
Expression Analysis of FGF/FGFR and FOX Family Proteins in Mucosal Tissue Obtained from Orofacial Cleft-Affected Children




Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Profile of Study Participants
2.2. Data and Sample Collection
2.3. Routine Histological Investigation
2.4. Immunohistochemistry (IHC)
2.5. Chromogenic In-Situ Hybridization (CISH)
2.6. Visualization and Statistical Analysis
3. Results
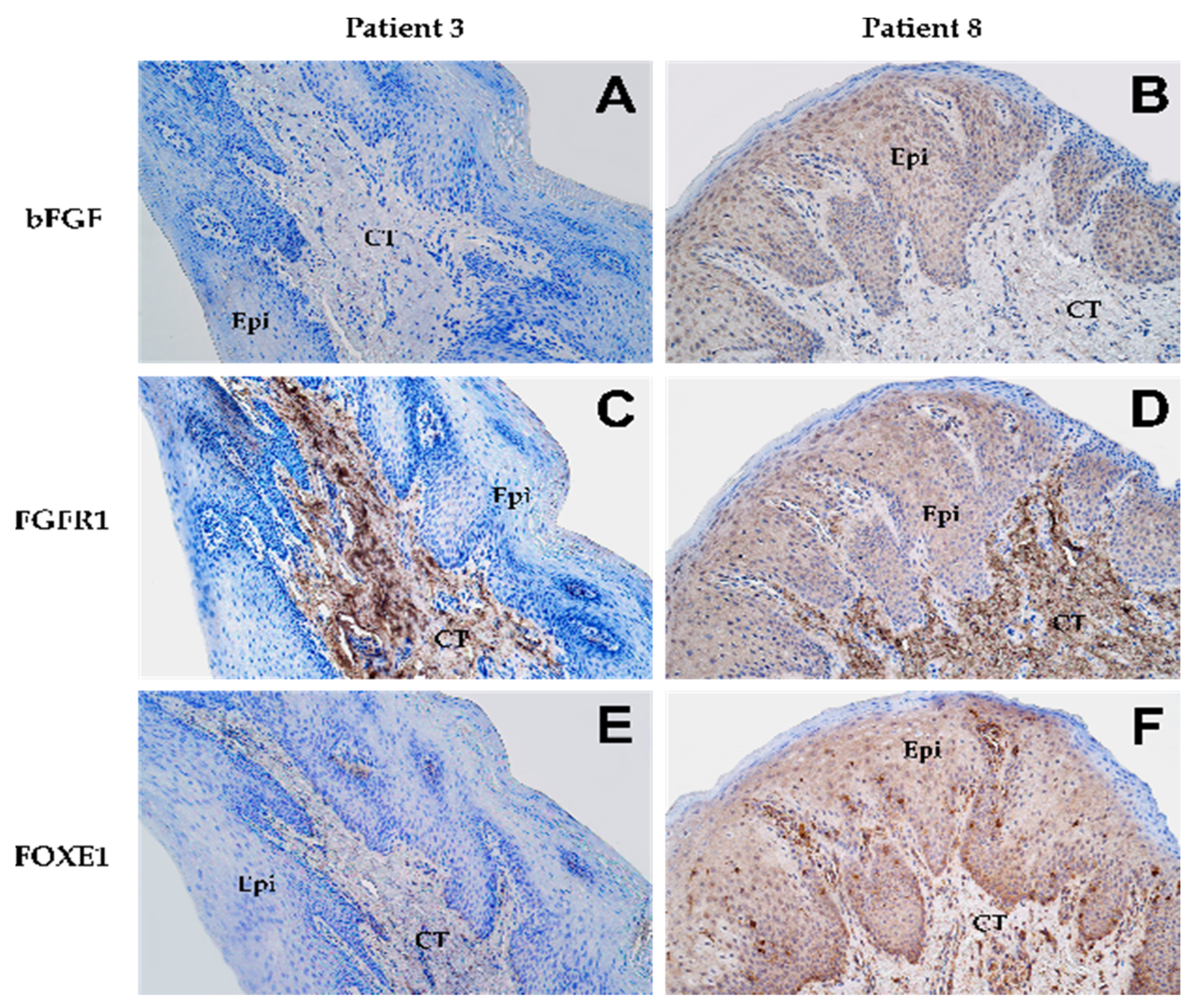
3.1. Immunohistochemistry Analysis
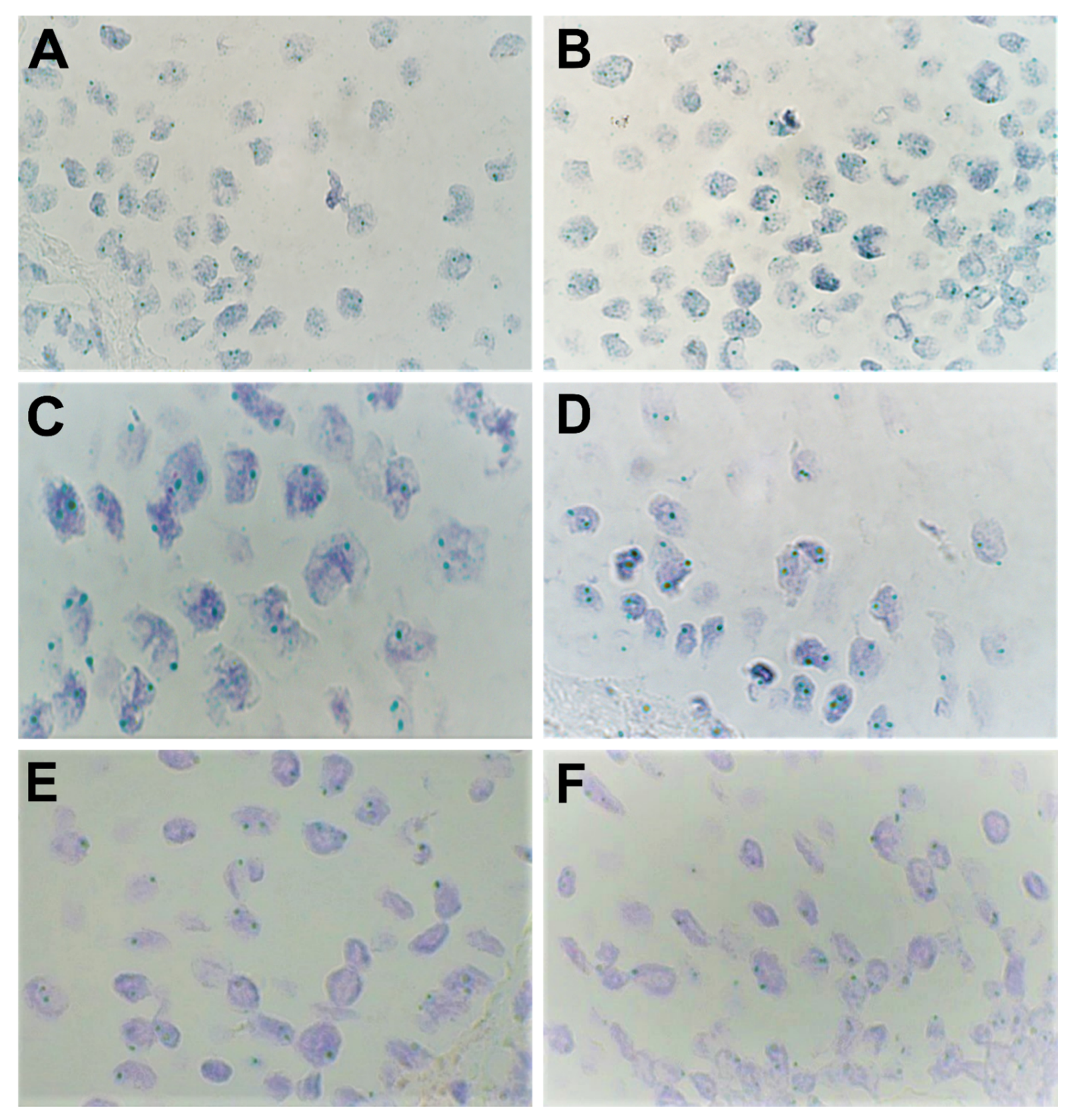
3.2. Chromogenic In-situ Hybridization Analysis
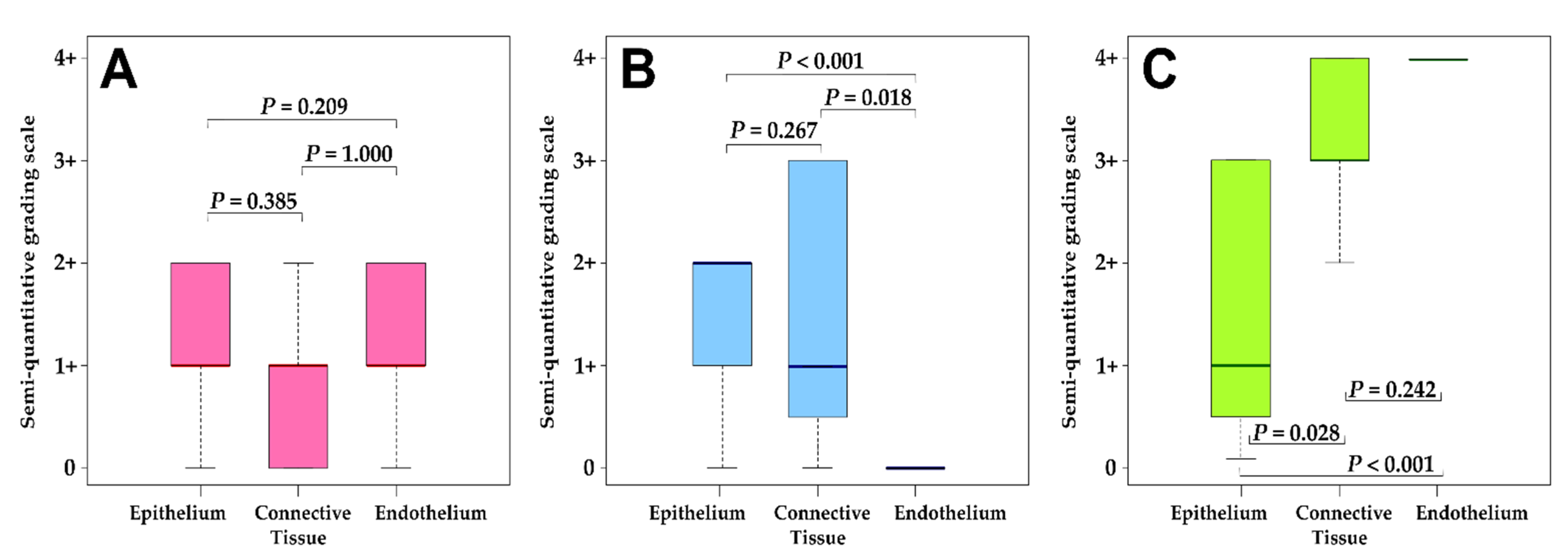
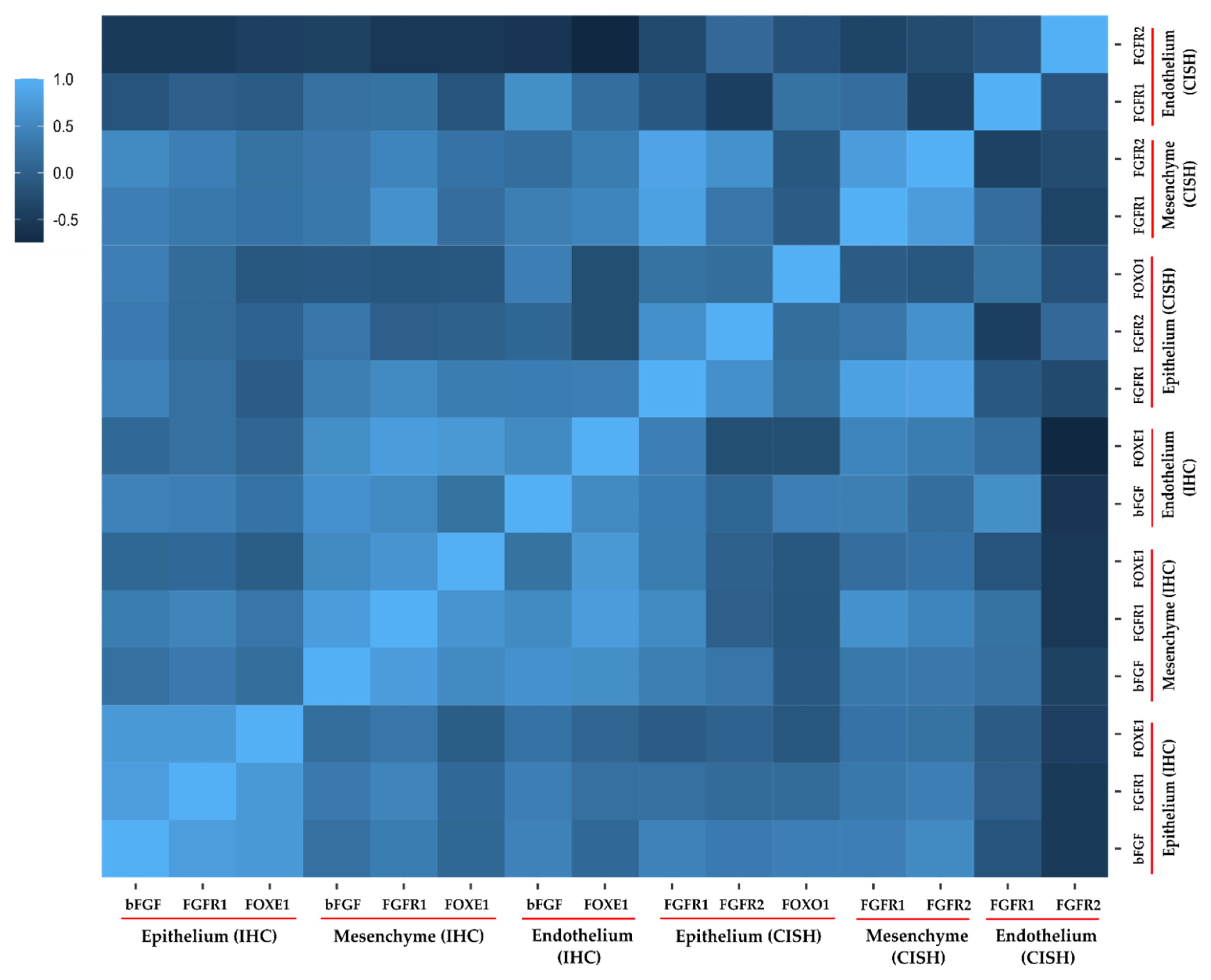
3.3. Correlation Analysis.
4. Discussion
4.1. Fibroblast Growth Factor Receptor 1 (FGFR1)
4.2. Fibroblast Growth Factor Receptor 2 (FGFR2)
4.3. Basic Fibroblast Growth Factor/Fibroblast Growth Factor 2 (bFGF/FGF2)
4.4. Potential Roles Mediated By FGF/FGFR Family Genes in Cleft Lip/Palate Pathogenesis
4.5. Forkhead Box Protein E1/Thyroid Transcription Factor 2 (FOXE1/TTF2)
4.6. Forkhead Box Protein O1 (FOXO1)
4.7. Clinical Diagnostic Techniques and Advances for Cleft-Affected Patients
4.8. Surgical Management of Cleft-Affected Patients
4.9. Relevance and Limitations of the Present Study
5. Conclusions
- Elevated expression of FGFR1 in cleft epithelium indicates its role in mediating cellular proliferation and local site inflammation. No to low expression in the endothelium indicates its role in fibrosis. Coupled together, this indicates that FGFR1 expression can help in predicting the sequalae and intensity of post-operative complications like scarring.
- bFGF (or FGF2) elevation may induce local site inflammation (via FGFR1) which chronically leads to creation and promotion of an environment suitable for angiogenic activity (via FGFR2). Additionally, over-amplification of FGFR2 in some patients points to its possible disordered role in epithelial–mesenchymal transition in cleft patients.
- High expression of FOXE1 possibly exerts a pro-inflammatory effect via involvement of the NR4A2/VEGF pathway (also induced by bFGF), while the lack or low level of amplification of FOXO1 can lead to retention of the midline epithelium coupled with increased endothelial oxidative stress and tissue inflammation.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mathes, D. Grabb and Smith’s plastic surgery. JAMA 2008, 299, 2450. [Google Scholar] [CrossRef]
- Vyas, T.; Gupta, P.; Kumar, S.; Gupta, R.; Gupta, T.; Singh, H.P. Cleft of lip and palate: A review. J. Fam. Med. Prim. Care 2020, 9, 2621–2625. [Google Scholar] [CrossRef]
- Mitchell, J.C.; Wood, R.J. Management of cleft lip and palate in primary care. J. Pediatr. Health Care 2000, 14, 13–19. [Google Scholar] [CrossRef]
- American Cleft Palate-Craniofacial Association (ACPA). Parameters for evaluation and treatment of patients with cleft lip/palate or other craniofacial differences. Cleft Palate Craniofacial J. 2018, 55, 137–156. [Google Scholar] [CrossRef]
- Banerjee, M.; Dhakar, A. Epidemiology-clinical profile of cleft lip and palate among children in india and its surgical consideration. CIBTech J. Surg. 2013, 2, 45–51. [Google Scholar]
- Muhamad, A.H.; Azzaldeen, A.; Watted, N. Cleft lip and palate: A comprehensive review. Int. J. Basic Appl. Med. Sci. 2014, 4, 338–355. [Google Scholar]
- Ferguson, M.W. Palate development. Development 1988, 103, 41–60. [Google Scholar] [CrossRef]
- Wang, C.; Chang, J.Y.; Yang, C.; Huang, Y.; Liu, J.; You, P.; McKeehan, W.L.; Wang, F.; Li, X. Type 1 fibroblast growth factor receptor in cranial neural crest cell-derived mesenchyme is required for palatogenesis. J. Biol. Chem. 2013, 288, 22174–22183. [Google Scholar] [CrossRef] [PubMed]
- Snyder-Warwick, A.K.; Perlyn, C.A. Coordinated events: FGF signaling and other related pathways in palatogenesis. J. Craniofacial Surg. 2012, 397–400. [Google Scholar] [CrossRef]
- Wilkie, A.O.; Morriss-Kay, G.M. Genetics of craniofacial development and malformation. Nat. Rev. Genet. 2001, 2, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Dixon, M.J.; Marazita, M.L.; Beaty, T.H.; Murray, J.C. Cleft lip and palate: Understanding genetic and environmental influences. Nat. Rev. Genet. 2011, 12, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.M.; Brandon, C.A.; McHenry, T.H.; Neiswanger, K.; Deleyiannis, F.W.; De Salamanca, J.E.; Castilla, E.E.; Czeizel, A.E.; Vieira, A.R.; Marazita, M.L. Rethinking isolated cleft palate: Evidence of occult lip defects in a subset of cases. Am. J. Med. Genet. A 2008, 146A, 1670–1675. [Google Scholar] [CrossRef] [PubMed]
- Scapoli, L.; Palmieri, A.; Martinelli, M.; Pezzetti, F.; Carinci, P.; Tognon, M.; Carinci, F. Strong evidence of linkage disequilibrium between polymorphisms at the IRF6 locus and nonsyndromic cleft lip with or without cleft palate, in an Italian population. Am. J. Human Genet. 2005, 76, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Mangold, E.; Ludwig, K.U.; Birnbaum, S.; Baluardo, C.; Ferrian, M.; Herms, S.; Reutter, H.; de Assis, N.A.; Al Chawa, T.; Mattheisen, M.; et al. Genome-wide association study identifies two susceptibility loci for nonsyndromic cleft lip with or without cleft palate. Nat. Genet. 2010, 42, 24–26. [Google Scholar] [CrossRef]
- Beaty, T.H.; Murray, J.C.; Marazita, M.L.; Munger, R.G.; Ruczinski, I.; Hetmanski, J.B.; Liang, K.Y.; Wu, T.; Murray, T.; Fallin, M.D.; et al. A genome-wide association study of cleft lip with and without cleft palate identifies risk variants near MAFB and ABCA4. Nat. Genet. 2010, 42, 525–529. [Google Scholar] [CrossRef]
- Osoegawa, K.; Vessere, G.M.; Utami, K.H.; Mansilla, M.A.; Johnson, M.K.; Riley, B.M.; L’Heureux, J.; Pfundt, R.; Staaf, J.; Van Der Vliet, W.A.; et al. Identification of novel candidate genes associated with cleft lip and palate using array comparative genomic hybridisation. J. Med. Genet. 2008, 45, 81–86. [Google Scholar] [CrossRef]
- Nie, X.; Luukko, K.; Kettunen, P. FGF signalling in craniofacial development and developmental disorders. Oral. Dis. 2006, 12, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Menezes, R.; Letra, A.; Ruff, J.; Granjeiro, J.M.; Vieira, A.R. Studies of genes in the FGF signaling pathway and oral clefts with or without dental anomalies. Am. J. Med. Genet. A 2008, 146A, 1614–1617. [Google Scholar] [CrossRef][Green Version]
- Marazita, M.L.; Lidral, A.C.; Murray, J.C.; Field, L.L.; Maher, B.S.; McHenry, T.G.; Cooper, M.E.; Govil, M.; Daack-Hirsch, S.; Riley, B.; et al. Genome scan, fine-mapping, and candidate gene analysis of non-syndromic cleft lip with or without cleft palate reveals phenotype-specific differences in linkage and association results. Human Hered. 2009, 68, 151–170. [Google Scholar] [CrossRef] [PubMed]
- Moreno, L.M.; Mansilla, M.A.; Bullard, S.A.; Cooper, M.E.; Busch, T.D.; Machida, J.; Johnson, M.K.; Brauer, D.; Krahn, K.; Daack-Hirsch, S.; et al. FOXE1 association with both isolated cleft lip with or without cleft palate, and isolated cleft palate. Human Mol. Genet. 2009, 18, 4879–4896. [Google Scholar] [CrossRef]
- van den Boogaard, M.J.; Dorland, M.; Beemer, F.A.; van Amstel, H.K. MSX1 mutation is associated with orofacial clefting and tooth agenesis in humans. Nat. Genet. 2000, 24, 342–343. [Google Scholar] [CrossRef] [PubMed]
- Jezewski, P.A.; Vieira, A.R.; Nishimura, C.; Ludwig, B.; Johnson, M.; O’Brien, S.E.; Daack-Hirsch, S.; Schultz, R.E.; Weber, A.; Nepomucena, B.; et al. Complete sequencing shows a role for MSX1 in non-syndromic cleft lip and palate. J. Med. Genet. 2003, 40, 399–407. [Google Scholar] [CrossRef]
- Suzuki, S.; Marazita, M.L.; Cooper, M.E.; Miwa, N.; Hing, A.; Jugessur, A.; Natsume, N.; Shimozato, K.; Ohbayashi, N.; Suzuki, Y.; et al. Mutations in BMP4 are associated with subepithelial, microform, and overt cleft lip. Am. J. Human Genet. 2009, 84, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; Williams, L.T. Structural and functional diversity in the FGF receptor multigene family. Adv. Cancer Res. 1993, 60, 1–41. [Google Scholar] [PubMed]
- Su, N.; Jin, M.; Chen, L. Role of FGF/FGFR signaling in skeletal development and homeostasis: Learning from mouse models. Bone Res. 2014, 2, 14003. [Google Scholar] [CrossRef] [PubMed]
- Roscioli, T.; Flanagan, S.; Kumar, P.; Masel, J.; Gattas, M.; Hyland, V.; Glass, I. Clinical findings in a patient with FGFR1 P252R mutation and comparison with the literature. Am. J. Med. Genet. 2000, 93, 22–28. [Google Scholar] [CrossRef]
- Jackson, B.C.; Carpenter, C.; Nebert, D.W.; Vasiliou, V. Update of human and mouse forkhead box (FOX) gene families. Human Genom. 2010, 4, 345. [Google Scholar] [CrossRef]
- Ali, A.H.M.; Yahya, A.Q.; Mohammed, H.L. Chromogenic in-situ hybridization technique versus immunohistochemistry in assessment of HER2/neu Status in 448 Iraqi patients with invasive breast carcinoma. Open Access Maced. J. Med. Sci. 2019, 7, 1917–1925. [Google Scholar] [CrossRef]
- Reliable and Simple Detection of Genomic Alterations Using Light Microscopy. ZytoDotR 2CTM-2-Color CISH for the Detection of Genomic Alterations. A User Manual Provided by ZytoVision GmbH-Fischkai 1, 27572 Bremerhaven- Germany. ZytoVision Molecular Diagnostics Simplified. 2019, pp. 182–183. Available online: www.Zytovision.com (accessed on 25 March 2021).
- Pilmane, M.; Rumba, I.; Sundler, F.; Luts, A. Patterns of distribution and occurrence of neuroendocrine elements in lungs of humans with chronic lung disease. Proc. Latv. Acad. Sci. 1998, 52, 144–152. [Google Scholar]
- van de Vijver, M.; Bilous, M.; Hanna, W.; Hofmann, M.; Kristel, P.; Penault-Llorca, F.; Rüschoff, J. Chromogenic in-situ hybridisation for the assessment of HER2 status in breast cancer: An international validation ring study. Breast Cancer Res. 2007, 9, R68. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, T.; Wu, T.; Hetmanski, J.B.; Ruczinski, I.; Schwender, H.; Liang, K.Y.; Murray, T.; Fallin, M.D.; Redett, R.J.; et al. The FGF and FGFR gene family and risk of cleft lip with or without cleft palate. Cleft Palate Craniofacial J. 2013, 50, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Welsh, I.C.; Hagge-Greenberg, A.; O’Brien, T.P. A dosage-dependent role for Spry2 in growth and patterning during palate development. Mech. Dev. 2007, 124, 746–761. [Google Scholar] [CrossRef]
- Li, J.; Shi, S.; Srivastava, S.; Kitada, M.; Nagai, T.; Nitta, K.; Kohno, M.; Kanasaki, K.; Koya, D. FGFR1 is critical for the anti-endothelial mesenchymal transition effect of N-acetyl-seryl-aspartyl-lysyl-proline via induction of the MAP4K4 pathway. Cell Death Dis. 2017, 8, e2965. [Google Scholar] [CrossRef] [PubMed]
- Oladipupo, S.S.; Smith, C.; Santeford, A.; Park, C.; Sene, A.; Wiley, L.A.; Osei-Owusu, P.; Hsu, J.; Zapata, N.; Liu, F.; et al. Endothelial cell FGF signaling is required for injury response but not for vascular homeostasis. Proc. Natl. Acad. Sci. USA 2014, 111, 13379–13384. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Funato, N.; Baba, Y.; Kuroda, T. Evidence for fibroblast growth factor receptors in myofibroblasts during palatal mucoperiosteal repair. Arch. Oral. Biol. 2003, 48, 213–221. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, F.; Li, H.; Xu, S.; Xu, W.; Pan, X.; Hu, Y.; Mao, L.; Qian, S.; Pan, J. Inhibition of fibroblast growth factor receptor by AZD4547 protects against inflammation in septic mice. Inflammation 2019, 42, 1957–1967. [Google Scholar] [CrossRef]
- Wang, C.; Li, Y.; Li, H.; Zhang, Y.; Ying, Z.; Wang, X.; Zhang, T.; Zhang, W.; Fan, Z.; Li, X.; et al. Disruption of FGF Signaling ameliorates inflammatory response in hepatic stellate cells. Front. Cell Dev. Biol. 2020, 8, 601. [Google Scholar] [CrossRef]
- Pilmane, M.; Sidhoma, E.; Akota, I.; Kazoka, D. Characterization of cytokines and proliferation marker Ki67 in cleft affected lip tissue. Medicina (Kaunas) 2019, 55, 518. [Google Scholar] [CrossRef]
- Soltani, A.M.; Francis, C.S.; Motamed, A.; Karatsonyi, A.L.; Hammoudeh, J.A.; Sanchez-Lara, P.A.; Reinisch, J.F.; Urata, M.M. Hypertrophic scarring in cleft lip repair: A comparison of incidence among ethnic groups. Clin. Epidemiol. 2012, 4, 187–191. [Google Scholar] [CrossRef]
- Pilmane, M.; Jain, N.; Jain, S.; Akota, I.; Kroiča, J. Quantification of cytokines in lip tissue from infants affected by congenital cleft lip and palate. Children (Basel) 2021, 8, 140. [Google Scholar] [CrossRef]
- Rice, R.; Spencer-Dene, B.; Connor, E.C.; Gritli-Linde, A.; McMahon, A.P.; Dickson, C.; Thesleff, I.; Rice, D.P. Disruption of Fgf10/Fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. J. Clin. Investig. 2004, 113, 1692–1700. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, R.; Deng, X.; Takamori, K.; Xu, X.; Urata, M.; Bringas, P., Jr.; Chai, Y. Epithelial-specific requirement of FGFR2 signaling during tooth and palate development. J. Exp. Zool. B Mol. Dev. Evol. 2009, 312B, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Ranieri, D.; Rosato, B.; Nanni, M.; Magenta, A.; Belleudi, F.; Torrisi, M.R. Expression of the FGFR2 mesenchymal splicing variant in epithelial cells drives epithelial-mesenchymal transition. Oncotarget 2016, 7, 5440–5460. [Google Scholar] [CrossRef]
- Ranieri, D.; Belleudi, F.; Magenta, A.; Torrisi, M.R. HPV16 E5 expression induces switching from FGFR2b to FGFR2c and epithelial-mesenchymal transition. Int. J. Cancer 2015, 137, 61–72. [Google Scholar] [CrossRef]
- Wahl, S.M.; Wong, H.; McCartney-Francis, N. Role of growthfactors in inflammation and repair. J. Cell Biochem. 1989, 40, 193–199. [Google Scholar] [CrossRef]
- Choi, W.; Kawanabe, H.; Sawa, Y.; Taniguchi, K.; Ishikawa, H. Effects of bFGF on suppression of collagen type I accumulation and scar tissue formation during wound healing after mucoperiosteal denudation of rat palate. Acta Odontol. Scand. 2008, 66, 31–37. [Google Scholar] [CrossRef]
- Ribatti, D.; Nico, B.; Vacca, A.; Roncali, L.; Presta, M. Endogenous and exogenous fibroblast growth factor-2 modulate wound healing in the chick embryo chorio-allantoic membrane. Angiogenesis 1999, 3, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, H.; Kurokawa, T.; Hanada, K.; Hiyama, Y.; Tamura, M.; Ogata, E.; Matsumoto, T. Stimulation of fracture repair by recombinant human basic fibroblast growth factor in normal and streptozotocin-diabetic rats. Endocrinology 1994, 135, 774–781. [Google Scholar] [CrossRef]
- Koike, Y.; Yozaki, M.; Utani, A.; Murota, H. Fibroblast growth factor 2 accelerates the epithelial–mesenchymal transition in keratinocytes during wound healing process. Sci. Rep. 2020, 10, 18545. [Google Scholar] [CrossRef]
- Zittermann, S.I.; Issekutz, A.C. Basic fibroblast growth factor (bFGF, FGF-2) potentiates leukocyte recruitment to inflammation by enhancing endothelial adhesion molecule expression. Am. J. Pathol. 2006, 168, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, S.; Tsunoda, T.; Onuma, E.; Majima, T.; Kagiyama, M.; Kikuchi, K. VEGF, basic-FGF, and TGF-beta in Crohn’s disease and ulcerative colitis: A novel mechanism of chronic intestinal inflammation. Am. J. Gastroenterol. 2001, 96, 822–828. [Google Scholar] [CrossRef]
- Shi, Y.J.; Shi, M.; Xiao, L.J.; Li, L.; Zou, L.H.; Li, C.Y.; Zhang, Q.J.; Zhou, L.F.; Ji, X.C.; Huang, H.; et al. Inhibitive Effects of FGF2/FGFR1 Pathway on Astrocyte-Mediated Inflammation in vivo and in vitro After Infrasound Exposure. Front. Neurosci. 2018, 12, 582. [Google Scholar] [CrossRef]
- Wang, C.; Ke, Y.; Liu, S.; Pan, S.; Liu, Z.; Zhang, H.; Fan, Z.; Zhou, C.; Liu, J.; Wang, F. Ectopic fibroblast growth factor receptor 1 promotes inflammation by promoting nuclear factor-kappaB signaling in prostate cancer cells. J. Biol. Chem. 2018, 293, 14839–14849. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.T.; Lee, J.G.; Na, M.; Kay, E.P. FGF-2 induced by interleukin-1 beta through the action of phosphatidylinositol 3-kinase mediates endothelial mesenchymal transformation in corneal endothelial cells. J. Biol. Chem. 2004, 279, 32325–32332. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, F.; Torcia, M.; Lucibello, M.; Morbidelli, L.; Ziche, M.; Platt, J.; Fabiani, S.; Brett, J.; Stern, D. Interferon-alpha and interleukin 2 synergistically enhance basic fibroblast growth factor synthesis and induce release, promoting endothelial cell growth. J. Clin. Investig. 1993, 91, 2504–2512. [Google Scholar] [CrossRef]
- Kuwabara, K.; Ogawa, S.; Matsumoto, M.; Koga, S.; Clauss, M.; Pinsky, D.J.; Lyn, P.; Leavy, J.; Witte, L.; Joseph-Silverstein, J. Hypoxia-mediated induction of acidic/basic fibroblast growth factor and platelet-derived growth factor in mononuclear phagocytes stimulates growth of hypoxic endothelial cells. Proc. Natl. Acad. Sci. USA 1995, 92, 4606–4610. [Google Scholar] [CrossRef]
- Presta, M.; Andrés, G.; Leali, D.; Dell’Era, P.; Ronca, R. Inflammatory cells and chemokines sustain FGF2-induced angiogenesis. Eur. Cytokine Netw. 2009, 20, 39–50. [Google Scholar] [CrossRef]
- Küchler, E.C.; Silva, L.A.D.; Nelson-Filho, P.; Sabóia, T.M.; Rentschler, A.M.; Granjeiro, J.M.; Oliveira, D.; Tannure, P.N.; Silva, R.A.D.; Antunes, L.S.; et al. Assessing the association between hypoxia during craniofacial development and oral clefts. J. Appl. Oral. Sci. 2018, 26, e20170234. [Google Scholar] [CrossRef]
- Conte, C.; Riant, E.; Toutain, C.; Pujol, F.; Arnal, J.F.; Lenfant, F.; Prats, A.C. FGF2 translationally induced by hypoxia is involved in negative and positive feedback loops with HIF-1alpha. PLoS ONE 2008, 3, e3078. [Google Scholar] [CrossRef]
- Calvani, M.; Rapisarda, A.; Uranchimeg, B.; Shoemaker, R.H.; Melillo, G. Hypoxic induction of an HIF-1alpha-dependent bFGF autocrine loop drives angiogenesis in human endothelial cells. Blood 2006, 107, 2705–2712. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shworak, N.W.; Simons, M. Increased responsiveness of hypoxic endothelial cells to FGF2 is mediated by HIF-1 alpha-dependent regulation of enzymes involved in synthesis of heparan sulfate FGF2-binding sites. J. Cell Sci. 2002, 115, 1951. [Google Scholar] [CrossRef]
- Wang, L.; Xiong, M.; Che, D.; Shaochun, L.; Chunrong, H.; Xiaojing, Z. The effect of hypoxia on expression of basic fibroblast growth factor in pulmonary vascular pericytes. J. Tongji Med. Univ. 2000, 20, 265. [Google Scholar] [PubMed]
- Yang, J.; Zhang, D.; Yu, Y.; Zhang, R.J.; Hu, X.L.; Huang, H.F.; Lu, Y.C. Binding of FGF2 to FGFR2 in an autocrine mode in trophectoderm cells is indispensable for mouse blastocyst formation through PKC-p38 pathway. Cell Cycle 2015, 14, 3318–3330. [Google Scholar] [CrossRef]
- Breviario, F.; d’Aniello, E.M.; Golay, J.; Peri, G.; Bottazzi, B.; Bairoch, A.; Saccone, S.; Marzella, R.; Predazzi, V.; Rocchi, M. Interleukin-1-induciblegenes in endothelial cells. Cloning of a new gene related to C-reactive protein and serum amyloid P component. J. Biol. Chem. 1992, 267, 22190. [Google Scholar] [CrossRef]
- Perollet, C.; Han, Z.C.; Savona, C.; Caen, J.P.; Bikfalvi, A. Platelet factor 4 modulates fibroblast growth factor 2 (FGF-2) activity and inhibits FGF-2dimerization. Blood 1998, 91, 3289. [Google Scholar] [CrossRef]
- Dathan, N.; Parlato, R.; Rosica, A.; De Felice, M.; Di Lauro, R. Distribution of the titf2/foxe1 gene product is consistent with an important role in the development of foregut endoderm, palate, and hair. Dev. Dyn. 2002, 224, 450–456. [Google Scholar] [CrossRef]
- Venza, I.; Visalli, M.; Parrillo, L.; De Felice, M.; Teti, D.; Venza, M. MSX1 and TGF-beta3 are novel target genes functionally regulated by FOXE1. Human Mol Genet. 2011, 20, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Adab, K.; Sayne, J.R.; Carlson, D.S.; Opperman, L.A. Tgf-beta1, Tgf-beta2, Tgf-beta3 and Msx2 expression is elevated during frontonasal suture morphogenesis and during active postnatal facial growth. Orthod. Craniofacal Res. 2002, 5, 227–237. [Google Scholar] [CrossRef]
- Fitzpatrick, D.R.; Denhez, F.; Kondaiah, P.; Akhurst, R.J. Differential expression of TGF beta isoforms in murine palatogenesis. Development 1990, 109, 585–595. [Google Scholar] [CrossRef]
- Fernández, L.P.; López-Márquez, A.; Martínez, A.M.; Gómez-López, G.; Santisteban, P. New insights into FoxE1 functions: Identification of direct FoxE1 targets in thyroid cells. PLoS ONE 2013, 8, e62849. [Google Scholar] [CrossRef] [PubMed]
- Maijenburg, M.W.; Gilissen, C.; Melief, S.M.; Kleijer, M.; Weijer, K.; Ten Brinke, A.; Roelofs, H.; Van Tiel, C.M.; Veltman, J.A.; de Vries, C.J.; et al. Nuclear receptors Nur77 and Nurr1 modulate mesenchymal stromal cell migration. Stem Cells Dev. 2012, 21, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Ozturk, F.; Pandey, S.; Guda, C.B.; Nawshad, A. Implications of TGFβ on transcriptome and cellular biofunctions of palatal mesenchyme. Front Physiol. 2012, 3, 85. [Google Scholar] [CrossRef]
- McEvoy, A.N.; Murphy, E.A.; Ponnio, T.; Conneely, O.M.; Bresnihan, B.; FitzGerald, O.; Murphy, E.P. Activation of nuclear orphan receptor {NURR1} transcription by {NF-kappa} B and cyclic adenosine 5′-monophosphate response element-binding protein in rheumatoid arthritis synovial tissue. J. Immunol. 2002, 168, 2979–2987. [Google Scholar] [CrossRef] [PubMed]
- McCoy, J.M.; Walkenhorst, D.E.; McCauley, K.S.; Elaasar, H.; Everett, J.R.; Mix, K.S. Orphan nuclear receptor NR4A2 induces transcription of the immunomodulatory peptide hormone prolactin. J. Inflamm. 2015, 12, 13. [Google Scholar] [CrossRef]
- Ke, C.Y.; Xiao, W.L.; Chen, C.M.; Lo, L.J.; Wong, F.H. IRF6 is the mediator of TGFβ3 during regulation of the epithelial mesenchymal transition and palatal fusion. Sci. Rep. 2015, 5, 12791. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Liu, F.; Xiong, Z.; Huo, J.; Li, W.; Jiang, B.; Mao, W.; He, B.; Wang, X.; Li, G. The cleft palate candidate gene BAG6 supports FoxO1 acetylation to promote FasL-mediated apoptosis during palate fusion. Exp. Cell Res. 2020, 396, 112310. [Google Scholar] [CrossRef]
- Kwak, J.H.; Kim, S.I.; Kim, J.K.; Choi, M.E. BAT3 interacts with transforming growth factor-beta (TGF-beta) receptors and enhances TGF-beta1-induced type I collagen expression in mesangial cells. J. Biol. Chem. 2008, 283, 19816–19825. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, K.; Happel, K.; Eelen, G.; Schoors, S.; Oellerich, M.F.; Lim, R.; Zimmermann, B.; Aspalter, I.M.; Franco, C.A.; Boettger, T.; et al. FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature 2016, 529, 216–220. [Google Scholar] [CrossRef]
- Matsuda, C.; Matsui, Y.; Ohno, K.; Michi, K. Salivary gland aplasia with cleft lip and palate: A case report and review of the literature. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 1999, 87, 594–599. [Google Scholar] [CrossRef]
- Ashley, L.M.; Richardson, G.E. Multiple congenital anomalies in astillborn infant. Anat. Rec. 1943, 86, 457–471. [Google Scholar] [CrossRef]
- Salgarelli, A.C.; Capparè, P.; Bellini, P.; Collini, M. Usefulness of fine-needle aspiration in parotid diagnostics. Oral Maxillofacial Surg. 2009, 13, 185–190. [Google Scholar] [CrossRef]
- Fakhouri, W.D.; Rhea, L.; Du, T.; Sweezer, E.; Morrison, H.; Fitzpatrick, D.; Yang, B.; Dunnwald, M.; Schutte, B.C. MCS9.7 enhancer activity is highly, but not completely, associated with expression of Irf6 and p63. Dev. Dyn. 2012, 241, 340–349. [Google Scholar] [CrossRef]
- Laugel-Haushalter, V.; Langer, A.; Marrie, J.; Fraulob, V.; Schuhbaur, B.; Koch-Phillips, M.; Dollé, P.; Bloch-Zupan, A. From the transcription of genes involved in ectodermal dysplasias to the understanding of associated dental anomalies. Mol. Syndromol. 2012, 3, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Tamasas, B.; Cox, T.C. Massively increased caries susceptibility in an Irf6 cleft lip/palate model. J. Dent. Res. 2017, 96, 315–322. [Google Scholar] [CrossRef] [PubMed]
- De Moerlooze, L.; Spencer-Dene, B.; Revest, J.; Hajihosseini, M.; Rosewell, I.; Dickson, C. An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development 2000, 127, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Sberna, M.T.; Rizzo, G.; Zacchi, E.; Capparè, P.; Rubinacci, A. A preliminary study of the use of peripheral quantitative computed tomography for investigating root canal anatomy. Int. Endod. J. 2009, 42, 66–75. [Google Scholar] [CrossRef]
- Ferrini, F.; Sannino, G.; Chiola, C.; Capparé, P.; Gastaldi, G.; Gherlone, E.F. Influence of Intra-Oral Scanner (I.O.S.) on The Marginal Accuracy of CAD/CAM Single Crowns. Int. J. Environ. Res. Public Health 2019, 16, 544. [Google Scholar] [CrossRef]
- Cattoni, F.; Teté, G.; Calloni, A.M.; Manazza, F.; Gastaldi, G.; Capparè, P. Milled versus moulded mock-ups based on the superimposition of 3D meshes from digital oral impressions: A comparative in vitro study in the aesthetic area. BMC Oral Health 2019, 19, 230. [Google Scholar] [CrossRef]
- Joda, T.; Zarone, F.; Ferrari, M. The complete digital workflow in fixed prosthodontics: A systematic review. BMC Oral Health 2017, 17, 124. [Google Scholar] [CrossRef]
- Patzelt, S.B.; Emmanouilidi, A.; Stampf, S.; Strub, J.R.; Att, W. Accuracy of full-arch scans using intraoral scanners. Clin. Oral Investig. 2014, 18, 1687–1694. [Google Scholar] [CrossRef]
- Wermker, K.; Jung, S.; Joos, U.; Kleinheinz, J. Dental implants in cleft lip, alveolus, and palate patients: A systematic review. Int. J. Oral Maxillofac. Implant. 2014, 29, 384–390. [Google Scholar] [CrossRef]
- Anastassov, G.E.; Joos, U. Comprehensive management of cleft lip and palate deformities. J. Oral Maxillofacial Surg. 2001, 59, 1062–1075. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.; Broyles, J.; Redett, R. Cleft lip and palate. Eplasty 2013, 13, ic25. [Google Scholar] [PubMed]
- Harris, P.A.; Oliver, N.K.; Slater, P.; Murdoch, L.; Moss, A.L. Safety of neonatal cleft lip repair. J. Plast. Surg. Hand Surg. 2010, 44, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Goodacre, T.E.; Hentges, F.; Moss, T.L.; Short, V.; Murray, L. Does repairing a cleft lip neonatally have any effect on the longer-term attractiveness of the repair? Cleft Palate Craniofacial J. 2004, 41, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.F.; Cole, T.J.; Mars, M. Hard palate repair timing and facial growth in unilateral cleft lip and palate: A longitudinal study. Cleft Palate Craniofacial J. 2006, 43, 547–556. [Google Scholar] [CrossRef]
- Crespi, R.; Capparè, P.; Gherlone, E. Sinus floor elevation by osteotome: Hand mallet versus electric mallet. A prospective clinical study. Int. J. Oral Maxillofac. Implant. 2012, 27, 1144–1150. [Google Scholar]
- Vinci, R.; Teté, G.; Lucchetti, F.R.; Capparé, P.; Gherlone, E.F. Implant survival rate in calvarial bone grafts: A retrospective clinical study with 10 year follow-up. Clin. Implant. Dent. Relat. Res. 2019, 21, 662–668. [Google Scholar] [CrossRef]
- Bruschi, G.B.; Crespi, R.; Capparè, P.; Bravi, F.; Bruschi, E.; Gherlone, E. Localized management of sinus floor technique for implant placement in fresh molar sockets. Clin. Implant. Dent. Relat. Res. 2013, 15, 243–250. [Google Scholar] [CrossRef]
- Kim, S.W.; Lee, I.K.; Yun, K.I.; Kim, C.H.; Park, J.U. Adult stem cells derived from human maxillary sinus membrane and their osteogenic differentiation. Int. J. Oral Maxillofac. Implant. 2009, 24, 991–998. [Google Scholar]
- Capparè, P.; Tetè, G.; Sberna, M.T.; Panina-Bordignon, P. The emerging role of stem cells in regenerative dentistry. Curr. Gene Ther. 2020, 20, 259–268. [Google Scholar] [CrossRef]
- Capparé, P.; Teté, G.; Romanos, G.E.; Nagni, M.; Sannino, G.; Gherlone, E.F. The ‘All-on-four’ protocol in HIV-positive patients: A prospective, longitudinal 7-year clinical study. Int. J. Oral Implantol. (Berl.) 2019, 12, 501–510. [Google Scholar]
- Gherlone, E.F.; Capparé, P.; Tecco, S.; Polizzi, E.; Pantaleo, G.; Gastaldi, G.; Grusovin, M.G. A prospective longitudinal study on implant prosthetic rehabilitation in controlled HIV-positive patients with 1-year follow-up: The role of CD4+ level, smoking habits, and oral hygiene. Clin. Implant. Dent. Relat. Res. 2016, 18, 955–964. [Google Scholar] [CrossRef]
- Gherlone, E.F.; Capparé, P.; Tecco, S.; Polizzi, E.; Pantaleo, G.; Gastaldi, G.; Grusovin, M.G. Implant prosthetic rehabilitation in controlled HIV-positive patients: A prospective longitudinal study with 1-year follow-up. Clin. Implant. Dent. Relat. Res. 2016, 18, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Tecco, S.; Grusovin, M.G.; Sciara, S.; Bova, F.; Pantaleo, G.; Capparé, P. The association between three attitude-related indexes of oral hygiene and secondary implant failures: A retrospective longitudinal study. Int. J. Dent. Hyg. 2018, 16, 372–379. [Google Scholar] [CrossRef]
- Gherlone, E.F.; Capparé, P.; Pasciuta, R.; Grusovin, M.G.; Mancini, N.; Burioni, R. Evaluation of resistance against bacterial microleakage of a new conical implant-abutment connection versus conventional connections: An in vitro study. New Microbiol. 2016, 39, 49–56. [Google Scholar]
- Bruschi, G.B.; Crespi, R.; Capparè, P.; Grande, N.; Bruschi, E.; Gherlone, E. Radiographic evaluation of crestal bone levels of delayed implants at medium-term follow-up. Int. J. Oral Maxillofac. Implant. 2014, 29, 441–447. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fujiwara, K.; Yamada, T.; Mishima, K.; Imura, H.; Sugahara, T. Morphological and immunohistochemical studies on cleft palates induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin in mice. Congenit Anom. (Kyoto) 2008, 48, 68–73. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Number | Age (in Months) | Gender | Clinical Diagnosis * | Material Collected | Remarks |
|---|---|---|---|---|---|
| 1 | 3.5 | M | Cheilognathouranoschisis sinistra | Lip mucosa | Mother reported use of paracetamol during pregnancy; father was smoker and partially alcoholic. Epilepsy in the family tree. Child was born overweight. |
| 2 | 4 | M | Cheilognathouranoschisis sinistra | Lip mucosa | There was a reported threat of miscarriage in the 36th gestational week; history of clefts in the family tree. |
| 3 | 4 | F | Cheilognathouranoschisis dextra | Lip mucosa | - |
| 4 | 4 | F | Cheilognathouranoschisis sinistra | Lip mucosa | Born in the 42nd gestational week; mother reported use of paracetamol during pregnancy. |
| 5 | 4 | M | Cheilognathouranoschisis sinistra | Lip mucosa | Born in the 41st gestational week; mother reported use of paracetamol during pregnancy. |
| 6 | 4 | M | Cheilognathouranoschisis dextra | Lip mucosa | History of arrhythmogenic right ventricular dysplasia (ARVD) during the first trimester; mother reported use of Neuromidin, Ibumetin, and Theraflu. |
| 7 | 4.5 | M | Cheilognathouranoschisis sinistra | Lip mucosa | History of Down syndrome in the family tree. |
| 8 | 5 | M | Cheilognathouranoschisis sinistra | Lip mucosa | History of clefts in the family tree; mother reported use of Amoxiclav during pregnancy. |
| 9 | 8 | M | Cheilognathouranoschisis sinistra | Lip mucosa | Both parents were regular smokers. |
| 10 | 13 | M | Cheilognathouranoschisis bilateralis | Lip mucosa | The child had multiple anomalies, including heart failure. |
| 11 | 4 | M | Cheilognathouranoschisis sinistra | Vomer mucosa | History of heavy toxicosis during the pregnancy; there was a threat of miscarriage in the 36th gestational week. |
| 12 | 18 | M | Cheilognathouranoschisis sinistra | Vomer mucosa | Mother was reported to suffer from high emotional stress. |
| Assigned Value | In-Lab Criteria Used for Assignment of Value | Interpretation |
|---|---|---|
| Immunohistochemistry (IHC) | ||
| 0 | No cells with a positive reaction were detected in the visual field | - |
| + | Few cells with a positive reaction were detected in the visual field | - |
| ++ | Moderate number of cells with a positive reaction were detected in the visual field | - |
| +++ | Numerous cells with a positive reaction were detected in the visual field | - |
| ++++ | Abundant cells with a positive reaction were detected in the visual field | - |
| Chromogenic In-Situ Hybridization (CISH) | ||
| 0 | 1 to 5 green signals (copies) per nucleus detected in the cells | No amplification |
| + | 5 to 6 green signals (copies) per nucleus detected in the cells | Low-level amplification |
| ++ | 6 to 10 green signals (copies) per nucleus detected in the cells | Moderate-level amplification |
| +++ | >10 green signals (copies) detected in the cells | High-level amplification |
| ++++ | Large cluster of green signals (copies) per nucleus detected in the cells | High-level amplification |
| Patient Number | Epithelium | Connective Tissue | Endothelium | ||||||
|---|---|---|---|---|---|---|---|---|---|
| bFGF | FGFR1 | FOXE1 | bFGF | FGFR1 | FOXE1 | bFGF | FGFR1 | FOXE1 | |
| 1 | + | ++ | + | + | 0 | ++++ | + | 0 | ++++ |
| 2 | + | + | + | + | + | ++++ | + | 0 | ++++ |
| 3 | 0 | 0 | 0 | 0 | +++ | + | 0 | 0 | ++ |
| 4 | + | + | ++ | + | + | ++++ | + | 0 | ++++ |
| 5 | ++ | ++ | + | + | + | ++++ | ++ | 0 | ++++ |
| 6 | + | ++ | +++ | + | + | +++ | ++ | 0 | ++++ |
| 7 | ++ | ++ | +++ | 0 | 0 | ++ | + | 0 | +++ |
| 8 | ++ | ++ | +++ | 0 | +++ | +++ | + | 0 | ++++ |
| 9 | + | ++ | + | + | + | +++ | + | 0 | ++++ |
| 10 | + | + | 0 | + | 0 | +++ | ++ | 0 | ++++ |
| 11 | ++ | ++ | +++ | ++ | + | +++ | ++ | 0 | ++++ |
| 12 | 0 | 0 | 0 | 0 | 0 | +++ | + | 0 | ++++ |
| Mean | 1.17 | 1.42 | 1.50 | 0.75 | 1.00 | 3.08 | 1.25 | 0.00 | 3.75 |
| SD * | 0.72 | 0.79 | 1.24 | 0.62 | 0.45 | 0.90 | 0.62 | 0.00 | 0.62 |
| CV% ** | 62.0 | 56.0 | 83.0 | 83.0 | 45.0 | 29.0 | 50.0 | 0.00 | 17.0 |
| Patient Number | Epithelium | Connective Tissue | Endothelium | ||||||
|---|---|---|---|---|---|---|---|---|---|
| FGFR1 | FGFR2 | FOXO1 | FGFR1 | FGFR2 | FOXO1 | FGFR1 | FGFR2 | FOXO1 | |
| 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2 | ++ | ++ | 0 | ++ | + | 0 | 0 | 0 | 0 |
| 3 | 0 | + | 0 | 0 | 0 | 0 | 0 | + | 0 |
| 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 5 | ++ | ++ | + | + | + | 0 | 0 | 0 | 0 |
| 6 | 0 | 0 | 0 | 0 | 0 | 0 | + | 0 | 0 |
| 7 | + | + | + | 0 | 0 | 0 | 0 | 0 | 0 |
| 8 | + | 0 | 0 | ++ | + | 0 | 0 | 0 | 0 |
| 9 | + | + | 0 | + | + | 0 | 0 | 0 | 0 |
| 10 | + | 0 | + | + | 0 | 0 | + | 0 | 0 |
| 11 | + | ++ | 0 | + | + | 0 | 0 | 0 | 0 |
| 12 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Mean | 0.75 | 0.75 | 0.25 | 0.67 | 0.42 | 0.00 | 0.17 | 0.08 | 0.00 |
| SD * | 0.74 | 0.87 | 0.45 | 0.78 | 0.51 | 0.00 | 0.39 | 0.28 | 0.00 |
| CV% ** | 99.0 | 116.0 | 180.0 | 116.0 | 121.0 | 0.00 | 229.0 | 350.0 | 0.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pilmane, M.; Jain, N.; Vitenberga-Verza, Z. Expression Analysis of FGF/FGFR and FOX Family Proteins in Mucosal Tissue Obtained from Orofacial Cleft-Affected Children. Biology 2021, 10, 423. https://doi.org/10.3390/biology10050423
Pilmane M, Jain N, Vitenberga-Verza Z. Expression Analysis of FGF/FGFR and FOX Family Proteins in Mucosal Tissue Obtained from Orofacial Cleft-Affected Children. Biology. 2021; 10(5):423. https://doi.org/10.3390/biology10050423
Chicago/Turabian StylePilmane, Māra, Nityanand Jain, and Zane Vitenberga-Verza. 2021. "Expression Analysis of FGF/FGFR and FOX Family Proteins in Mucosal Tissue Obtained from Orofacial Cleft-Affected Children" Biology 10, no. 5: 423. https://doi.org/10.3390/biology10050423
APA StylePilmane, M., Jain, N., & Vitenberga-Verza, Z. (2021). Expression Analysis of FGF/FGFR and FOX Family Proteins in Mucosal Tissue Obtained from Orofacial Cleft-Affected Children. Biology, 10(5), 423. https://doi.org/10.3390/biology10050423