
RNA-Binding Proteins Hold Key Roles in Function, Dysfunction, and Disease


Abstract
Simple Summary
Abstract
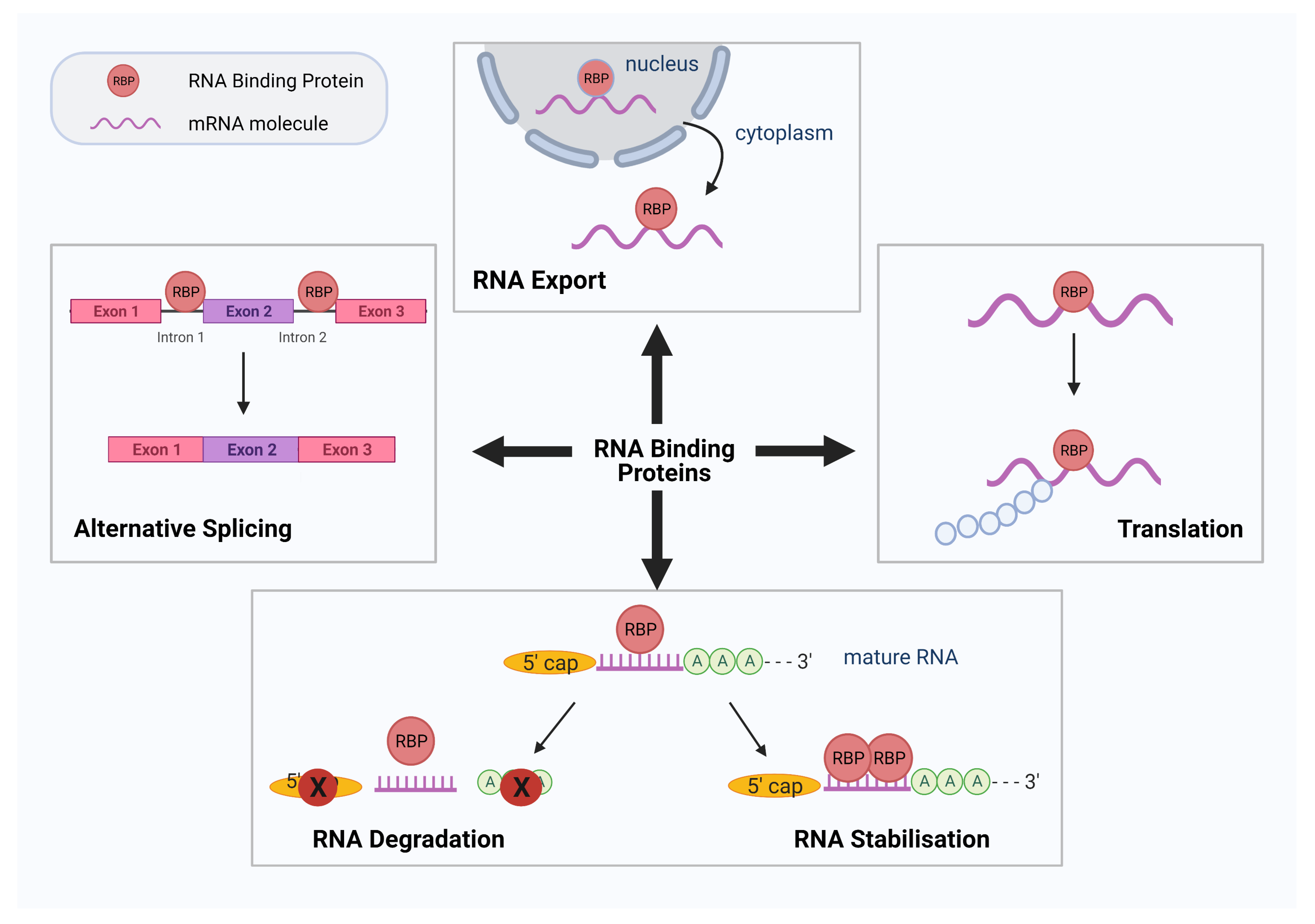
1. Introduction
2. RBPs in the Pathogenesis of Diabetes and Cardiovascular Disease
3. RBPs and Their Role in Cancer Development and Progression
4. RBPs and Their Role in Neurodegenerative Disease
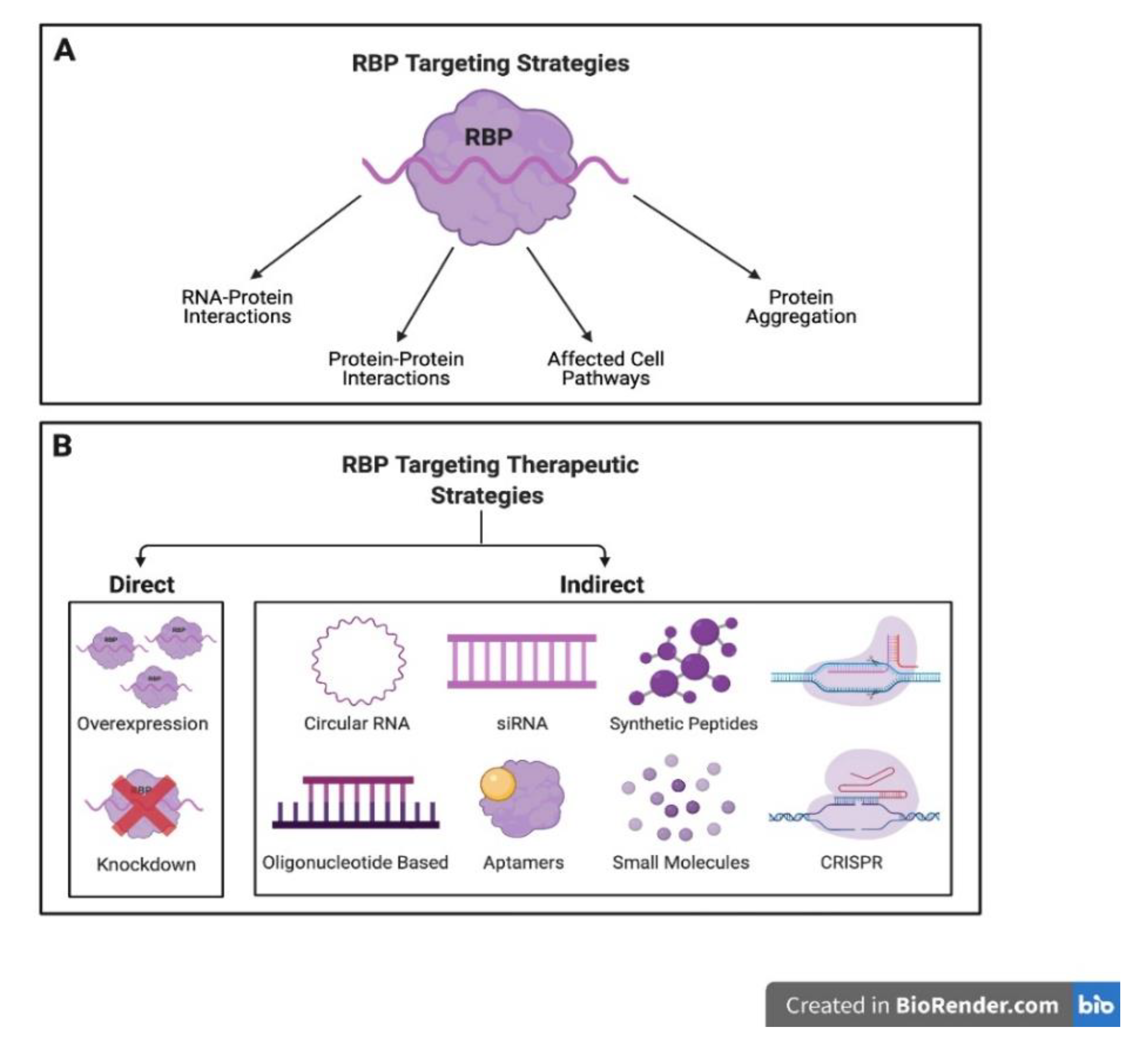
5. RBP-Based Therapeutics and Future Directions
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Liu-Yesucevitz, L.; Bassell, G.J.; Gitler, A.D.; Hart, A.C.; Klann, E.; Richter, J.D.; Warren, S.T.; Wolozin, B. Local RNA translation at the synapse and in disease. J. Neurosci. 2011, 31, 16086–16093. [Google Scholar] [CrossRef] [PubMed]
- Van Nostrand, E.L.; Freese, P.; Pratt, G.A.; Wang, X.; Wei, X.; Xiao, R.; Blue, S.M.; Chen, J.Y.; Cody, N.A.L.; Dominguez, D.; et al. A large-scale binding and functional map of human RNA-binding proteins. Nature 2020, 583, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Quattrone, A.; Dassi, E. The Architecture of the Human RNA-Binding Protein Regulatory Network. iScience 2019, 21, 706–719. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.S.; von Lindern, M. RNA Binding Proteins and Regulation of mRNA Translation in Erythropoiesis. Front. Physiol. 2018, 9, 910. [Google Scholar] [CrossRef] [PubMed]
- Dhatariya, K. Diabetes: The place of new therapies. Adv. Endocrinol. Metab. 2018, 10, 2042018818807599. [Google Scholar] [CrossRef]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef]
- Leon, B.M.; Maddox, T.M. Diabetes and cardiovascular disease: Epidemiology, biological mechanisms, treatment recommendations and future research. World J. Diabetes 2015, 6, 1246–1258. [Google Scholar] [CrossRef]
- Mobasseri, M.; Shirmohammadi, M.; Amiri, T.; Vahed, N.; Hosseini Fard, H.; Ghojazadeh, M. Prevalence and incidence of type 1 diabetes in the world: A systematic review and meta-analysis. Health Promot. Perspect 2020, 10, 98–115. [Google Scholar] [CrossRef]
- Wilcox, G. Insulin and insulin resistance. Clin. Biochem. Rev. 2005, 26, 19–39. [Google Scholar]
- Whelan, J.T.; Hollis, S.E.; Cha, D.S.; Asch, A.S.; Lee, M.H. Post-transcriptional regulation of the Ras-ERK/MAPK signaling pathway. J. Cell. Physiol. 2012, 227, 1235–1241. [Google Scholar] [CrossRef]
- Scott, L.J.; Mohlke, K.L.; Bonnycastle, L.L.; Willer, C.J.; Li, Y.; Duren, W.L.; Erdos, M.R.; Stringham, H.M.; Chines, P.S.; Jackson, A.U.; et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 2007, 316, 1341–1345. [Google Scholar] [CrossRef] [PubMed]
- Nutter, C.A.; Jaworski, E.A.; Verma, S.K.; Deshmukh, V.; Wang, Q.; Botvinnik, O.B.; Lozano, M.J.; Abass, I.J.; Ijaz, T.; Brasier, A.R.; et al. Dysregulation of RBFOX2 Is an Early Event in Cardiac Pathogenesis of Diabetes. Cell Rep. 2016, 15, 2200–2213. [Google Scholar] [CrossRef] [PubMed]
- Brosseau, J.P.; Lucier, J.F.; Nwilati, H.; Thibault, P.; Garneau, D.; Gendron, D.; Durand, M.; Couture, S.; Lapointe, E.; Prinos, P.; et al. Tumor microenvironment-associated modifications of alternative splicing. RNA 2014, 20, 189–201. [Google Scholar] [CrossRef]
- Jeyabal, P.; Thandavarayan, R.A.; Joladarashi, D.; Suresh Babu, S.; Krishnamurthy, S.; Bhimaraj, A.; Youker, K.A.; Kishore, R.; Krishnamurthy, P. MicroRNA-9 inhibits hyperglycemia-induced pyroptosis in human ventricular cardiomyocytes by targeting ELAVL1. Biochem. Biophys. Res. Commun. 2016, 471, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Huang, Z.; Tang, A.; Wu, X.; Aube, J.; Xu, L.; Xing, C.; Huang, Y. Inhibition of RNA-binding protein HuR reduces glomerulosclerosis in experimental nephritis. Clin. Sci. 2020, 134, 1433–1448. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Zhao, Z. Emerging role of HuR in inflammatory response in kidney diseases. Acta Biochim. Biophys. Sin. 2017, 49, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Lee, D.-Y.; Féliers, D.; Abboud, H.E.; Bhat, M.A.; Gorin, Y. Interplay between RNA-binding protein HuR and Nox4 as a novel therapeutic target in diabetic kidney disease. Mol. Metab. 2020, 36, 100968. [Google Scholar] [CrossRef]
- Govindaraju, S.; Lee, B.S. Adaptive and maladaptive expression of the mRNA regulatory protein HuR. World J. Biol. Chem. 2013, 4, 111–118. [Google Scholar] [CrossRef]
- Strawbridge, R.J.; Dupuis, J.; Prokopenko, I.; Barker, A.; Ahlqvist, E.; Rybin, D.; Petrie, J.R.; Travers, M.E.; Bouatia-Naji, N.; Dimas, A.S.; et al. Genome-Wide Association Identifies Nine Common Variants Associated With Fasting Proinsulin Levels and Provides New Insights Into the Pathophysiology of Type 2 Diabetes. Diabetes 2011, 60, 2624–2634. [Google Scholar] [CrossRef]
- Lai, W.S.; Carballo, E.; Thorn, J.M.; Kennington, E.A.; Blackshear, P.J. Interactions of CCCH Zinc Finger Proteins with mRNA: Binding of Tristetraprolin-Related Zinc Finger Proteins to Au-Rich Elements and Destabilization of mRNA. J. Biol. Chem. 2000, 275, 17827–17837. [Google Scholar] [CrossRef]
- Congrains, A.; Kamide, K.; Ohishi, M.; Rakugi, H. ANRIL: Molecular Mechanisms and Implications in Human Health. Int. J. Mol. Sci. 2013, 14, 1278–1292. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Eleftheriadou, M.; Kelaini, S.; Morrison, T.; González, M.V.; Caines, R.; Edwards, N.; Yacoub, A.; Edgar, K.; Moez, A.; et al. Targeting QKI-7 in vivo restores endothelial cell function in diabetes. Nat. Commun. 2020, 11, 3812. [Google Scholar] [CrossRef]
- He, R.-Z.; Luo, D.-X.; Mo, Y.-Y. Emerging roles of lncRNAs in the post-transcriptional regulation in cancer. Genes Dis. 2019, 6, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Froberg, J.E.; Lee, J.T. Long noncoding RNAs: Fresh perspectives into the RNA world. Trends Biochem. Sci. 2014, 39, 35–43. [Google Scholar] [CrossRef]
- Liu, J.Y.; Yao, J.; Li, X.M.; Song, Y.C.; Wang, X.Q.; Li, Y.J.; Yan, B.; Jiang, Q. Pathogenic role of lncRNA-MALAT1 in endothelial cell dysfunction in diabetes mellitus. Cell Death Dis. 2014, 5, e1506. [Google Scholar] [CrossRef]
- Puthanveetil, P.; Chen, S.; Feng, B.; Gautam, A.; Chakrabarti, S. Long non-coding RNA MALAT1 regulates hyperglycaemia induced inflammatory process in the endothelial cells. J. Cell. Mol. Med. 2015, 19, 1418–1425. [Google Scholar] [CrossRef]
- Yan, B.; Yao, J.; Liu, J.-Y.; Li, X.-M.; Wang, X.-Q.; Li, Y.-J.; Tao, Z.-F.; Song, Y.-C.; Chen, Q.; Jiang, Q. lncRNA-MIAT Regulates Microvascular Dysfunction by Functioning as a Competing Endogenous RNA. Circ. Res. 2015, 116, 1143–1156. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, D.; Ji, T.-F.; Shi, L.; Yu, J.-L. Overexpression of lncRNA ANRIL up-regulates VEGF expression and promotes angiogenesis of diabetes mellitus combined with cerebral infarction by activating NF-κB signaling pathway in a rat model. Oncotarget 2017, 8, 17347–17359. [Google Scholar] [CrossRef] [PubMed]
- Qiu, G.-Z.; Tian, W.; Fu, H.-T.; Li, C.-P.; Liu, B. Long noncoding RNA-MEG3 is involved in diabetes mellitus-related microvascular dysfunction. Biochem. Biophys. Res. Commun. 2016, 471, 135–141. [Google Scholar] [CrossRef]
- Qin, H.; Ni, H.; Liu, Y.; Yuan, Y.; Xi, T.; Li, X.; Zheng, L. RNA-binding proteins in tumor progression. J. Hematol. Oncol. 2020, 13, 90. [Google Scholar] [CrossRef]
- Wang, J.; Liu, Q.; Shyr, Y. Dysregulated transcription across diverse cancer types reveals the importance of RNA-binding protein in carcinogenesis. BMC Genom. 2015, 16, S5. [Google Scholar] [CrossRef]
- Li, B.; Liu, J.; Jia, Y.; Qin, T.; Xu, Z.; Zhang, Y.; Huang, G.; Xiao, Z. Clinical Features and Biological Implications of U2AF1 Mutations in Myelodysplastic Syndromes. Blood 2017, 130, 586. [Google Scholar] [CrossRef]
- Palangat, M.; Anastasakis, D.G.; Fei, D.L.; Lindblad, K.E.; Bradley, R.; Hourigan, C.S.; Hafner, M.; Larson, D.R. The splicing factor U2AF1 contributes to cancer progression through a noncanonical role in translation regulation. Genes Dev. 2019, 33, 482–497. [Google Scholar] [CrossRef] [PubMed]
- Quesada, V.; Conde, L.; Villamor, N.; Ordóñez, G.R.; Jares, P.; Bassaganyas, L.; Ramsay, A.J.; Beà, S.; Pinyol, M.; Martínez-Trillos, A.; et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat. Genet. 2011, 44, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lawrence, M.S.; Wan, Y.; Stojanov, P.; Sougnez, C.; Stevenson, K.; Werner, L.; Sivachenko, A.; DeLuca, D.S.; Zhang, L.; et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N. Engl. J. Med. 2011, 365, 2497–2506. [Google Scholar] [CrossRef]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef]
- Dang, H.; Takai, A.; Forgues, M.; Pomyen, Y.; Mou, H.; Xue, W.; Ray, D.; Ha, K.C.H.; Morris, Q.D.; Hughes, T.R.; et al. Oncogenic Activation of the RNA Binding Protein NELFE and MYC Signaling in Hepatocellular Carcinoma. Cancer Cell 2017, 32, 101–114.e108. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Babu, K.R.; Lim, C.Y.; Kwok, Z.H.; Li, J.; Zhou, S.; Yang, H.; Tay, Y. A comprehensive expression landscape of RNA-binding proteins (RBPs) across 16 human cancer types. RNA Biol. 2020, 17, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Modic, M.; Ule, J.; Sibley, C.R. CLIPing the brain: Studies of protein–RNA interactions important for neurodegenerative disorders. Mol. Cell. Neurosci. 2013, 56, 429–435. [Google Scholar] [CrossRef]
- Kang, D.; Lee, Y.; Lee, J.-S. RNA-Binding Proteins in Cancer: Functional and Therapeutic Perspectives. Cancers 2020, 12, 2699. [Google Scholar] [CrossRef] [PubMed]
- Van Kouwenhove, M.; Kedde, M.; Agami, R. MicroRNA regulation by RNA-binding proteins and its implications for cancer. Nat. Rev. Cancer 2011, 11, 644–656. [Google Scholar] [CrossRef]
- Balzeau, J.; Menezes, M.R.; Cao, S.; Hagan, J.P. The LIN28/let-7 Pathway in Cancer. Front. Genet. 2017, 8, 31. [Google Scholar] [CrossRef]
- Degrauwe, N.; Suvà, M.L.; Janiszewska, M.; Riggi, N.; Stamenkovic, I. IMPs: An RNA-binding protein family that provides a link between stem cell maintenance in normal development and cancer. Genes Dev. 2016, 30, 2459–2474. [Google Scholar] [CrossRef] [PubMed]
- Jonas, K.; Calin, G.A.; Pichler, M. RNA-Binding Proteins as Important Regulators of Long Non-Coding RNAs in Cancer. Int. J. Mol. Sci. 2020, 21, 2969. [Google Scholar] [CrossRef]
- Nagaoka, K.; Udagawa, T.; Richter, J.D. CPEB-mediated ZO-1 mRNA localization is required for epithelial tight-junction assembly and cell polarity. Nat. Commun. 2012, 3, 675. [Google Scholar] [CrossRef]
- Nagaoka, K.; Fujii, K.; Zhang, H.; Usuda, K.; Watanabe, G.; Ivshina, M.; Richter, J.D. CPEB1 mediates epithelial-to-mesenchyme transition and breast cancer metastasis. Oncogene 2016, 35, 2893–2901. [Google Scholar] [CrossRef]
- Garneau, N.L.; Wilusz, J.; Wilusz, C.J. The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol. 2007, 8, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-Y.A.; Shyu, A.-B. Deadenylation and P-bodies. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2013; Volume 768, pp. 183–195. [Google Scholar] [CrossRef] [PubMed]
- Perron, G.; Jandaghi, P.; Solanki, S.; Safisamghabadi, M.; Storoz, C.; Karimzadeh, M.; Papadakis, A.I.; Arseneault, M.; Scelo, G.; Banks, R.E.; et al. A General Framework for Interrogation of mRNA Stability Programs Identifies RNA-Binding Proteins that Govern Cancer Transcriptomes. Cell Rep. 2018, 23, 1639–1650. [Google Scholar] [CrossRef] [PubMed]
- Truitt, M.L.; Ruggero, D. Erratum: New frontiers in translational control of the cancer genome. Nat. Rev. Cancer 2017, 17, 332. [Google Scholar] [CrossRef]
- Hsieh, A.C.; Ruggero, D. Targeting Eukaryotic Translation Initiation Factor 4E (eIF4E) in Cancer. Clin. Cancer Res. 2010, 16, 4914–4920. [Google Scholar] [CrossRef]
- Nilsen, T.W.; Graveley, B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature 2010, 463, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Biamonti, G.; Catillo, M.; Pignataro, D.; Montecucco, A.; Ghigna, C. The alternative splicing side of cancer. Semin. Cell Dev. Biol. 2014, 32, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Dvinge, H.; Kim, E.; Abdel-Wahab, O.; Bradley, R.K. RNA splicing factors as oncoproteins and tumour suppressors. Nat. Rev. Cancer 2016, 16, 413–430. [Google Scholar] [CrossRef]
- Wang, J.; Liu, T.; Wang, M.; Lv, W.; Wang, Y.; Jia, Y.; Zhang, R.; Liu, L. SRSF1-dependent alternative splicing attenuates BIN1 expression in non-small cell lung cancer. J. Cell. Biochem. 2020, 121, 946–953. [Google Scholar] [CrossRef]
- Venkat, S.; Tisdale, A.A.; Schwarz, J.R.; Alahmari, A.A.; Maurer, H.C.; Olive, K.P.; Eng, K.H.; Feigin, M.E. Alternative polyadenylation drives oncogenic gene expression in pancreatic ductal adenocarcinoma. Genome Res. 2020, 30, 347–360. [Google Scholar] [CrossRef]
- Erson-Bensan, A.E.; Can, T. Alternative Polyadenylation: Another Foe in Cancer. Mol. Cancer Res. 2016, 14, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Langa, K.M.; Larson, E.B.; Crimmins, E.M.; Faul, J.D.; Levine, D.A.; Kabeto, M.U.; Weir, D.R. A Comparison of the Prevalence of Dementia in the United States in 2000 and 2012. JAMA Intern. Med. 2017, 177, 51–58. [Google Scholar] [CrossRef]
- Nussbacher, J.K.; Tabet, R.; Yeo, G.W.; Lagier-Tourenne, C. Disruption of RNA Metabolism in Neurological Diseases and Emerging Therapeutic Interventions. Neuron 2019, 102, 294–320. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.C.; Ng, C.S.; Xiang, P.; Liu, H.; Zhang, K.; Mohamud, Y.; Luo, H. Dysregulation of RNA-Binding Proteins in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2020, 13, 78. [Google Scholar] [CrossRef]
- Mackenzie, I.R.A.; Rademakers, R.; Neumann, M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010, 9, 995–1007. [Google Scholar] [CrossRef]
- Abramzon, Y.A.; Fratta, P.; Traynor, B.J.; Chia, R. The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front. Neurosci. 2020, 14. [Google Scholar] [CrossRef]
- Liu, E.Y.; Cali, C.P.; Lee, E.B. RNA metabolism in neurodegenerative disease. Dis. Models Mech. 2017, 10, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.O.; Pioro, E.P.; Boehringer, A.; Chia, R.; Feit, H.; Renton, A.E.; Pliner, H.A.; Abramzon, Y.; Marangi, G.; Winborn, B.J.; et al. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat. Neurosci. 2014, 17, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Schultz, P.G.; Johnson, K.A. Mechanistic studies of a small-molecule modulator of SMN2 splicing. Proc. Natl. Acad. Sci. USA 2018, 115, E4604–E4612. [Google Scholar] [CrossRef] [PubMed]
- Buckanovich, R.J.; Posner, J.B.; Darnell, R.B. Nova, the paraneoplastic Ri antigen, is homologous to an RNA-binding protein and is specifically expressed in the developing motor system. Neuron 1993, 11, 657–672. [Google Scholar] [CrossRef]
- Ule, J.; Jensen, K.B.; Ruggiu, M.; Mele, A.; Ule, A.; Darnell, R.B. CLIP identifies Nova-regulated RNA networks in the brain. Science 2003, 302, 1212–1215. [Google Scholar] [CrossRef] [PubMed]
- Milne, C.A.; Hodgkin, J. ETR-1, a homologue of a protein linked to myotonic dystrophy, is essential for muscle development in Caenorhabditis elegans. Curr. Biol. 1999, 9, 1243–1246. [Google Scholar] [CrossRef]
- Belzil, V.V.; Gendron, T.F.; Petrucelli, L. RNA-mediated toxicity in neurodegenerative disease. Mol. Cell. Neurosci. 2013, 56, 406–419. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, N.C.; Wang, Y.D.; Scarborough, E.A.; Moore, J.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.; Molliex, A.; et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013, 495, 467–473. [Google Scholar] [CrossRef]
- Li, Y.R.; King, O.D.; Shorter, J.; Gitler, A.D. Stress granules as crucibles of ALS pathogenesis. J. Cell Biol. 2013, 201, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Courchaine, E.M.; Lu, A.; Neugebauer, K.M. Droplet organelles? EMBO J. 2016, 35, 1603–1612. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Calidas, D.; Schmidt, H.; Lu, T.; Rasoloson, D.; Seydoux, G. Spatial patterning of P granules by RNA-induced phase separation of the intrinsically-disordered protein MEG-3. Elife 2016, 5, e21337. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.; Kato, M.; Wu, L.C.; Lin, Y.; Ding, M.; Zhang, Y.; Yu, Y.; McKnight, S.L. The LC Domain of hnRNPA2 Adopts Similar Conformations in Hydrogel Polymers, Liquid-like Droplets, and Nuclei. Cell 2015, 163, 829–839. [Google Scholar] [CrossRef]
- Vanderweyde, T.; Yu, H.; Varnum, M.; Liu-Yesucevitz, L.; Citro, A.; Ikezu, T.; Duff, K.; Wolozin, B. Contrasting pathology of the stress granule proteins TIA-1 and G3BP in tauopathies. J. Neurosci. 2012, 32, 8270–8283. [Google Scholar] [CrossRef]
- Gibbings, D.J.; Ciaudo, C.; Erhardt, M.; Voinnet, O. Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat. Cell Biol. 2009, 11, 1143–1149. [Google Scholar] [CrossRef]
- Wolozin, B.; Apicco, D. RNA binding proteins and the genesis of neurodegenerative diseases. Adv. Exp. Med. Biol. 2015, 822, 11–15. [Google Scholar] [CrossRef]
- Yang, C.; Kelaini, S.; Caines, R.; Margariti, A. RBPs Play Important Roles in Vascular Endothelial Dysfunction under Diabetic Conditions. Front. Physiol. 2018, 9, 1310. [Google Scholar] [CrossRef]
- Aslam, N.; Zaheer, I. The biosynthesis characteristics of TTP and TNF can be regulated through a posttranscriptional molecular loop. J. Biol. Chem. 2011, 286, 3767–3776. [Google Scholar] [CrossRef]
- Zhang, Y.; Si, Y.; Ma, N.; Mei, J. The RNA-binding protein PCBP2 inhibits Ang II-induced hypertrophy of cardiomyocytes though promoting GPR56 mRNA degeneration. Biochem. Biophys. Res. Commun. 2015, 464, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Gomes, C.P.C.; Schroen, B.; Kuster, G.M.; Robinson, E.L.; Ford, K.; Squire, I.B.; Heymans, S.; Martelli, F.; Emanueli, C.; Devaux, Y. Regulatory RNAs in Heart Failure. Circulation 2020, 141, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Mohibi, S.; Chen, X.; Zhang, J. Cancer the‘RBP’eutics–RNA-binding proteins as therapeutic targets for cancer. Pharmacol. Ther. 2019, 203, 107390. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.A.; Castanotto, D. FDA-Approved Oligonucleotide Therapies in 2017. Mol. Ther. 2017, 25, 1069–1075. [Google Scholar] [CrossRef]
- Duffy, A.G.; Makarova-Rusher, O.V.; Ulahannan, S.V.; Rahma, O.E.; Fioravanti, S.; Walker, M.; Abdullah, S.; Raffeld, M.; Anderson, V.; Abi-Jaoudeh, N.; et al. Modulation of tumor eIF4E by antisense inhibition: A phase I/II translational clinical trial of ISIS 183750—An antisense oligonucleotide against eIF4E—In combination with irinotecan in solid tumors and irinotecan-refractory colorectal cancer. Int. J. Cancer 2016, 139, 1648–1657. [Google Scholar] [CrossRef]
- Huang, Y.-J.; Xiao, S.; Sun, Z.-L.; Zeng, J.-G.; Liu, Y.-S.; Liu, Z.-Y. Identification of allocryptopine and protopine metabolites in rat liver S9 by high-performance liquid chromatography/quadrupole-time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2016, 30, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Mongelard, F.; Bouvet, P. AS-1411, a guanosine-rich oligonucleotide aptamer targeting nucleolin for the potential treatment of cancer, including acute myeloid leukemia. Curr. Opin. Mol. 2010, 12, 107–114. [Google Scholar]
- Kristensen, L.S.; Hansen, T.B.; Venø, M.T.; Kjems, J. Circular RNAs in cancer: Opportunities and challenges in the field. Oncogene 2018, 37, 555–565. [Google Scholar] [CrossRef]
- Abdelmohsen, K.; Panda, A.C.; Munk, R.; Grammatikakis, I.; Dudekula, D.B.; De, S.; Kim, J.; Noh, J.H.; Kim, K.M.; Martindale, J.L.; et al. Identification of HuR target circular RNAs uncovers suppression of PABPN1 translation by CircPABPN1. RNA Biol. 2017, 14, 361–369. [Google Scholar] [CrossRef]
- Tian, X.; Gu, T.; Patel, S.; Bode, A.M.; Lee, M.-H.; Dong, Z. CRISPR/Cas9—An evolving biological tool kit for cancer biology and oncology. NPJ Precis. Oncol. 2019, 3, 8. [Google Scholar] [CrossRef]
- Gorkovskiy, A.; Reidy, M.; Masison, D.C.; Wickner, R.B. Hsp104 disaggregase at normal levels cures many [PSI+] prion variants in a process promoted by Sti1p, Hsp90, and Sis1p. Proc. Natl. Acad. Sci. USA 2017, 114, E4193–E4202. [Google Scholar] [CrossRef]
- Jackrel, M.E.; DeSantis, M.E.; Martinez, B.A.; Castellano, L.M.; Stewart, R.M.; Caldwell, K.A.; Caldwell, G.A.; Shorter, J. Potentiated Hsp104 variants antagonize diverse proteotoxic misfolding events. Cell 2014, 156, 170–182. [Google Scholar] [CrossRef]
- Chen, H.-J.; Mitchell, J.C.; Novoselov, S.; Miller, J.; Nishimura, A.L.; Scotter, E.L.; Vance, C.A.; Cheetham, M.E.; Shaw, C.E. The heat shock response plays an important role in TDP-43 clearance: Evidence for dysfunction in amyotrophic lateral sclerosis. Brain 2016, 139, 1417–1432. [Google Scholar] [CrossRef] [PubMed]
- Barmada, S.J.; Serio, A.; Arjun, A.; Bilican, B.; Daub, A.; Ando, D.M.; Tsvetkov, A.; Pleiss, M.; Li, X.; Peisach, D.; et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat. Chem. Biol. 2014, 10, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.-F.; Guo, B.-S.; Liu, Y.-C.; Wu, C.-C.; Yang, C.-H.; Tsai, K.-J.; Shen, C.-K.J. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc. Natl. Acad. Sci. USA 2012, 109, 15024–15029. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| RNA Binding Protein | Functions in Pathology | Disease Outcomes | |
|---|---|---|---|
| RNA Binding Fox-1 Homolog 2 | RBFOX2 | Regulation of alternative splicing | Diabetic cardiomyopathy via alternative splicing defects of genes important for healthy cardiac regulation |
| Human Antigen R/ELAV Like RNA Binding Protein 1 | HuR/ELAV1 | Inducement of RNA stabilization and promotion of mRNA translation via binding to 3′UTR AREs | Diabetic nephropathy via binding of target genes such as SNAIL and FOS which contribute to EMT and nephropathy in diabetic conditions |
| Tristetraprolin | TTP | Inducement of RNA destabilisation and decay via binding to 3′UTR AREs | Atherosclerosis progression, and inflammation in TTP-deficient ECs |
| Quaking | QKI | Enablement of mRNA degradation | Diabetic EC dysfunction via degradation of targets such as VE-cadherin |
| U2 Small Nuclear RNA Auxiliary Factor 1 | U2AF1 | Mutations associated with disruption to pre-mRNA alternative splicing | Cancer progression via differential splicing of cancer-relevant gene targets in MDS |
| Mutated Splicing Factor 3b Subunit 1 | SF3B1 | Mutations associated with disruption to pre-mRNA alternative splicing | Cancer progression in CLL |
| Negative Elongation Factor E | NELFE | Inducement of mRNA stabilisation of protooncogenes | Cancer progression by stabilization of MYC-associated genes and MYC signalling in HCC |
| Lin-28 Homolog A | LIN28 | Blocking of miRNA processing and maturation | Cancer development and progression via promotion of several cellular functions involved in cell proliferation, invasion, and angiogenesis |
| Cytoplasmic Polyadenylation Element Binding Protein 1 | CPEB1 | Enablement of mRNA localization Inducement of alternative polyadenylation | Cancer progression via promotion of cancer cell migration CPEB1 deficiency associated with cancer development |
| Insulin Like Growth Factor 2 MRNA Binding Protein 1 | IGF2BP | Inducement of mRNA stability, translocation, and translation | Cancer progression via stabilization and translation of cancer-relevant mRNA |
| Eukaryotic Translation Initiation Factor 4E | eIF4E | Regulation of mRNA translation | Promotion of tumorigenesis by translation of protooncogenes, and malignancy-related factors |
| Serine/Arginine-Rich Splicing Factor 1 | SRSF1 | Regulation of alternative splicing | Cancer progression via splicing of protooncogenes and tumor suppressor genes |
| Ataxin 2 | ATXN2 | Mutations in genes elevated in neurodegenerative disorder | Progression and development of neurodegenerative disorder ALS |
| Heterogenous Nuclear Ribonucleoprotein A1 | hnRNPA1 | ||
| Matrin 3 | MATR3 | ||
| TIA1 Cytotoxic Granule Associated RNA Binding Protein | TIA-1 | ||
| TAR DNA-binding protein 43 | TDP-43 | Fragmentation and formation of inclusion bodies | Promotion of neurodegenerative disease advancement in ALS |
| FUS RNA Binding Protein | FUS | Regulation of RNA translocation, and localization in stress granules | Neuronal disease onset by stress granule aggregation |
| Neuro-oncological ventral antigen 1 and 2 | Nova 1 and 2 | Regulation of alternative splicing | POMA onset by autoantibody secretion |
| Far Upstream Element Binding Protein 1 | FUBP1 | Regulation of alternative splicing | Involvement in SMA by increasing FUBP1 affinity to SNF1 pre-mRNA. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kelaini, S.; Chan, C.; Cornelius, V.A.; Margariti, A. RNA-Binding Proteins Hold Key Roles in Function, Dysfunction, and Disease. Biology 2021, 10, 366. https://doi.org/10.3390/biology10050366
Kelaini S, Chan C, Cornelius VA, Margariti A. RNA-Binding Proteins Hold Key Roles in Function, Dysfunction, and Disease. Biology. 2021; 10(5):366. https://doi.org/10.3390/biology10050366
Chicago/Turabian StyleKelaini, Sophia, Celine Chan, Victoria A Cornelius, and Andriana Margariti. 2021. "RNA-Binding Proteins Hold Key Roles in Function, Dysfunction, and Disease" Biology 10, no. 5: 366. https://doi.org/10.3390/biology10050366
APA StyleKelaini, S., Chan, C., Cornelius, V. A., & Margariti, A. (2021). RNA-Binding Proteins Hold Key Roles in Function, Dysfunction, and Disease. Biology, 10(5), 366. https://doi.org/10.3390/biology10050366