Abstract
Porous carbon fibers play a pivotal role in electrochemistry due to their unique structural and textural properties, offering a promising avenue for diverse applications ranging from energy storage to electrocatalysis. In this study, we investigate the intricate relationship between the electrochemical responses of porous carbon fibers synthesized using the Design of Experiments protocol and their textural properties, aiming to elucidate key insights for material design and optimization. Through comprehensive correlation analyses, we uncover notable associations between oxygen reduction reaction mass activities and capacitances measured at different polarization rates, highlighting the significance of pore accessibility in dictating electrochemical performance. While direct correlations with specific surface area and total pore volume for mass activities were not observed, our findings reveal significant trends regarding capacitance retention. Specifically, materials with an elevated specific surface area and total pore volume demonstrate enhanced capacitance retention, particularly under varying charging and discharging rates. These results underscore the importance of optimizing specific surface area and pore volume to maximize capacitive performance across diverse operating conditions. Our study provides valuable guidance for developing porous carbon fibers tailored for superior electrochemical performance in various applications.
1. Introduction
The increasing energy demands of our society, coupled with the anticipated depletion of fossil fuel reserves, requires advancing effective energy conversion and storage technologies. Electrochemical power sources reliant on carbon, such as lithium ion, lithium–sulfur, sodium ion supercapacitors, metal ion capacitors, etc., are at the forefront of addressing this challenge, offering a wide range of energy and power densities suitable for diverse mobile and stationary applications [1].
Among a myriad of carbon materials, porous carbon fibers present a class of materials that offer unique property profiles and have garnered significant interest [2,3]. Cellulose has been identified as an excellent precursor, offering sustainability as well as technical benefits compared to fossil-based precursors like polyacrylonitrile (PAN) or pitch [4]. Despite a large variety of available natural cellulosic fibers, man-made cellulose fibers like lyocell or viscose are considered to be superior due to their higher homogeneity and the possibility to tailor fiber properties [5]. Cellulose-based porous carbon fibers can offer unique properties and a wide range of potential applications [6,7,8]. They exhibit excellent mechanical performance, high surface area, tunable pore size distribution, and low electrical resistance along the fiber axis [3]. Depending on the designed properties of cellulose-based porous carbon fibers, they can be used in various applications such as energy storage devices [9,10], catalysis [11], adsorption [12,13,14], and environmental remediation [15,16], among others.
Several methods have been developed for the synthesis of cellulose-based porous carbon fibers, including electrospinning [17,18], template synthesis [19], direct carbonization of cellulose precursors [20], or various chemical and physical activation treatments [10,21,22,23]. Each method offers unique advantages in terms of control over pore structure and scalability, all of which can determine the applicability of a material. Special attention has to be paid to retaining the fibrous morphology of the precursor in order to fully exploit its benefits compared to conventional particulate porous carbons. Breitenbach et al. have shown that KOH activation leads to the destruction of the fiber structure and thus decreases the conductivity of cellulose-based porous carbons compared to their CO2 or steam-activated counterparts [10]. Additionally, the tunable pore structure of porous carbon fibers enables precise control over pore size distribution and provides the ability to adjust the pore sizes to the size of the potential reactants, thus offering versatility in accommodating various reactant molecules and optimizing ion transport kinetics. Their inherent chemical stability ensures enduring catalytic activity and prolonged cycling stability in energy storage applications. However, one of the main hurdles is the precise control of the surface chemistry and electronic properties of cellulose-based materials. While their structural characteristics, such as pore network, large surface area, and inherently present structural defect, provide ample active sites for ORR, double-layer formation, and pseudo-faradaic processes, their inherent hydrophilicity and insulating nature [24] can impede electron transfer kinetics and hinder performance in catalytic and energy storage applications. Connecting these seemingly opposing properties can provide sufficient evidence for factors guiding ORR and charge storage on carbon materials. Furthermore, the intricate interplay between pore structure and mass transport kinetics presents another challenge in maximizing catalytic and charge storage performance [25]. In one our previous studies focusing on activated carbon fibers as capacitor electrodes and ORR catalysts, we could show that the tunable pore structure of cellulose-based carbons offers opportunities for optimizing molecular/ion diffusion and accessibility to active sites, but achieving the desired balance between pore size, distribution, and connectivity remains a complex task [26]. Developing a comprehensive understanding and predictive models of how pore structure influences reaction kinetics is essential for guiding the design and synthesis of cellulose-based carbons with superior performance. Addressing the missing link in connecting the properties of cellulose-based materials with catalytic and charge storage performance necessitates interdisciplinary collaboration and innovative approaches. Advanced characterization techniques, computational modeling, and materials design strategies can provide insights into the underlying mechanisms governing energy conversion and storage processes on cellulose-based carbons and can guide the development of tailored solutions to enhance their performance.
In this study, we examine the correlation between the oxygen reduction and capacitance properties of specifically designed carbon fibers with a broad spectrum of specific surfaces. Viscose-based activated carbon fibers were produced and their electrochemical properties were systematically investigated, mutually linked, and correlated to the textural properties of presented carbon fibers.
2. Materials and Methods
2.1. Material Synthesis
Cellulose fibers (1.7 dtex, 38 mm) prepared in a viscose process by Lenzing AG (Lenzing, Austria) were used as the precursor. Prior to use, the fibers were washed with distilled water, put in a spin dryer for 15 min, and then dried in a drying cabinet at 90 °C for 24 h. A moisture analyzer (MX-50, A&D Company, Tokyo, Japan) was used for the determination of residual moisture. Then, 1 g of the cellulose fibers was dried at 105 °C until a constant mass was achieved. A mass loss rate lower than 0.05% min−1 was used as the termination criterion.
For the preparation of carbon fibers, 100–400 g of the washed cellulose fibers was carbonized in a chamber furnace (HTK8, Carbolite Gero GmbH, Neuhausen, Germany). The chamber was evacuated, and a nitrogen flow of 250 L h−1 was established. The temperature program consisted of a ramp of 1.0 °C min−1 up to 850 °C followed by an isothermal step of 30 min. Afterwards, the chamber was cooled to room temperature under a nitrogen atmosphere.
Activation was performed in an RSR-B 120/500/11 rotary kiln (Nabertherm GmbH, Lilienthal, Germany), with 10 g of carbonized viscose fibers placed in the middle of the quartz glass reactor. Prior to the start of the heat treatment, the setup was purged with N2 for 25 min at a flow rate of 100 L h−1. The sample was then heated under an N2 flow rate of 50 L h−1 until the desired activation temperature was reached (Supplementary Information, Table S1) and kept isothermally for 30 min. CO2 flow was then introduced at the desired rate for a specified time (Supplementary Information, Table S1). The process was completed by cooling the kiln to room temperature under N2 flow. Activation of the carbonized viscose fibers was carried out using a Design of Experiments (DoE) Central Composite Design (CCD) approach. Three parameters—activation temperature, activation time, and CO2 flow rate—were varied at three different levels as summarized in Table S1 (Supplementary Information). No significant variations were observed in the carbon yield and porosity parameters, so no replicates of the center point Run10 are included in the following sections. The activation temperature was limited to 870 °C to prevent complete sample consumption. An activation time of 105 min and a CO2 flow rate of 45 L h−1 were determined as suitable in the preliminary trials and thus chosen as the center point.
2.2. Material Characterization
A PhenomProX scanning electron microscope (SEM) from Thermo Fisher Scientific (Waltham, MA, USA) was utilized to examine the samples’ morphology and elemental composition via Energy-Dispersive X-ray Analysis (EDX).
To analyze the specific surface area and textural structure of the activated carbon samples, N2 isothermal adsorption at −196.15 °C was performed using an Autosorb-iQ gas sorption system (Anton Paar QuantaTec Inc., Graz, Austria). Prior to analysis, the samples were degassed for at least 120 min at 200 °C. The specific surface area was determined using the Brunauer–Emmett–Teller (BET) method [27], while pore size distribution (PSD) calculations were conducted using the non-local density functional theory (NLDFT) [28].
2.3. Electrochemical Measurement
Thin film electrodes were created using the drop casting method with carbon ink. To prepare the ink, 5 mg of the desired, finely ground material was dispersed in a mixture of 400 μL ethanol, 590 μL deionized water, and 10 μL of 0.5 wt% Nafion solution in ethanol. This mixture was then homogenized by ultrasonic treatment for 30 min. Films were formed by evaporating the solvent from a 10 μL droplet of the carbon ink applied to a glassy carbon (GC) disk electrode with a geometric surface area of 0.196 cm² under a mild nitrogen stream. The specific mass loading was 250 μg cm−2, which was used to calculate the material’s capacitance. Electrochemical tests were conducted in a standard three-electrode configuration, with a high surface area platinum counter electrode and a saturated calomel electrode (SCE) as the reference. Cyclic voltammetry was used to measure the capacitance of the materials in 6 mol dm−3 aqueous KOH, 0.5 mol dm−3 aqueous H2SO4, and 2 mol dm−3 aqueous KNO3 solutions using an Ivium VO1107 potentiostat/galvanostat (Ivium Technologies, Eindhoven, The Netherlands). The gravimetric capacitances (C, in F g−1) were calculated using the following formula:
C = Q/(2 × ΔV × m) = (∫idt)/(2 × ΔV × m)
In the equation above, Q is the charge obtained by integrating positive and negative sweeps in cyclic voltammograms, ΔV is the potential window used to record the CV, and m is the mass of the active material deposited on the GC electrode surface. Before the experiment, the electrolyte was purged with N2 for 15 min, and a gentle N2 flow was maintained just below the electrolyte surface during the measurements. All measurements were carried out at room temperature, specifically 25.0 ± 0.5 °C.
The electrode preparation for the ORR (oxygen reduction reaction) measurements followed the same procedure as for the capacitance measurements. However, the rotating disc electrode (RDE) technique was utilized, and the measurements were conducted in an O2-saturated 0.1 mol dm−3 KOH solution. This method is the standard for assessing the ORR activity of carbon materials and has been employed in our laboratory for the past decade. To prevent catalyst contamination with traces of Pt, which dissolve during the experiment, graphite was used as the counter electrode instead of a Pt electrode. The baseline results indicate that the electrochemically accessible surface in the electrolyte varies from sample to sample, roughly correlating with the trends observed in measured capacitance.
3. Results
3.1. Physicochemical Properties
The main characterization results of the presented set of materials have been published in our previous work [29], and here we present them for the sake of completeness. Table 1 summarizes the main textural and chemical properties of the investigated materials, showing a wide range of total pore volumes and specific surface areas tuned by designed carbonization and activation protocols.

Table 1.
Elemental composition of studied carbon materials obtained using EDX (average of four individual spot measurements, implicit errors smaller than ±0.2 at.%).
After carbonization and activation, the fiber widths averaged around 8 μm and had distinctive flower-like cross-sections [29], which are a consequence of the spinning process during precursor production. Following the milling step, the fiber lengths ranged from 100 to 150 μm, with numerous smaller debris particles measuring a few microns in length. While this observation may appear trivial, it holds significant importance for further material performance analysis. Specifically, across the entire range of designed carbon fibers, morphology did not appear to be a contributing factor to intra-series variations in electrochemical performance.
Using energy dispersive X-ray spectroscopy (EDX), the chemical composition of the produced activated carbon fibers was analyzed (refer to Table 1). Carbon was identified as the predominant element in all the samples, accompanied by approximately 7 percent atomic oxygen and traces of sodium and sulfur, likely stemming from the precursor material. The consistent chemical composition across samples is not surprising, given that the carbonization temperature was uniform and the maximum activation temperature closely aligned with the carbonization temperature.
Uniform elemental distribution was observed in all the samples and was attributed to the homogeneous chemical composition of the precursor and the absence of any impregnation agent that could induce phase separation during carbonization/activation. Notably, not only was the elemental distribution consistent at low magnification but also along individual fibers [29], with variations below 1 at.% along approximately 35 μm of fiber length.
The textural characteristics of the samples are presented in Table 1, encompassing total pore volume (Vtot), average pore diameter (dmean), and specific surface area determined using the BET method (SBET). The produced activated carbon fibers were predominantly microporous, with only sample Run1 exhibiting pores transitioning significantly into the mesopore domain, defined as pores with a diameter exceeding 2 nm. These properties exhibit considerable variation, differing by approximately one order of magnitude, indicative of the successful manipulation of the textural properties through the Design of Experiments (DoE) protocol. The results suggest that activation temperature, followed by activation time, plays a pivotal role in yielding samples with higher SBET values. In other words, higher temperatures (870 °C over 670 and 770 °C) and longer activation times (180 over 30 and 105 min) result in higher SBET values (see Table S1 for synthetic conditions and Table 1 for SBET values).
3.2. Capacitive Properties
In the first set of experiments, we systematically addressed the capacitive properties of the studied materials, focusing on their capacitances in aqueous solutions. The results are summarized in Tables S2–S4 (see Supplementary File), for the KOH, KNO3, and H2SO4 solutions, respectively. As can be seen, the variety of textural properties reflects the capacitive behavior of the studied materials, where the capacitances range from approx. 120 F g−1 to 10 F g−1. Moreover, there is a pronounced effect of the supporting electrolyte on the capacitive response, as we generally see that the capacitances are the highest in the KOH solution for all the samples. The capacitances measured in the KNO3 and H2SO4 solutions are generally smaller than those measured in the KOH but vary from sample to sample, and there is no obvious trend to extract at which point the supporting solution of those two gives higher capacitance.
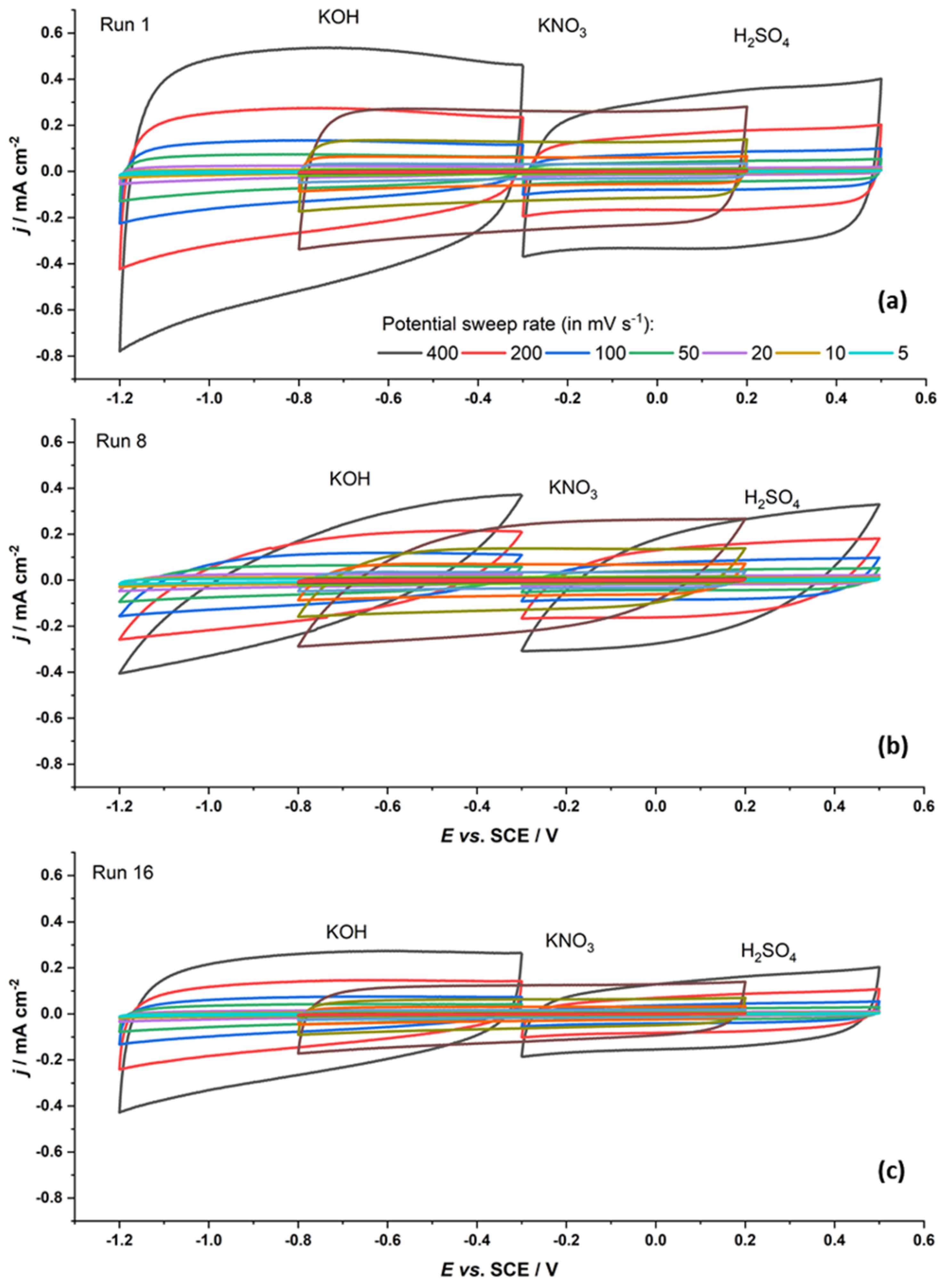
Not only do the values of measured capacitances vary over the series, but the shape of the recorded capacitive responses also varies. For example, Figure 1 shows the capacitive responses of the Run 1, Run 8, and Run 16 samples. If we consider the data in Table 1, we see that the chemical composition of these materials is basically the same as that of the others in the series, but their textural properties vary. The Run 1 sample has a very high SBET and average pore diameter. Its capacitive response is large, while the recorded cyclic voltammograms are rectangular in shape. The shape of the cyclic voltammograms for the Run 16 sample is similar, but smaller, which can be connected to its specific surface, as it is significantly smaller compared to the Run 1 sample. This sample also has a somewhat lower total pore volume than the Run 1 sample and also much narrower pores. However, the pore diameter itself is likely not the key factor. Namely, the Run 8 sample has a very distorted capacitive response but the same pore diameter as the Run 16 sample. Thus, the deformation of the cyclic voltammograms of the Run 8 sample is likely due to the smaller SBET and smaller total pore volume.
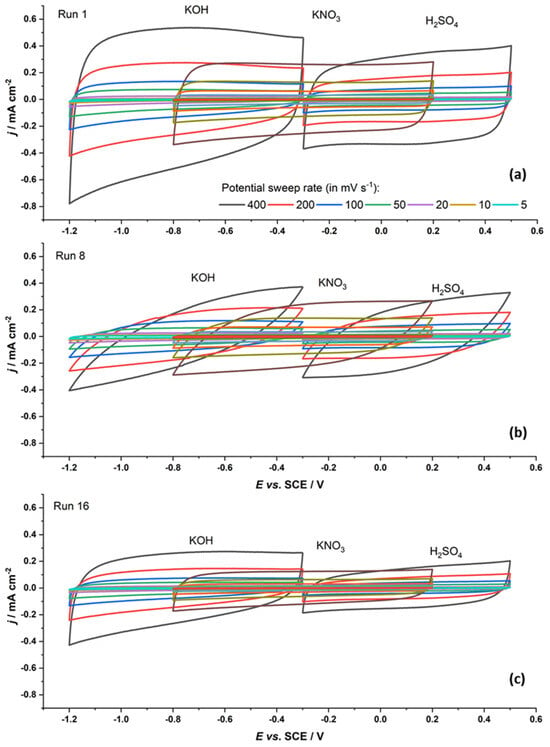
Figure 1.
Cyclic voltammograms of the Run 1 (a), Run 8 (b), and Run 16 (c) samples (400 mV s−1 to 5 mV s−1, color legend provided in (a) is the same for all three samples) in three different solutions.
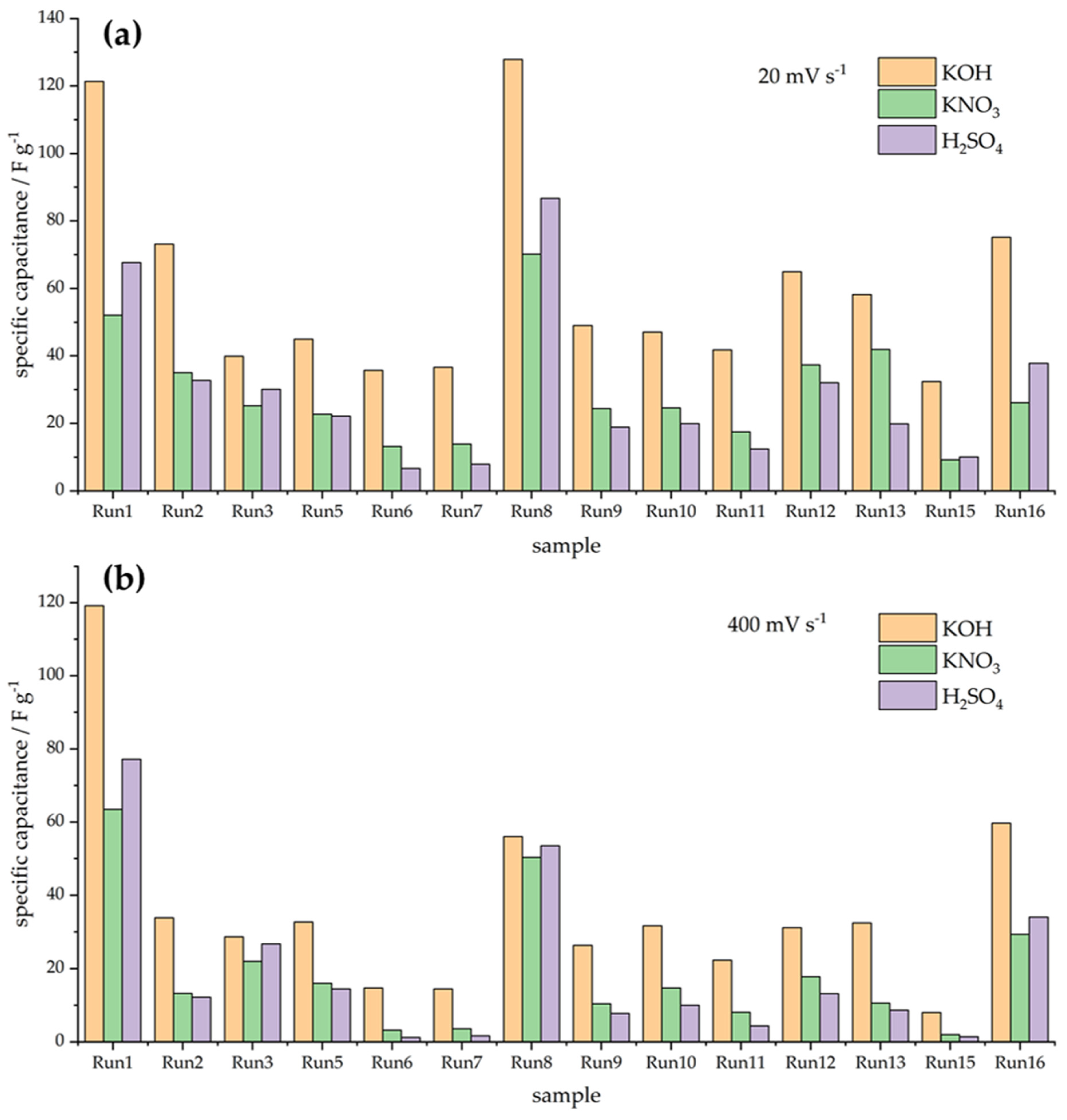
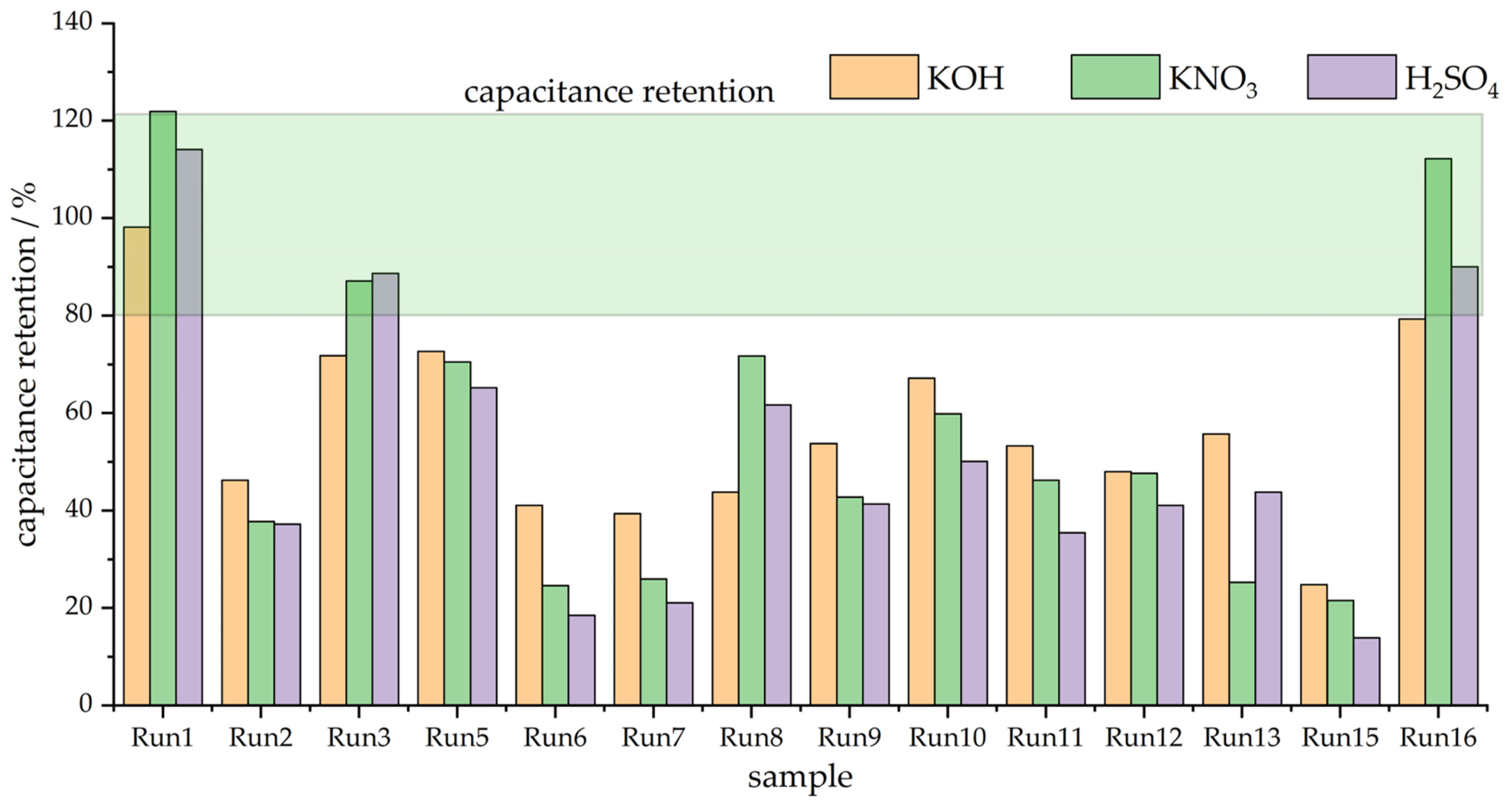
In order to clearly depict the variability in capacitive responses over the series of investigated materials, Figure 2 exhibits the capacitances measured in three electrolytes at the potential scan rates of 20 and 400 mV s−1. Obviously, the variations in the measured capacitances are quite large, and it is also clear that the capacitance retention differs from sample to sample. Moreover, it is very difficult to clearly link the capacitive behavior with the variations in textural properties. To further address capacitance retention when the scan rate is increased, we calculated the percentage of capacitance measured at 20 mV s−1 when the scan rate was increased to 400 mV s−1. The results are presented in Figure 3.
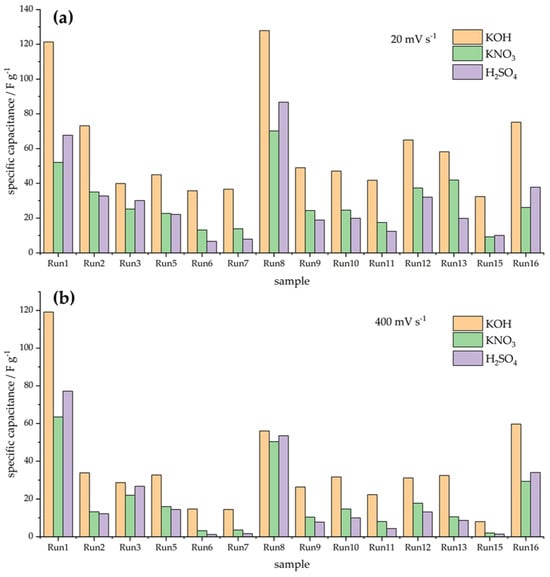
Figure 2.
Measured capacitances at 20 mV s−1 (a) and 400 mV s−1 (b) in three different solutions.
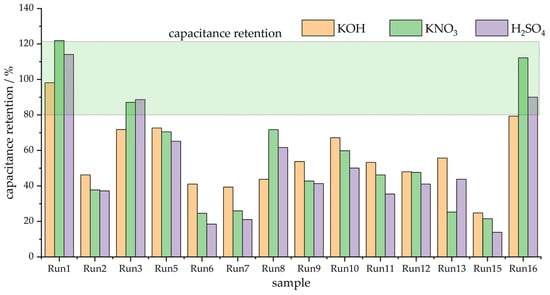
Figure 3.
Capacitance retention (400 mV s−1 vs. 20 mV s−1) in three different solutions.
As can be seen, the capacitance retention is below 80% of the capacitance measured at 20 mV s−1, with the exception of three samples—Run 1, Run 3, and Run 16. These samples are the ones with the highest SBET values over the entire series (Table 1), which points out that the specific surface area, although perhaps not tightly connected with the capacitance value, could be linked to capacitance retention, i.e., the rate capability of the capacitive carbon materials. We shall address this issue in more detail in the Discussion section.
3.3. ORR Measurements
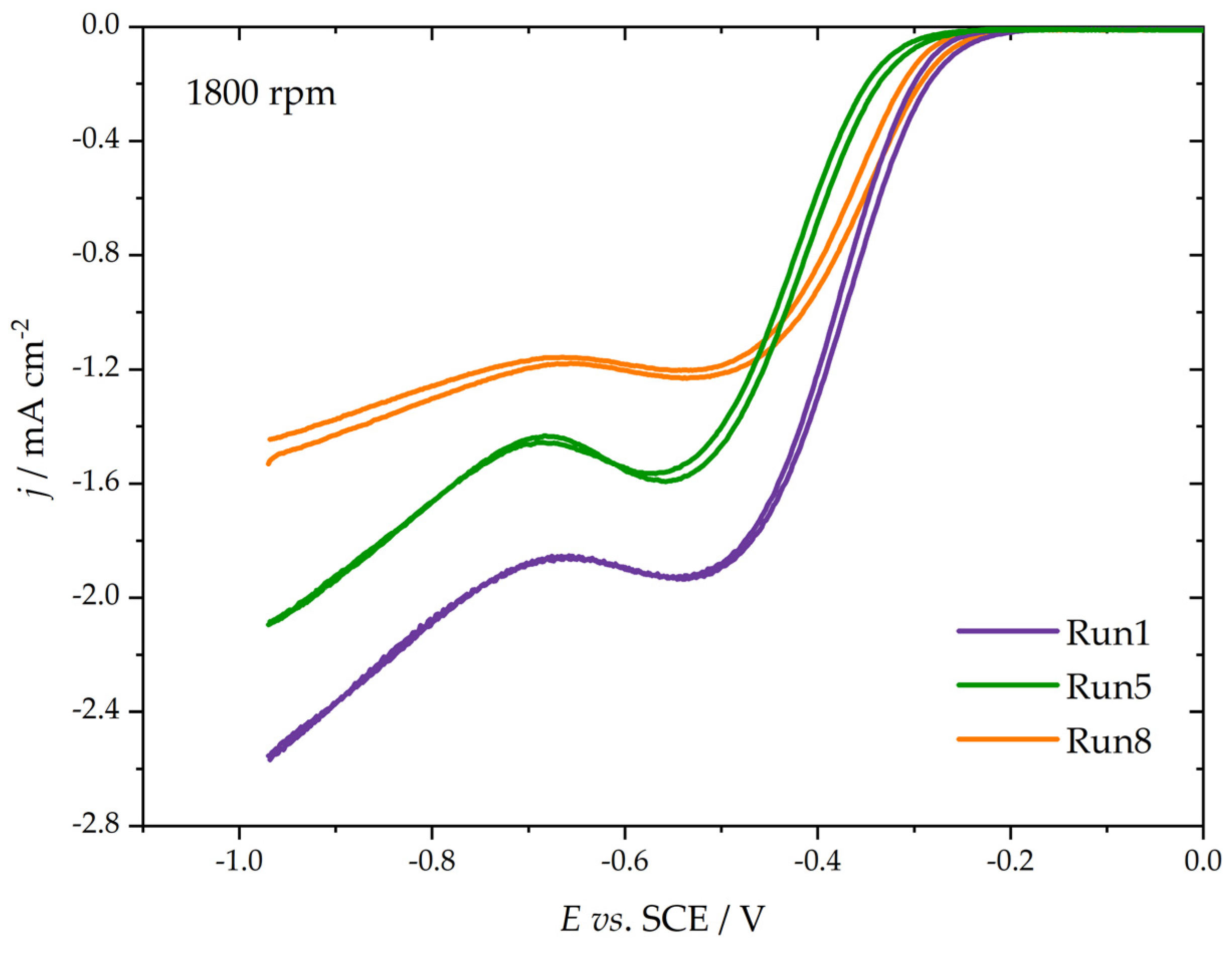
The electrocatalytic performance of the studied materials was analyzed by using an oxygen reduction reaction as a probe, utilizing a thin film RDE technique. Here, we focused on the ORR onset and kinetic currents measured close to the ORR onset. As carbon materials are generally active for ORR in alkaline media, the ORR kinetics was investigated only in the KOH solution (0.1 mol dm−3). Figure 4 shows the background-corrected ORR polarization curves for three samples (Run 1, Run 5, and Run 8). For all three samples, the oxygen reduction reaction started at potentials between −0.4 and −0.2 V vs. SCE (i.e., 0.6 and 0.8 V vs. reversible hydrogen electrode). As the ORR overpotential increased, there was an increase in the measured current, which was limited by the diffusion of dissolved O2, but no flat plateau was observed. Rather, there was an increase in the ORR current at very-deep negative potentials. By the values of measured currents, one can judge that the ORR commenced as a 2e− process, and that the number of consumed electrons per O2 increased to higher values as the ORR overpotential became more negative. This is quite common behavior for carbon materials without significant amounts of heteroatoms, like nitrogen, boron, or phosphorus, incorporated into the carbon structure. Moreover, there is no pronounced hysteresis between the cathodic and anodic scans, which is common for metallic catalysts [30], suggesting that the surface oxidation during anodic excursions of the carbon fibers investigated here was minor.
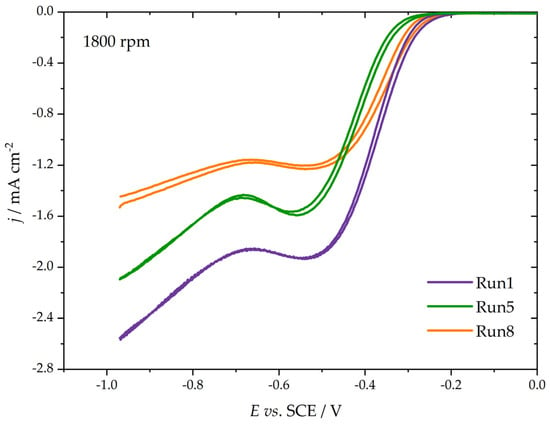
Figure 4.
ORR currents of three selected samples (indicated in the figure) were measured at a common electrode rotation rate of 1800 rpm (polarization rate 20 mV s−1).
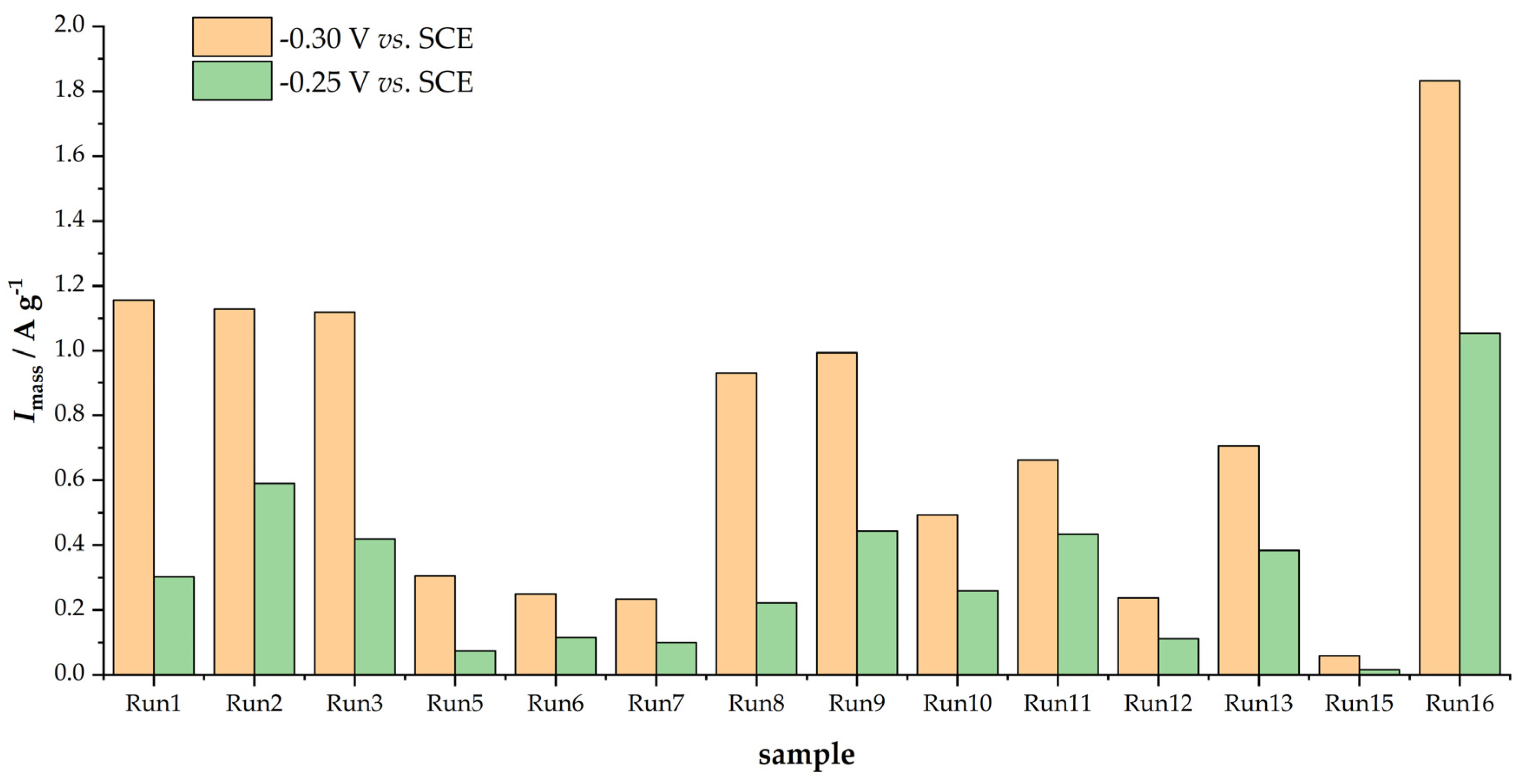
As Figure 4 shows, the Tafel slopes are also different (the slope of the polarization curve), leading to distinct changes in kinetic currents with the increase in the ORR overpotential. To evaluate the activity, we calculated mass-specific kinetic currents (Imass) by dividing the measured kinetic current densities by the carbon loading on the glassy carbon electrode surface (250 µg cm−2) at two different potentials, −0.30 and −0.25 V vs. SCE. The results are summarized in Figure 5.
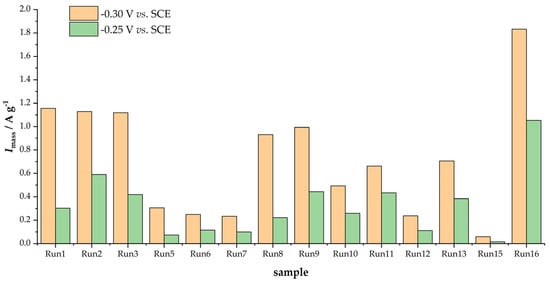
Figure 5.
ORR mass activities of investigated samples at −0.25 and −0.30 V vs. SCE, determined from anodic scan.
The obtained results show that the ORR activities of the studied materials could differ by a factor of 20 as a result of the carbonization and activation protocol used to convert viscose fibers into activated carbons. Moreover, the sample with the highest SBET (Run 1) was not the most active one, which was sample Run 16. By comparing the overall trends in capacitances (Figure 2) and ORR activities (Figure 5), one can see that high values of both quantities are obtained at the beginning of the series, its middle, and at the end of the series. Thus, in the next section, we discuss these links in more detail.
4. Discussion
4.1. Capacitive vs. Electrocatalytic Properties
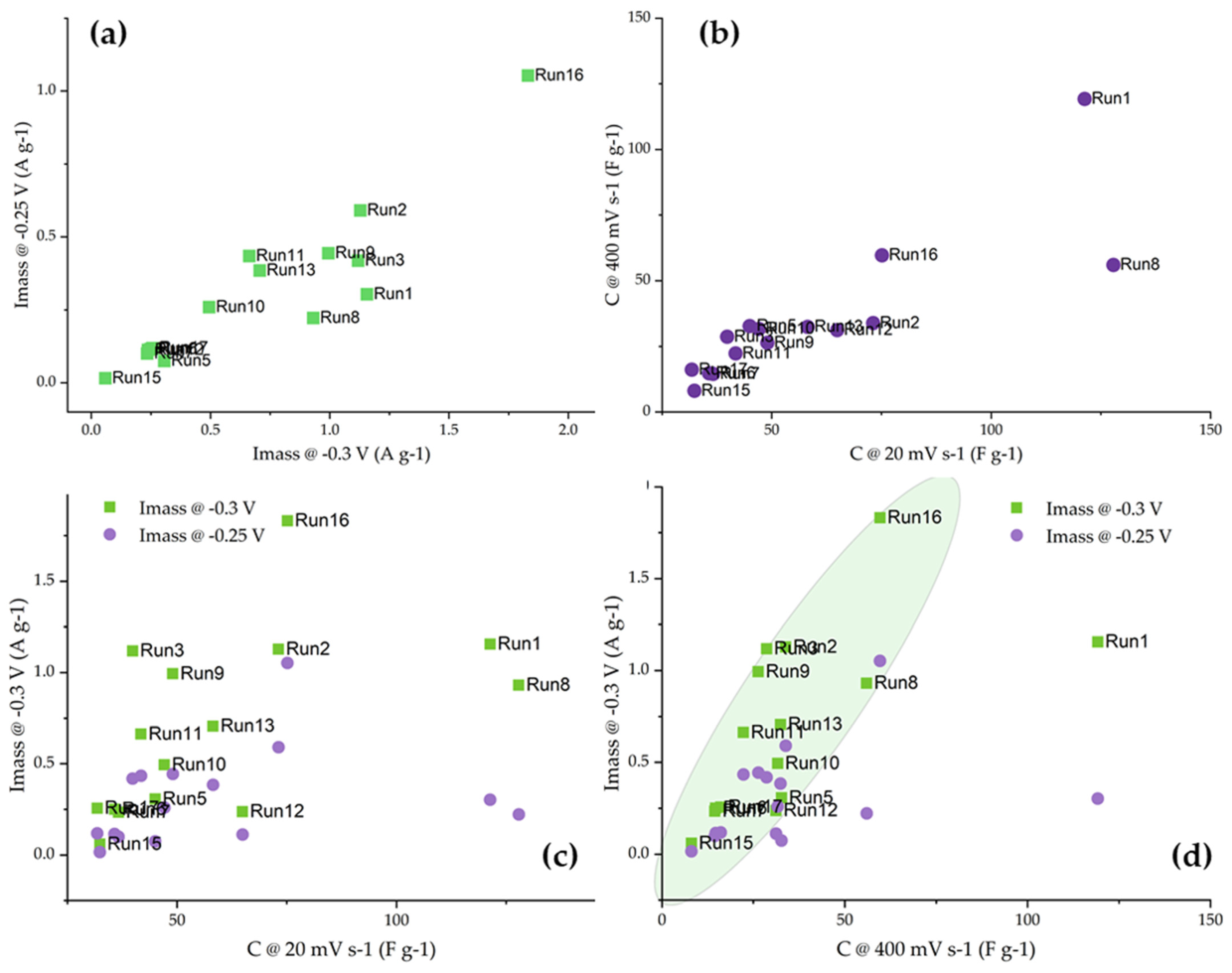
First, we address the question of the mutual connection between the electrochemical responses of the studied activated carbon fibers. By correlating mass activities measured at −0.25 and −0.30 V vs. SCE (Figure 6a), it can be seen that the correlation is quite good, with several samples (Run 1, Run 3, and Run 8) skipping the line as a result of the slower increase in the mass activity with the increase in the ORR overpotential. Also, the capacitances measured at 20 and 400 mV s−1 are quite well correlated, with only sample Run 8 being an outlier. Interestingly, this sample had a relatively high total pore volume and a small specific surface, suggesting that the pore accessibility to the electrolyte could be different compared to the other samples. However, in the next iteration, we correlated the capacitances measured in KOH with the measured ORR mass activities.
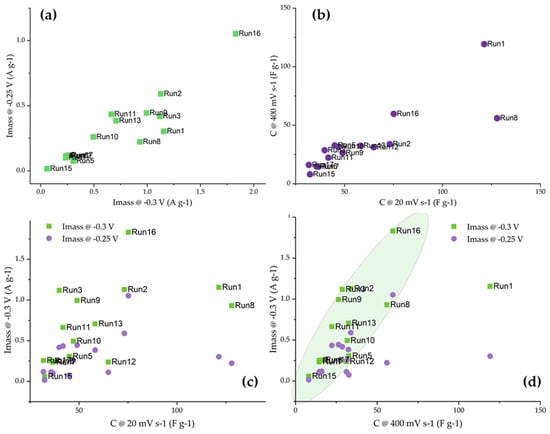
Figure 6.
(a) Mass activities and (b) capacitances at 20 and 400 mV s−1 in KOH solutions mutually correlated. (c) Mass activities correlated to specific capacitance at 20 mV s−1, and (d) mass activities correlated to specific capacitance at 400 mV s−1.
When capacitance was measured at low polarization rates, the capacitances were generally higher as the ions had more time to reach the pores in the inner parts of the materials. However, for ORR, external surfaces and mesopores are dominant [31]. The same is true for the capacitances measured at high polarization rates, as in this case, fast charging and discharging of the electrical double layer did not allow the inner pores to contribute to the capacitance. We believe that this is the reason that a very poor correlation between the measured capacitances at 20 mV s−1 and the mass activities was observed (Figure 6c). On the other hand, there was a general trend between the capacitances measured at 400 mV s−1 and the mass activities measured at −0.3 V vs. SCE. The sample Run 1 was an obvious outlier, which can be connected to the very large pore diameters and SBET contributing to high capacitance retention at high polarization rates.
4.2. Linking to Materials Properties
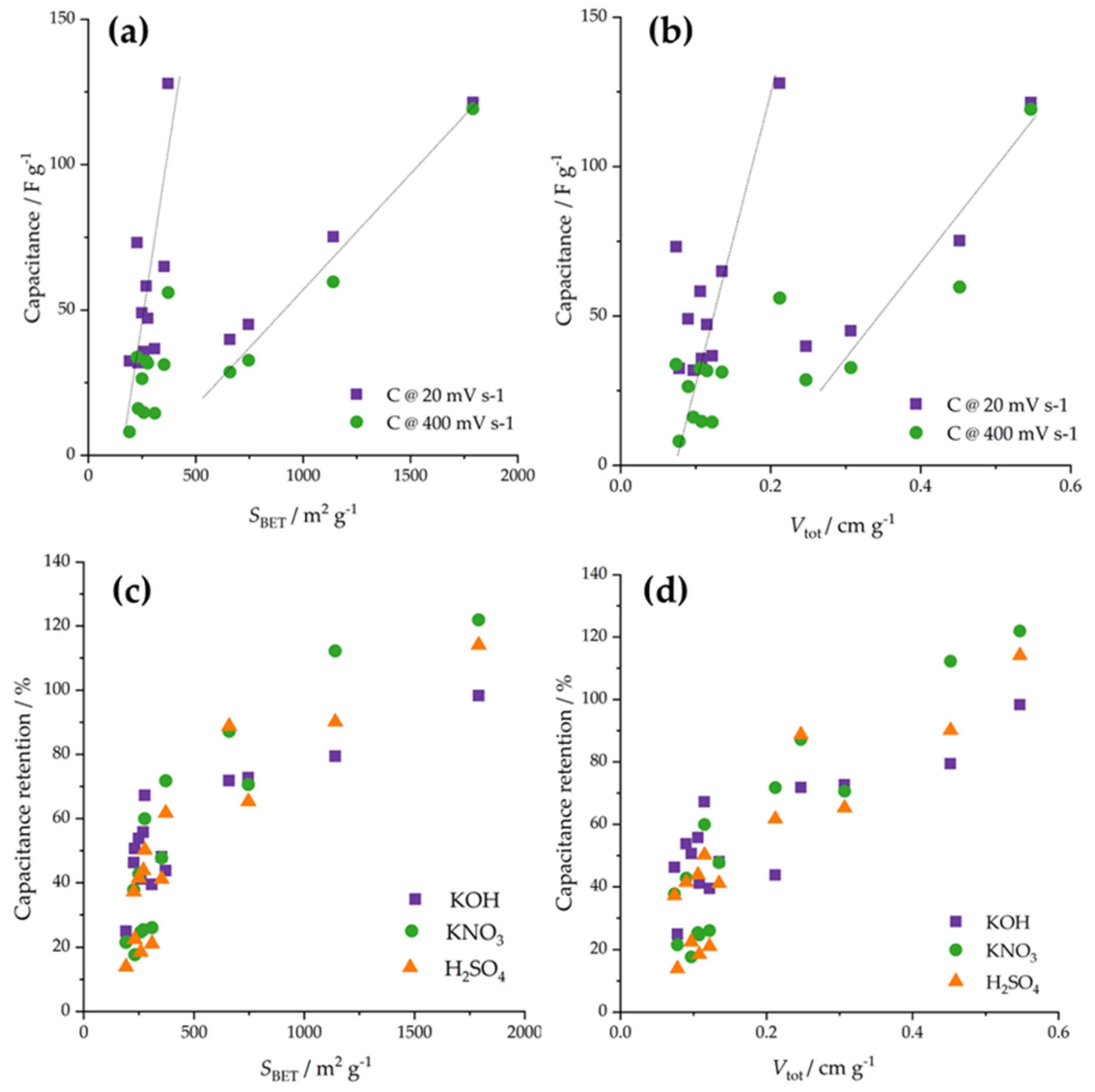
Next, we tried to link the electrochemical response to the properties of the studied materials. As the chemical composition was similar for the entire series of activated carbon fibers, we focused on textural properties, in particular, total pore volume and specific surface, having in mind previous discussions regarding the effects of pore diameters. Importantly, we did not find any correlations of mass activities with total pore volumes and specific surfaces, neither separately nor using their linear combination in the sense of a multiple linear regression approach. However, several interesting observations can be outlined. First, when the capacitances measured at 20 and 400 mV s−1 are correlated to specific surfaces (Figure 7a), it appears that there are two correlation lines, one assembling low specific surface materials and one connecting higher specific surface materials. Similar splitting but with lower correlations was observed for total pore volume (Figure 7b). This could mean that after reaching a critical specific surface of pore volume, the specific surface and pore volumes start to dominate the capacitive response. However, we still note that this is not the case for the ORR activity, of which a link to textural properties remains elusive.
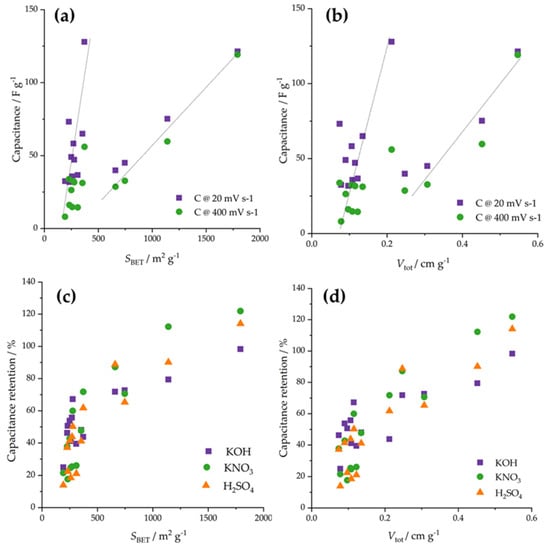
Figure 7.
(a) Correlation between the specific surface and the capacitances measured at 20 and 400 mV s−1. (b) Correlation between the total pore volume and the capacitances measured at 20 and 400 mV s−1. (c) Correlation between capacitance retention and the specific surface. (d) Correlation between capacitance retention and the total pore volume.
By further focusing on the capacitive properties, we analyzed the link between capacitance retention (Figure 3) and the specific surface (Figure 7c), on the one hand, and the total pore volume (Figure 7d), on the other hand. Obviously, there are overall trends connecting capacitance retention with the specific surface and the total pore volume. Namely, as the specific surface (or total pore volume) increased, the capacitance retention also increased. This is, we believe, a very important finding, suggesting that if a material is to be used at conditions that require charging and discharging at various rates, the specific surface and pore volume should be high to maintain high performances irrespective of the charging/discharging rates. Moreover, this would also imply that such materials would fall in the range where the critical values of specific surface (above 600 m2 g−1) and total pore volume (above 0.25 cm3 g−1) are reached, also ensuring that their further increase would also increase the capacitances.
There are other important questions regarding the link between surface chemistry and the electrochemical behavior of carbon materials. While we focus primarily on the effects of textural properties here, linking different types of surface functional groups with electrochemical performance is exceptionally important, but also very difficult. This is primarily because it is practically impossible to prepare carbon materials with a single type of surface functional group. Rather, there is a variety of different functional groups, and this makes resolving the interlinks between the properties multidimensional and extremely complicated. There are attempts reported in the literature that aim to establish such links, but conclusions regarding the importance of different surface moieties are still elusive.
5. Conclusions
In summary, our study explored the relationship between the electrochemical behavior of activated carbon fibers and their textural properties, revealing key insights for material design. We found correlations between mass activities and capacitances at different polarization rates, highlighting the influence of pore accessibility on electrochemical performance. While direct correlations with specific surface area and total pore volume were not observed for the mass activities, we discovered significant trends regarding capacitance retention. Materials with a higher specific surface area and total pore volume exhibited improved capacitance retention, especially under varying charging and discharging rates. These findings underscore the importance of optimizing specific surface area and pore volume to enhance capacitive performance across diverse operating conditions. Moving forward, materials falling within critical ranges of specific surface area (>600 m2 g−1) and total pore volume (>0.25 cm3 g−1) hold promise for applications requiring superior electrochemical performance. This research provides valuable guidance for developing carbon-based materials tailored to various electrochemical applications. Considering that the materials described here are carbon fibers, they can be used in different forms. Either powders upon milling, like those demonstrated here, or carbon cloths can be produced and used as gas diffusion electrodes or self-standing electrodes for electrochemical capacitors.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/fib12060046/s1, Table S1: Experimental parameters for the activation of studied series of carbon materials. Carbonization was performed at 850 °C with a heating rate of 1 °C min−1. Missing sample numbers correspond to repetitions of center point Run10. Reproduced from Ref. [29] under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/). Table S2: Measured capacitances (F g−1) in KOH solution at different scan rate. Table S3: Measured capacitances (F g−1) in KNO3 solution at different scan rate. Table S4: Measured capacitances (F g−1) in H2SO4 solution at different scan rates.
Author Contributions
Conceptualization, C.U., S.B. and I.A.P.; methodology, S.B. and N.G.; validation, C.U., C.F. and I.A.P.; formal analysis, S.B. and N.G.; investigation, C.U., S.B. and N.G.; resources, C.F. and I.A.P.; writing—original draft preparation, C.U., N.G. and I.A.P.; writing—review and editing, all authors; visualization, N.G. and I.A.P.; supervision, C.F. and I.A.P.; project administration, C.U.; funding acquisition, C.U. and I.A.P. All authors have read and agreed to the published version of the manuscript.
Funding
S.B., C.U. and C.F. gratefully acknowledge financial support from the European Regional Development Fund (EFRE) and the province of Upper Austria through the program IBW 2021-2027 (Project Sus2C). Further support was received through the COMET Programme (Competence Centers for Excellent Technologies) funded by the Austrian ministries BMK, BMAW, and the federal states of Upper Austria, Lower Austria, and Carinthia, operated by the Austrian Research Promotion Agency (FFG). N.G. and I.A.P. acknowledge the support provided by the Serbian Ministry of Education, Science and Technological Development (contract number: 451-03-65/2024-03/200146).
Data Availability Statement
Data are available upon request from the corresponding author.
Conflicts of Interest
The authors declare that this study received funding from the organizations stated above. The funders were not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
References
- Liu, Y.; Ma, C.; Wang, K.; Chen, J.-S. Recent advances in porous carbons for electrochemical energy storage. N. Carbon Mater. 2023, 38, 1–17. [Google Scholar] [CrossRef]
- Suzuki, M. Activated carbon fiber: Fundamentals and applications. Carbon 1994, 32, 577–586. [Google Scholar] [CrossRef]
- Pandolfo, A.G.; Hollenkamp, A.F. Carbon properties and their role in supercapacitors. J. Power Sources 2006, 157, 11–27. [Google Scholar] [CrossRef]
- Kim, J.-H.; Jung, S.-C.; Lee, H.-M.; Kim, B.-J. Comparison of Pore Structures of Cellulose-Based Activated Carbon Fibers and Their Applications for Electrode Materials. Int. J. Mol. Sci. 2022, 23, 3680. [Google Scholar] [CrossRef] [PubMed]
- Gindl-Altmutter, W.; Czabany, I.; Unterweger, C.; Gierlinger, N.; Xiao, N.; Bodner, S.C.; Keckes, J. Structure and electrical resistivity of individual carbonised natural and man-made cellulose fibres. J. Mater. Sci. 2020, 55, 10271–10280. [Google Scholar] [CrossRef]
- Lin, Y.; Huang, C.; Huang, C.; Deng, Y.; Zou, X.; Ma, W.; Fang, G.; Ragauskas, A.J. Cellulose regulated lignin/cellulose-based carbon materials with hierarchical porous structure for energy storage. Adv. Compos. Hybrid Mater. 2024, 7, 600. [Google Scholar] [CrossRef]
- Kong, X.; Zhou, S.; Strømme, M.; Xu, C. All-cellulose-based freestanding porous carbon nanocomposites and their versatile applications. Compos. Part B Eng. 2022, 232, 109602. [Google Scholar] [CrossRef]
- Samir, A.; Ashour, F.H.; Hakim, A.A.A.; Bassyouni, M. Recent advances in biodegradable polymers for sustainable applications. NPJ Mater. Degrad. 2022, 6, 443. [Google Scholar] [CrossRef]
- Ani, P.C.; Nzereogu, P.U.; Agbogu, A.C.; Ezema, F.I.; Nwanya, A.C. Cellulose from waste materials for electrochemical energy storage applications: A review. Appl. Surf. Sci. Adv. 2022, 11, 100298. [Google Scholar] [CrossRef]
- Breitenbach, S.; Duchoslav, J.; Mardare, A.I.; Unterweger, C.; Stifter, D.; Hassel, A.W.; Fürst, C. Comparative Behavior of Viscose-Based Supercapacitor Electrodes Activated by KOH, H2O, and CO2. Nanomaterials 2022, 12, 677. [Google Scholar] [CrossRef]
- Adam, D. Cellulose: A new bio-support for aqueous phase catalysts. Nature 2001, 1, 21. [Google Scholar] [CrossRef]
- Rehman, A.; Nazir, G.; Yop Rhee, K.; Park, S.-J. A rational design of cellulose-based heteroatom-doped porous carbons: Promising contenders for CO2 adsorption and separation. Chem. Eng. J. 2021, 420, 130421. [Google Scholar] [CrossRef]
- Bilgin Simsek, E.; Novak, I.; Sausa, O.; Berek, D. Microporous carbon fibers prepared from cellulose as efficient sorbents for removal of chlorinated phenols. Res. Chem. Intermed. 2017, 43, 503–522. [Google Scholar] [CrossRef]
- Suhas; Gupta, V.K.; Carrott, P.J.M.; Singh, R.; Chaudhary, M.; Kushwaha, S. Cellulose: A review as natural, modified and activated carbon adsorbent. Bioresour. Technol. 2016, 216, 1066–1076. [Google Scholar] [CrossRef] [PubMed]
- Phan, N.H.; Rio, S.; Faur, C.; Le Coq, L.; Le Cloirec, P.; Nguyen, T.H. Production of fibrous activated carbons from natural cellulose (jute, coconut) fibers for water treatment applications. Carbon 2006, 44, 2569–2577. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, S.; Liu, J.; Sun, J.; Zhang, Z.; Zhu, Q. Sustainable cellulose nanomaterials for environmental remediation—Achieving clean air, water, and energy: A review. Carbohydr. Polym. 2022, 285, 119251. [Google Scholar] [CrossRef]
- Wang, Y.; Qu, Q.; Cui, J.; Lu, T.; Li, F.; Zhang, M.; Liu, K.; Zhang, Q.; He, S.; Huang, C. Design and fabrication of cellulose derived free-standing carbon nanofiber membranes for high performance supercapacitors. Carbohydr. Polym. Technol. Appl. 2021, 2, 100117. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, C.; Wang, Y. Recent progress in cellulose-based electrospun nanofibers as multifunctional materials. Nanoscale Adv. 2021, 3, 6040–6047. [Google Scholar] [CrossRef] [PubMed]
- Anusiya, G.; Jaiganesh, R. A review on fabrication methods of nanofibers and a special focus on application of cellulose nanofibers. Carbohydr. Polym. Technol. Appl. 2022, 4, 100262. [Google Scholar] [CrossRef]
- Shi, J.; Huang, T.; Wu, R.; Wu, J.; Li, Y.; Kuang, Y.; Xing, H.; Zhang, W. Direct carbonization of cellulose toward hydroxyl-rich porous carbons for pseudocapacitive energy storage. Int. J. Biol. Macromol. 2024, 264, 130460. [Google Scholar] [CrossRef]
- Sun, B.; Yuan, Y.; Li, H.; Li, X.; Zhang, C.; Guo, F.; Liu, X.; Wang, K.; Zhao, X.S. Waste-cellulose-derived porous carbon adsorbents for methyl orange removal. Chem. Eng. J. 2019, 371, 55–63. [Google Scholar] [CrossRef]
- Hina, K.; Zou, H.; Qian, W.; Zuo, D.; Yi, C. Preparation and performance comparison of cellulose-based activated carbon fibres. Cellulose 2018, 25, 607–617. [Google Scholar] [CrossRef]
- Hassan, M.F.; Sabri, M.A.; Fazal, H.; Hafeez, A.; Shezad, N.; Hussain, M. Recent trends in activated carbon fibers production from various precursors and applications—A comparative review. J. Anal. Appl. Pyrolysis 2020, 145, 104715. [Google Scholar] [CrossRef]
- Chen, Z.; Li, Z.; Lan, P.; Xu, H.; Lin, N. Hydrophobic and thermal-insulating aerogels based on rigid cellulose nanocrystal and elastic rubber. Carbohydr. Polym. 2022, 275, 118708. [Google Scholar] [CrossRef] [PubMed]
- Bandosz, T.J. Revealing the impact of small pores on oxygen reduction on carbon electrocatalysts: A journey through recent findings. Carbon 2022, 188, 289–304. [Google Scholar] [CrossRef]
- Breitenbach, S.; Gavrilov, N.; Pašti, I.; Unterweger, C.; Duchoslav, J.; Stifter, D.; Hassel, A.W.; Fürst, C. Biomass-Derived Carbons as Versatile Materials for Energy-Related Applications: Capacitive Properties vs. Oxygen Reduction Reaction Catalysis. J. Carbon Res. C 2021, 7, 55. [Google Scholar] [CrossRef]
- Bardestani, R.; Patience, G.S.; Kaliaguine, S. Experimental methods in chemical engineering: Specific surface area and pore size distribution measurements—BET, BJH, and DFT. Can. J. Chem. Eng. 2019, 97, 2781–2791. [Google Scholar] [CrossRef]
- Kupgan, G.; Liyana-Arachchi, T.P.; Colina, C.M. NLDFT Pore Size Distribution in Amorphous Microporous Materials. Langmuir 2017, 33, 11138–11145. [Google Scholar] [CrossRef] [PubMed]
- Tasić, T.; Milanković, V.; Batalović, K.; Breitenbach, S.; Unterweger, C.; Fürst, C.; Pašti, I.A.; Lazarević-Pašti, T. Application of Viscose-Based Porous Carbon Fibers in Food Processing-Malathion and Chlorpyrifos Removal. Foods 2023, 12, 2362. [Google Scholar] [CrossRef]
- Hodnik, N.; Baldizzone, C.; Cherevko, S.; Zeradjanin, A.; Mayrhofer, K.J.J. The Effect of the Voltage Scan Rate on the Determination of the Oxygen Reduction Activity of Pt/C Fuel Cell Catalyst. Electrocatalysis 2015, 6, 237–241. [Google Scholar] [CrossRef]
- Wan, K.; Long, G.-F.; Liu, M.-Y.; Du, L.; Liang, Z.-X.; Tsiakaras, P. Nitrogen-doped ordered mesoporous carbon: Synthesis and active sites for electrocatalysis of oxygen reduction reaction. Appl. Catal. B Environ. 2015, 165, 566–571. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).