.B. Impacts of G × E × M on nitrogen use efficiency in wheat and future prospects. Front. Plant Sci. 2020, 11, 1157. [Google Scholar] [CrossRef]
- Chen, M.; Chen, G.; Di, D.; Kronzucker, H.J.; Shi, W. Higher nitrogen use efficiency (NUE) in hybrid “super rice” links to improved morphological and physiological traits in seedling roots. J. Plant Physiol. 2020, 251, 153191. [Google Scholar] [CrossRef]
- Palenchar, P.M.; Kouranov, A.; Lejay, L.V.; Coruzzi, G.M. Genome-wide patterns of carbon and nitrogen regulation of gene expression validate the combined carbon and nitrogen (CN)-signaling hypothesis in plants. Genome Biol. 2004, 5, R91. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Guo, R.; Li, M.; Liu, Y.; Wang, X.; Fu, H.; Wang, S.; Liu, X.; Shi, L. Physiological characteristics and metabolomics reveal the tolerance mechanism to low nitrogen in Glycine soja leaves. Physiol. Plant 2020, 168, 819–834. [Google Scholar] [CrossRef]
- Zheng, Z.L. Carbon and nitrogen nutrient balance signaling in plants. Plant Signal. Behav. 2009, 4, 584–591. [Google Scholar] [CrossRef] [Green Version]
- Alisdair, N.N.; Fernie, A.R.; Stitt, M. Metabolic and signaling aspects underpinning the regulation of plant carbon nitrogen interactions. Mol. Plant 2010, 3, 973–996. [Google Scholar]
- Wang, M.; Shen, Q.; Xu, G.; Guo, S. New insight into the strategy for nitrogen metabolism in plant cells. Int. Rev. Cell Mol. Biol. 2014, 310, 1–37. [Google Scholar] [PubMed]
- Kishorekumar, R.; Bulle, M.; Wany, A.; Gupta, K.J. An overview of important enzymes involved in nitrogen assimilation of plants. Methods Mol. Biol. 2020, 2057, 1–13. [Google Scholar]
- Baslam, M.; Mitsui, T.; Sueyoshi, K.; Ohyama, T. Recent advances in carbon and nitrogen metabolism in C3 plants. Int. J. Mol. Sci. 2020, 22, 318. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.C.; Zhou, C.Z.; Burnap, R.L.; Peng, L. Carbon/nitrogen metabolic balance: Lessons from cyanobacteria. Trends Plant Sci. 2018, 23, 1116–1130. [Google Scholar] [CrossRef]
- Li, Q.; Ding, G.; Yang, N.; White, P.J.; Ye, X.; Cai, H.; Lu, J.; Shi, L.; Xu, F. Comparative genome and transcriptome analysis unravels key factors of nitrogen use efficiency in Brassica napus L. Plant Cell Environ. 2020, 43, 712–731. [Google Scholar] [CrossRef]
- Jain, M. Next-generation sequencing technologies for gene expression profiling in plants. Brief. Funct. Genomics 2012, 11, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, J.M.; Vidal, E.A.; Gutiérrez, R.A. Integration of local and systemic signaling pathways for plant N responses. Curr. Opin. Plant Biol. 2012, 15, 185–191. [Google Scholar] [CrossRef]
- Domínguez-Figueroa, J.; Carrillo, L.; Renau-Morata, B.; Yang, L.; Medina, J. The arabidopsis transcription factor CDF3 is involved in nitrogen responses and improves nitrogen use efficiency in tomato. Front. Plant Sci. 2020, 11, 601558. [Google Scholar] [CrossRef]
- Sinha, S.K.; Sevanthi, V.A.M.; Chaudhary, S.; Tyagi, P.; Venkadesan, S.; Rani, M.; Mandal, P.K. Transcriptome analysis of two rice varieties contrasting for nitrogen use efficiency under chronic n starvation reveals differences in chloroplast and starch metabolism-related genes. Genes 2018, 9, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sultana, N.; Islam, S.; Juhasz, A.; Yang, R.; She, M.; Alhabbar, Z.; Zhang, J.; Ma, W. Transcriptomic study for identification of major nitrogen stress responsive genes in australian bread wheat cultivars. Front. Genet. 2020, 11, 583785. [Google Scholar] [CrossRef]
- Wang, J.; Song, K.; Sun, L.; Qin, Q.; Sun, Y.; Pan, J.; Xue, Y. Morphological and transcriptome analysis of wheat seedlings response to low nitrogen stress. Plants 2019, 8, 98. [Google Scholar] [CrossRef] [Green Version]
- Curci, P.L.; Aiese, C.R.; Zuluaga, D.L.; Janni, M.; Sanseverino, W.; Sonnante, G. Transcriptomic response of durum wheat to nitrogen starvation. Sci. Rep. 2017, 7, 1176. [Google Scholar] [CrossRef]
- Munoz-Huerta, R.F.; Guevara-Gonzalez, R.G.; Contreras-Medina, L.M.; Torres-Pacheco, I.; Prado-Olivarez, J.; Ocampo-Velazquez, R.V. A review of methods for sensing the nitrogen status in plants: Advantages, disadvantages and recent advances. Sensors 2013, 13, 10823–10843. [Google Scholar] [CrossRef]
- Ding, J.; Liang, P.; Wu, P.; Zhu, M.; Li, C.; Zhu, X.; Gao, D.; Chen, Y.; Guo, W. Effects of waterlogging on grain yield and associated traits of historic wheat cultivars in the middle and lower reaches of the Yangtze River, China. F. Crop. Res. 2020, 246, 107695. [Google Scholar] [CrossRef]
- Ma, D.; Gao, H.; Du, C.; Li, L.; Sun, W.; Liu, S.; Wang, C.; Xie, Y.; Kang, G. Transcriptomic and metabolomics analysis of different endosperm region under nitrogen treatments. Int. J. Mol. Sci. 2019, 20, 4212. [Google Scholar] [CrossRef] [Green Version]
- Siren, J.; Valimaki, N.; Makinen, V. Indexing graphs for path queries with applications in genome research. IEEE-ACM T. Comput. Biol. Bioinform. 2014, 11, 375–388. [Google Scholar] [CrossRef]
- Jin, J.; Tian, F.; Yang, D.C.; Meng, Y.Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017, 45, D1040–D1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Cosgrove, D.J. Plant cell wall extensibility: Connecting plant cell growth with cell wall structure, mechanics, and the action of wall-modifying enzymes. J. Exp. Bot. 2016, 67, 463–476. [Google Scholar] [CrossRef]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; He, X.; Wu, D.; Zhu, B.; Cai, S.; Nadira, U.A.; Jabeen, Z.; Zhang, G. Comparative transcriptome profiling of two Tibetan wild barley genotypes in responses to low potassium. PLoS ONE 2014, 9, e100567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C (T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Becana, M.; Matamoros, M.A.; Udvardi, M.; Dalton, D.A. Recent insights into antioxidant defenses of legume root nodules. New Phytol. 2010, 188, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Mascia, M.; Sega, D.; Zamboni, A.; Varanini, Z. Nitrogen starvation differentially influences transcriptional and uptake rate profiles in roots of two maize inbred lines with different NUE. Int. J. Mol. Sci. 2019, 20, 4856. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Wang, Z.; Ma, Z.; Song, W.; Lu, W.; Xiao, K. Characterization on TaMPK14, an MAPK family gene of wheat, in modulating N-starvation response through regulating N uptake and ROS homeostasis. Plant Cell Rep. 2020, 39, 1285–1299. [Google Scholar] [CrossRef]
- Tan, L.; Gao, G.; Yu, C.; Zhu, A.; Chen, P.; Chen, K.; Chen, J.; Xiong, H. Transcriptome analysis of high-NUE (T29) and low-NUE (T13) genotypes identified different responsive patterns involved in nitrogen stress in ramie (Boehmeria nivea (L.) Gaudich). Plants 2020, 9, 767. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, A.; Dong, Q.; Wang, X.; Gui, H.; Zhang, H.; Zhang, X.; Song, M. Transcriptome analysis reveals differences in key genes and pathways regulating carbon and nitrogen metabolism in cotton genotypes under N starvation and resupply. Int. J. Mol. Sci. 2020, 21, 1500. [Google Scholar] [CrossRef] [Green Version]
- Masclaux-Daubresse, C.; Daniel-Vedele, F.; Dechorgnat, J.; Chardon, F.; Gaufichon, L.; Suzuki, A. Nitrogen uptake, assimilation and remobilization in plants: Challenges for sustainable and productive agriculture. Ann. Bot. 2010, 105, 1141–1157. [Google Scholar] [CrossRef] [Green Version]
- Tegeder, M.; Masclaux-Daubresse, C. Source and sink mechanisms of nitrogen transport and use. New Phytol. 2018, 217, 35–53. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.Y.; Cheng, Y.H.; Chen, K.E.; Tsay, Y.F. Nitrate transport, signaling, and use efficiency. Annu. Rev. Plant. Biol. 2018, 69, 85–122. [Google Scholar] [CrossRef]
- Corratge-Faillie, C.; Lacombe, B. Substrate (un) specificity of Arabidopsis NRT1/PTR FAMILY (NPF) proteins. J. Exp. Bot. 2017, 68, 3107–3113. [Google Scholar] [CrossRef]
- Zhong, C.; Bai, Z.G.; Zhu, L.F.; Zhang, J.H.; Zhu, C.Q.; Huang, J.L.; Jin, Q.Y.; Cao, X.C. Nitrogen-mediated alleviation of photosynthetic inhibition under moderate water deficit stress in rice (Oryza sativa L.). Environ. Exp. Bot. 2019, 157, 269–282. [Google Scholar] [CrossRef]
- Zhang, Z.; Xiong, S.; Wei, Y.; Meng, X.; Wang, X.; Ma, X. The role of glutamine synthetase isozymes in enhancing nitrogen use efficiency of N-efficient winter wheat. Sci. Rep. 2017, 7, 1000. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Yang, Z.; Wang, T.; Fu, Y.; Chen, Y.; Hu, B.; Yamagishi, J.; Ren, W. Environmental compensation effect and synergistic mechanism of optimized nitrogen management increasing nitrogen use efficiency in indica hybrid rice. Front. Plant Sci. 2019, 10, 245. [Google Scholar] [CrossRef]
- Miflin, B.J.; Habash, D.Z. The role of glutamine synthetase and glutamate dehydrogenase in nitrogen assimilation and possibilities for improvement in the nitrogen utilization of crops. J. Eex. Bot. 2002, 53, 979–987. [Google Scholar] [CrossRef] [Green Version]
- Mamedov, M.; Nadtochenko, V.; Semenov, A. Primary electron transfer processes in photosynthetic reaction centers from oxygenic organisms. Photosynth Res. 2015, 125, 51–63. [Google Scholar] [CrossRef]
- Amunts, A.; Toporik, H.; Borovikova, A.; Nelson, N. Structure determination and improved model of plant photosystem I. J. Biol. Chem. 2010, 285, 3478–3486. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Wang, H.; Yuan, Q.; Feng, Y. Structure and function of the photosystem supercomplexes. Front. Plant Sci. 2018, 9, 357. [Google Scholar] [CrossRef]
- Pan, X.; Ma, J.; Su, X.; Cao, P.; Chang, W.; Liu, Z.; Zhang, X.; Li, M. Structure of the maize photosystem I supercomplex with light-harvesting complexes I and II. Science 2018, 360, 1109–1113. [Google Scholar] [CrossRef] [Green Version]
- Esposito, S. Nitrogen assimilation, abiotic stress and glucose 6-phosphate dehydrogenase: The full circle of reductants. Plants 2016, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Xin, W.; Zhang, L.; Zhang, W.; Gao, J.; Yi, J.; Zhen, X.; Li, Z.; Zhao, Y.; Peng, C.; Zhao, C. An integrated analysis of the rice transcriptome and metabolome reveals differential regulation of carbon and nitrogen metabolism in response to nitrogen availability. Int. J. Mol. Sci. 2019, 20, 2349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, X.; Zeng, J.; Ye, L.; Chen, G.; Han, Z.; Shah, J.M.; Zhang, G. Transcriptome profiling analysis for two Tibetan wild barley genotypes in responses to low nitrogen. BMC Plant Biol. 2016, 16, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beleggia, R.; Omranian, N.; Holtz, Y.; Gioia, T.; Fiorani, F.; Nigro, F.M.; Pecchioni, N.; De Vita, P.; Schurr, U.; David, J.L.; et al. Comparative analysis based on transcriptomics and metabolomics data reveal differences between emmer and durum wheat in response to nitrogen starvation. Int. J. Mol. Sci. 2021, 22, 4790. [Google Scholar] [CrossRef]
- Hodges, M. Enzyme redundancy and the importance of 2-oxoglutarate in plant ammonium assimilation. J. Exp. Bot. 2002, 53, 905–916. [Google Scholar] [CrossRef] [Green Version]
- Ng, D.W.; Abeysinghe, J.K.; Kamali, M. Regulating the regulators: The control of transcription factors in plant defense signaling. Int. J. Mol. Sci. 2018, 19, 3737. [Google Scholar] [CrossRef] [Green Version]
- Kong, L.; Zhang, Y.; Du, W.; Xia, H.; Fan, S.; Zhang, B. Signaling responses to N starvation: Focusing on wheat and filling the putative gaps with findings obtained in other plants. A review. Front. Plant Sci. 2021, 12, 656696. [Google Scholar] [CrossRef]
- He, X.; Qu, B.; Li, W.; Zhao, X.; Teng, W.; Ma, W.; Ren, Y.; Li, B.; Li, Z.; Tong, Y. The nitrate-inducible nac transcription factor tanac2-5a controls nitrate response and increases wheat yield. Plant Physiol. 2015, 169, 1991–2005. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Xu, T.; Yin, Z.; Wu, W.; Geng, H.; Li, L.; Yang, M.; Cai, H.; Lian, X. Overexpression of OsMYB305 in rice enhances the nitrogen uptake under low-nitrogen condition. Front. Plant Sci. 2020, 11, 369. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NILs | Grain Yield (g plant−1) | Total N Accumulation (mg plant−1) | NUE (%) |
|---|---|---|---|
| 1Y | 9.43 ± 0.45 a | 231.09 ± 15.57 a | 49.33 ± 3.23 a |
| 1W | 7.72 ± 0.61 b | 157.73 ± 10.07 b | 33.41 ± 2.11 b |
| Group | Gene ID | Log2 (1Y/1W) | Description |
|---|---|---|---|
| NRT | TraesCS1B02G038700 | 1.24 | Protein NRT1/PTR FAMILY 6.2 |
| TraesCS1D02G147400 | −1.05 | Protein NRT1/PTR FAMILY 5.2 | |
| TraesCS1D02G269700 | 1.02 | Protein NRT1/PTR FAMILY 5.10 | |
| TraesCS3A02G081100 | −1.39 | Protein NRT1/PTR FAMILY 6.1 | |
| TraesCS3A02G418700 | 2.93 | Protein NRT1/PTR FAMILY 2.13 | |
| TraesCS3D02G414300 | 4.09 | Protein NRT1/PTR FAMILY 2.13 | |
| TraesCS4A02G225400 | −2.07 | Protein NRT1/PTR FAMILY 4.3 | |
| TraesCS5D02G419200 | −1.18 | Protein NRT1/PTR FAMILY 6.4 | |
| TraesCS7A02G365100 | 1.07 | Protein NRT1/PTR FAMILY 4.6 | |
| TraesCS7B02G201900 | −1.12 | Protein NRT1/PTR FAMILY 6.3 | |
| TraesCS7B02G262200 | 1.83 | Protein NRT1/PTR FAMILY 4.6 | |
| TraesCS7D02G297000 | −1.15 | Protein NRT1/PTR FAMILY 6.3 | |
| AMT | TraesCS1D02G296600 | 2.24 | Ammonium transporter 2 member 1 |
| TraesCS4A02G352900 | 2.74 | Ammonium transporter 3 member 2 | |
| TraesCS5B02G520200 | 1.71 | Ammonium transporter 3 member 2 | |
| TraesCS5D02G519400 | 2.54 | Ammonium transporter 3 member 2 | |
| GS | TraesCS6A02G298100 | 1.11 | Glutamine synthetase |
| TraesCS6B02G327500 | 1.08 | Glutamine synthetase | |
| GOGAT | TraesCS3D02G266400 | 1.87 | Glutamate synthase 1 [NADH], chloroplastic |
| GDH | TraesCS2A02G389900 | −1.45 | Glutamate dehydrogenase 2, mitochondrial |
| Group | Gene | Log2 (1Y/1W) | KEGG: Description |
|---|---|---|---|
| photosynthesis | TraesCS2A02G296400 | 2.68 | psaO; photosystem I |
| TraesCS2B02G312600 | 2.20 | psaO; photosystem I | |
| TraesCS2D02G294300 | 2.22 | psaO; photosystem I | |
| TraesCS2B02G395900 | 1.46 | psbQ; photosystem II | |
| TraesCS2D02G220800 | 1.18 | psbQ; photosystem II | |
| TraesCS3D02G366600 | −3.79 | petF; ferredoxin | |
| photosynthesis-antenna proteins | TraesCS6A02G094200 | 5.90 | Lhcb1; chlorophyll a/b binding protein 1 |
| TraesCS7A02G276400 | 3.43 | Lhcb1; chlorophyll a/b binding protein 1 | |
| TraesCS7D02G276300 | 4.21 | Lhcb1; chlorophyll a/b binding protein 1 | |
| Glycolysis | TraesCS1A02G122800 | 1.62 | hexokinase |
| TraesCS3B02G525400 | 1.41 | hexokinase | |
| TraesCS3D02G475600 | 1.87 | hexokinase | |
| TraesCS1D02G346200 | 1.05 | 6-phosphofructokinase | |
| TraesCS3B02G126400 | 1.94 | 6-phosphofructokinase | |
| TraesCS3B02G311900 | 1.04 | 6-phosphofructokinase | |
| TraesCS7B02G086800 | 5.07 | phosphoglycerate mutase | |
| TraesCS7D02G071200 | 1.44 | enolase | |
| TraesCS4D02G018900 | 3.69 | pyruvate kinase | |
| PPP | TraesCS3D02G491400 | 1.28 | 6-phosphogluconate dehydrogenase |
| TCA | TraesCS2B02G094300 | 1.19 | α-oxoglutarate dehydrogenase |
| TraesCS1A02G122500 | 1.91 | malate dehydrogenase |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Li, F.; Ding, Y.; Ma, Q.; Yi, Y.; Zhu, M.; Ding, J.; Li, C.; Guo, W.; Zhu, X. Transcriptome Analysis of Two Near-Isogenic Lines with Different NUE under Normal Nitrogen Conditions in Wheat. Biology 2021, 10, 787. https://doi.org/10.3390/biology10080787
Zhang X, Li F, Ding Y, Ma Q, Yi Y, Zhu M, Ding J, Li C, Guo W, Zhu X. Transcriptome Analysis of Two Near-Isogenic Lines with Different NUE under Normal Nitrogen Conditions in Wheat. Biology. 2021; 10(8):787. https://doi.org/10.3390/biology10080787
Chicago/Turabian StyleZhang, Xinbo, Fujian Li, Yonggang Ding, Quan Ma, Yuan Yi, Min Zhu, Jinfeng Ding, Chunyan Li, Wenshan Guo, and Xinkai Zhu. 2021. "Transcriptome Analysis of Two Near-Isogenic Lines with Different NUE under Normal Nitrogen Conditions in Wheat" Biology 10, no. 8: 787. https://doi.org/10.3390/biology10080787
APA StyleZhang, X., Li, F., Ding, Y., Ma, Q., Yi, Y., Zhu, M., Ding, J., Li, C., Guo, W., & Zhu, X. (2021). Transcriptome Analysis of Two Near-Isogenic Lines with Different NUE under Normal Nitrogen Conditions in Wheat. Biology, 10(8), 787. https://doi.org/10.3390/biology10080787