
A Blood–Bone–Tooth Model for Age Prediction in Forensic Contexts



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Population Sample
2.2. Sanger Sequencing of C1orf132 in Blood Samples from Living Individuals
2.3. Statistical Analyses
3. Results
3.1. Multi-Tissue BBT-APM using Sanger Sequencing
3.2. Multi-Tissue BBT-APM Using SNaPshot Methodology
3.3. Differences between Predicted and Chronological Ages with an Increase in Age
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goel, N.; Karira, P.; Garg, V.K. Role of DNA methylation in human age prediction. Mech. Ageing Dev. 2017, 166, 33–41. [Google Scholar] [CrossRef]
- Jung, S.E.; Lim, S.M.; Hong, S.R.; Lee, E.H.; Shin, K.J.; Lee, H.Y. DNA methylation of the ELOVL2, FHL2, KLF14, C1orf132/MIR29B2C, and TRIM59 genes for age prediction from blood, saliva, and buccal swab samples. Forensic Sci. Int. Genet. 2019, 38, 1–8. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [Green Version]
- Alsaleh, H.; McCallum, N.A.; Halligan, D.L.; Haddrill, P.R. A multi-tissue age prediction model based on DNA methylation analysis. Forensic Sci. Int. Genet. Suppl. Ser. 2017, 6, e62–e64. [Google Scholar] [CrossRef] [Green Version]
- Correia Dias, H.; Cordeiro, C.; Corte Real, F.; Cunha, E.; Manco, L. Age estimation based on DNA methylation using blood samples from deceased individuals. J. Forensic Sci. 2019, 65, 465–470. [Google Scholar] [CrossRef]
- Correia Dias, H.; Cunha, E.; Corte Real, F.; Manco, L. Age prediction in living: Forensic epigenetic age estimation based on blood samples. Leg. Med. 2020, 47, 101763. [Google Scholar] [CrossRef] [PubMed]
- Correia Dias, H.; Cordeiro, C.; Pereira, J.; Pinto, C.; Corte Real, F.; Cunha, E.; Manco, L. DNA methylation age estimation in blood samples of living and deceased individuals using a multiplex SNaPshot assay. Forensic Sci. Int. 2020, 311, 110267. [Google Scholar] [CrossRef]
- Correia Dias, H.; Corte Real, F.; Cunha, E.; Manco, L. DNA methylation age estimation from human bone and teeth. Aust. J. Forensic Sci. 2021, Ahead-of-print. 1–14. [Google Scholar] [CrossRef]
- Bocklandt, S.; Lin, W.; Sehl, M.E.; Sanchez, F.J.; Sinsheimer, J.S.; Horvath, S.; Vilain, E. Epigenetic predictor of age. PLoS ONE 2011, 6, e14821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garagnani, P.; Bacalini, M.G.; Pirazzini, C.; Gori, D.; Giuliani, C.; Mari, D.; Di Blasio, A.M.D.; Gentilini, D.; Vitale, G.; Collino, S.; et al. Methylation of ELOVL2 gene as a new epigenetic marker of age. Aging Cell 2012, 11, 1132–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekaert, B.; Kamalandua, A.; Zapico, S.C.; Van de Voorde, W.; Decorte, R. Improved age determination of blood and teeth samples using a selected set of DNA methylation markers. Epigenetics 2015, 10, 922–930. [Google Scholar] [CrossRef] [Green Version]
- Zbieć-Piekarska, R.; Spólnicka, M.; Kupiec, T.; Parys-Proszek, A.; Makowska, Z.; Pałeczka, A.; Kucharczyk, K.; Płoski, R.; Branicki, W. Development of a forensically useful age prediction method based on DNA methylation analysis. Forensic Sci. Int. Genet. 2015, 17, 173–179. [Google Scholar] [CrossRef]
- Naue, J.; Sänger, T.; Hoefsloot, H.C.J.; Lutz-Bonengel, S.; Kloosterman, A.D.; Verschure, P.J. Proof of concept study of age-dependent DNA methylation markers across different tissues by massive parallel sequencing. Forensic Sci. Int. Genet. 2018, 36, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Gopalan, S.; Gaige, J.; Henn, B.M. DNA methylation-based forensic age estimation in human bone. BioRxiv 2019, 801647. [Google Scholar] [CrossRef] [Green Version]
- Márquez-Ruiz, A.B.; González-Herrera, L.; Luna, J.D.; Valenzuela, A. DNA methylation levels and telomere length in human teeth: Usefulness for age estimation. Int. J. Leg. Med. 2020, 134, 451–459. [Google Scholar] [CrossRef]
- Eipel, M.; Mayer, F.; Arent, T.; Ferreira, M.R.P.; Birkhofer, C.; Costa, I.G.; Ritz-Timme, S.; Wagner, W. Epigenetic age predictions based on buccal swabs are more precise in combination with cell type—Specific DNA methylation signatures. Aging 2016, 8, 1034–1048. [Google Scholar] [CrossRef] [Green Version]
- Koch, M.C.; Wagner, W. Epigenetic-aging-signature to determine age in different tissues. Aging 2011, 3, 1018. [Google Scholar] [CrossRef] [Green Version]
- Illingworth, R.; Kerr, A.; Desousa, D.; Jørgensen, H.; Ellis, P.; Stalker, J.; Jackson, D.; Clee, C.; Plumb, R.; Rogers, J.; et al. A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol. 2008, 6, e22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zhu, J.; Tian, G.; Li, N.; Li, Q.; Ye, M.; Zheng, H.; Yu, J.; Wu, H.; Sun, J.; et al. The DNA methylome of human peripheral blood mononuclear cells. PLoS Biol. 2010, 8, e1000533. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Stamatoyannopoulos, J.A.; Costello, J.F.; Ren, B.; Milosavljevic, A.; Meissner, A.; Kellis, M.; Marra, M.A.; Beaudet, A.L.; Ecker, J.R.; et al. The NIH roadmap epigenomics mapping consortium. Nat. Biotechnol. 2010, 28, 1045–1048. [Google Scholar] [CrossRef] [Green Version]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.-B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell. 2013, 49, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Weidner, C.I.; Lin, Q.; Koch, C.M.; Eisele, L.; Beier, F.; Ziegler, P.; Bauerschlag, D.O.; Jöckel, K.H.; Erbel, R.; Mühleisen, T.W.; et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014, 15, R24. [Google Scholar] [CrossRef] [Green Version]
- Bekaert, B.; Kamalandua, A.; Zapico, S.C.; Van de Voorde, W.; Decorte, R. A selective set of DNA-methylation markers for age determination of blood, teeth and buccal samples. Forensic Sci. Int. Genet. Suppl. Ser. 2015, 5, e144–e145. [Google Scholar] [CrossRef] [Green Version]
- Zbieć-Piekarska, R.; Spólnicka, M.; Kupiec, T.; Makowska, Z.; Spas, A.; Parys-Proszek, A.; Kucharczyk, K.; Płoski, R.; Branicki, W. Examination of DNA methylation status of the ELOVL2 marker may be useful for human age prediction in forensic science. Forensic Sci. Int. Genet. 2015, 14, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Qu, H.; Wang, G.; Xie, B.; Shi, Y.; Yang, Y.; Zhao, Z.; Hu, L.; Fang, X.; Yan, J.; et al. A novel strategy for forensic age prediction by DNA methylation and support vector regression model. Nature 2015, 5, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Hamano, Y.; Manabe, S.; Morimoto, C.; Fujimoto, S.; Ozeki, M.; Tamaki, K. Forensic age prediction for dead or living samples by use of methylation-sensitive high resolution melting. Leg. Med. 2016, 21, 5–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giuliani, C.; Cilli, E.; Bacalini, M.G.; Pirazzini, C.; Sazzini, M.; Gruppioni, G.; Franceschi, C.; Garagnani, P.; Luiselli, D. Inferring chronological age from DNA methylation patterns of human teeth. Am. J. Phys. Anthropol. 2016, 159, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Freire-Aradas, A.; Phillips, C.; Mosquera-Miguel, A.; Girón-Santamaría, L.; Gómez-Tato, A.; de Cal, M.C.; Álvarez-Dios, J.; Ansede-Bermejo, J.; Torres-Español, M.; Schneider, P.M.; et al. Development of a methylation marker set for forensic age estimation using analysis of public methylation data and the Agena Bioscience EpiTYPER system. Forensic Sci. Int. Genet. 2016, 24, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Jung, S.-E.; Hong, S.R.; Lee, E.H.; Lee, J.H.; Lee, S.D.; Lee, H.Y. Independent validation of DNA-based approaches for age prediction in blood. Forensic Sci. Int. Genet. 2017, 29, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Thong, Z.; Chan, X.L.S.; Tan, J.Y.Y.; Loo, E.S.; Syn, C.K.C. Evaluation of DNA methylation-based age prediction on blood. Forensic Sci. Int. Genet. Suppl. Ser. 2017, 6, e249–e251. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.J.; Goodman, S.J.; Kobor, M.S. DNA methylation and healthy human aging. Aging Cell 2015, 14, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Heijmans, B.T.; Hjelmborg, J.V.; Soerensen, M.; Christensen, K.; Christiansen, L. Epigenetic drift in the aging genome: A ten-year follow-up in an elderly twin cohort. Int. J. Epidemiol. 2016, 45, 1146–1158. [Google Scholar] [CrossRef] [Green Version]
- Freire-Aradas, A.; Phillips, C.; Lareu, M.V. Forensic individual age estimation with DNA: From initial approaches to methylation tests. Forensic Sci. Rev. 2017, 29, 121–144. [Google Scholar] [PubMed]
- Hofreiter, N.; Jaenicke, V.; Serre, D.; von Haeseler, A.; Paabo, S. DNA sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA. Nucleic Acids Res. 2001, 29, 4793–4799. [Google Scholar] [CrossRef] [PubMed]
- Brotherton, P.; Endicott, P.; Sanchez, J.J.; Beaumont, M.; Barnett, R.; Austin, J.; Cooper, A. Novel high-resolution characterization of ancient DNA reveals C > U-type base modification events as the sole cause of post mortem miscoding lesions. Nucleic Acids Res. 2007, 35, 5717–5728. [Google Scholar] [CrossRef] [Green Version]
- Dahl, C.; Grønbæk, K.; Guldberg, P. Advances in DNA methylation: 5-hydroxymethylcytosine revisited. Clin. Chim. Acta 2011, 412, 831–836. [Google Scholar] [CrossRef]
- Shen, L.; Zhang, Y. 5-Hydroxymethylcytosine: Generation, fate, and genomic Distribution. Curr. Opin. Cell Biol. 2013, 25, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Briggs, A.W.; Stenzel, U.; Meyer, M.; Krause, J.; Kircher, M.; Paabo, S. Removal of deaminated cytosines and detection of in vivo methylation in ancient DNA. Nucleic Acids Res. 2010, 38, e87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llamas, B.; Holland, M.L.; Chen, K.; Cropley, J.E.; Cooper, A.; Suter, C.M. High-resolution analysis of cytosine methylation in ancient DNA. PLoS ONE 2012, 7, e30226. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, J.S.; Valen, E.; Velazquez, A.M.; Parker, B.J.; Rasmussen, M.; Lindgreen, S.; Lilje, B.; Tobin, D.J.; Kelly, T.K.; Vang, S.; et al. Genome-wide nucleosome map and cytosine methylation levels of an ancient human genome. Genome Res. 2014, 24, 454–466. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Locus | CpG Site | Location | Multi-Tissue: Type of Samples Included | N | R | R2 | Corrected R2 | SE | p-Value | MAD |
|---|---|---|---|---|---|---|---|---|---|---|
| Simple linear regression | ||||||||||
| ELOVL2 | CpG6 | Chr6:11044644 | Blood * + Bones + Teeth | 185 | 0.759 | 0.576 | 0.573 | 14.70 | 6.87 × 10−36 | 12.01 |
| FHL2 | CpG1 | Chr2:105399282 | Blood * + Bones + Teeth | 185 | 0.692 | 0.479 | 0.476 | 16.29 | 1.11 × 10−27 | 13.16 |
| EDARADD | CpG3 | Chr1:236394382 | Blood * + Bones + Teeth | 185 | −0.682 | 0.465 | 0.462 | 16.51 | 1.21 × 10−26 | 13.52 |
| C1orf132 | CpG1 | Chr1:207823681 | Blood * + Bones + Teeth | 185 | −0.654 | 0.428 | 0.425 | 17.07 | 5.67 × 10−24 | 13.23 |
| PDE4C | CpG2 | Chr19:18233133 | Blood * + Bones + Teeth | 185 | 0.613 | 0.376 | 0.372 | 17.83 | 1.79 × 10−20 | 13.58 |
| Multiple linear regression | ||||||||||
| APM (EDARADD CpG3, FHL2 CpG5, FHL2 CpG11, ELOVL2 CpG5, PDE4C CpG5, PDE4C CpG9, C1orf132 CpG3) | Blood * + Bones + Teeth | 185 | 0.940 | 0.883 | 0.878 | 7.86 | 7.36 × 10−79 | 6.06 | ||
| Locus | Location | Multi-Tissue: Type of Samples Included | N | R | R2 | Corrected R2 | SE | p-Value | MAD |
|---|---|---|---|---|---|---|---|---|---|
| Simple linear regression | |||||||||
| ELOVL2 | Chr6:11044628 | Blood * + Bones + Teeth | 168 | 0.772 | 0.597 | 0.594 | 13.896 | 1.54 × 10−34 | 10.95 |
| FHL2 | Chr2:105399282 | Blood * + Bones + Teeth | 168 | 0.686 | 0.471 | 0.468 | 15.885 | 1.36 × 10−24 | 12.63 |
| KLF14 | Chr7:130734355 | Blood * + Bones + Teeth | 168 | 0.677 | 0.459 | 0.456 | 16.091 | 6.57 × 10−24 | 12.74 |
| C1orf132 | Chr1:207823681 | Blood * + Bones + Teeth | 168 | −0.693 | 0.480 | 0.477 | 15.779 | 2.49 × 10−25 | 12.10 |
| TRIM59 | Chr3:160450189 | Blood * + Bones + Teeth | 168 | 0.584 | 0.341 | 0.337 | 17.780 | 1.17 × 10−16 | 13.64 |
| Multiple linear regression | |||||||||
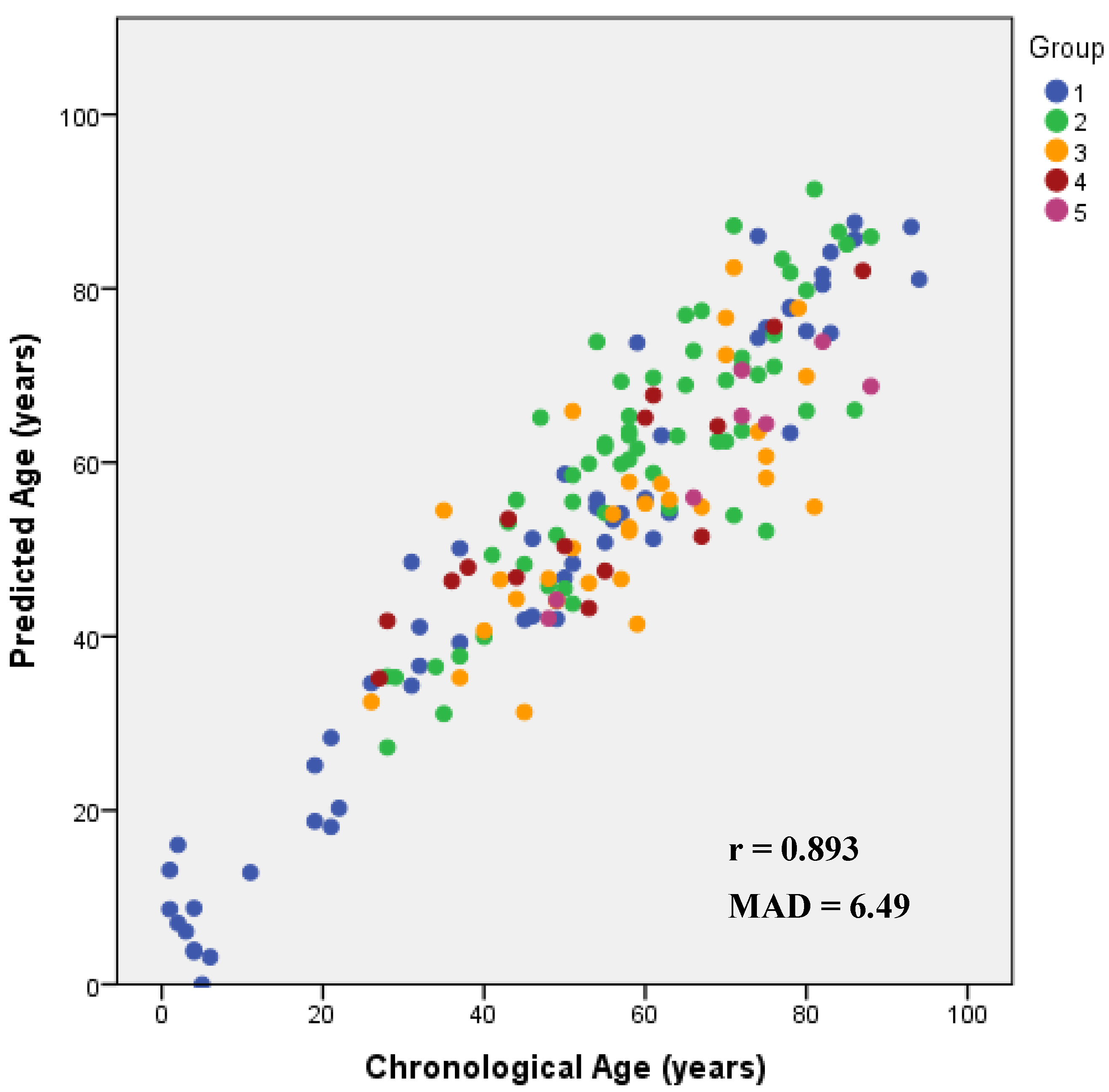
| APM (ELOVL2, KLF14 and C1orf132) | Blood * + Bones + Teeth | 168 | 0.922 | 0.850 | 0.847 | 8.53 | 3.14 × 10−67 | 6.49 | |
| Method | Sanger Sequencing | SNaPshot | |||
|---|---|---|---|---|---|
| Age Range | N | MAD (Years) | N | MAD (Years) | |
| <30 years | 33 | 4.73 | 23 | 5.51 | |
| 31–55 years | 58 | 6.37 | 56 | 6.23 | |
| 56–79 years | 74 | 5.67 | 68 | 6.74 | |
| >80 years | 20 | 8.81 | 21 | 7.37 | |
| Method | Sanger Sequencing | SNaPshot |
|---|---|---|
| CpGs and genes included in the APM | 7 CpGs located at 5 genes (EDARADD CpG3, FHL2 CpG5, FHL2 CpG11, ELOVL2 CpG5, PDE4C CpG5, PDE4C CpG9, C1orf132 CpG3) | 3 CpGs located at 3 genes (ELOVL2, KLF14, C1orf132) |
| Age correlation value | 0.940 | 0.922 |
| Variance in age explained | 87.8% | 84.7% |
| Accuracy (MAD) | 6.06 years | 6.49 years |
| Results | Using the Sanger sequencing methodology, more CpGs and genes were included in the APM, but higher age correlation, higher explained variance in age, and a better accuracy in age prediction (lower MAD value) were obtained. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Correia Dias, H.; Manco, L.; Corte Real, F.; Cunha, E. A Blood–Bone–Tooth Model for Age Prediction in Forensic Contexts. Biology 2021, 10, 1312. https://doi.org/10.3390/biology10121312
Correia Dias H, Manco L, Corte Real F, Cunha E. A Blood–Bone–Tooth Model for Age Prediction in Forensic Contexts. Biology. 2021; 10(12):1312. https://doi.org/10.3390/biology10121312
Chicago/Turabian StyleCorreia Dias, Helena, Licínio Manco, Francisco Corte Real, and Eugénia Cunha. 2021. "A Blood–Bone–Tooth Model for Age Prediction in Forensic Contexts" Biology 10, no. 12: 1312. https://doi.org/10.3390/biology10121312
APA StyleCorreia Dias, H., Manco, L., Corte Real, F., & Cunha, E. (2021). A Blood–Bone–Tooth Model for Age Prediction in Forensic Contexts. Biology, 10(12), 1312. https://doi.org/10.3390/biology10121312