Porous ZnO/2–Hydroxyethyl Methacrylate Eluting Coatings for Ureteral Stent Applications


Abstract
1. Introduction
2. Materials and Methods
2.1. Preparation of Porous ZnO Thin Films
2.2. Preparation of PolyHEMA (pHEMA) and Poly(HEMA-co-AA)
2.3. Preparation of ZnO/Polymer Bilayer Coatings
2.4. Calcein Loading and Release Experiments
2.5. Characterization Setup
3. Results and Discussion
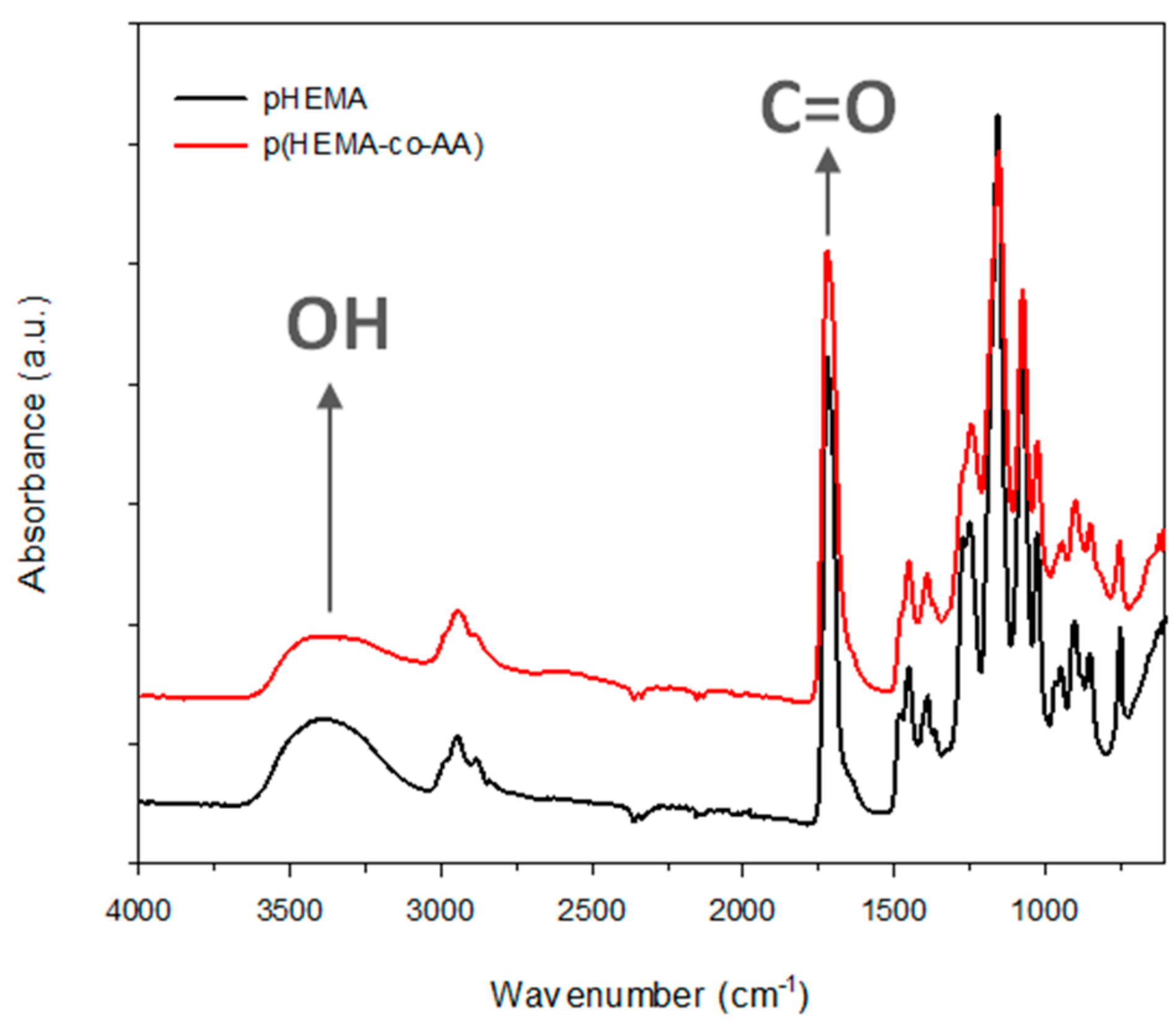
3.1. Characterization of PolyHEMA (pHEMA) and Poly(HEMA-co-AA)
3.2. Characterization of Porous ZnO/Polymer Bilayer Systems
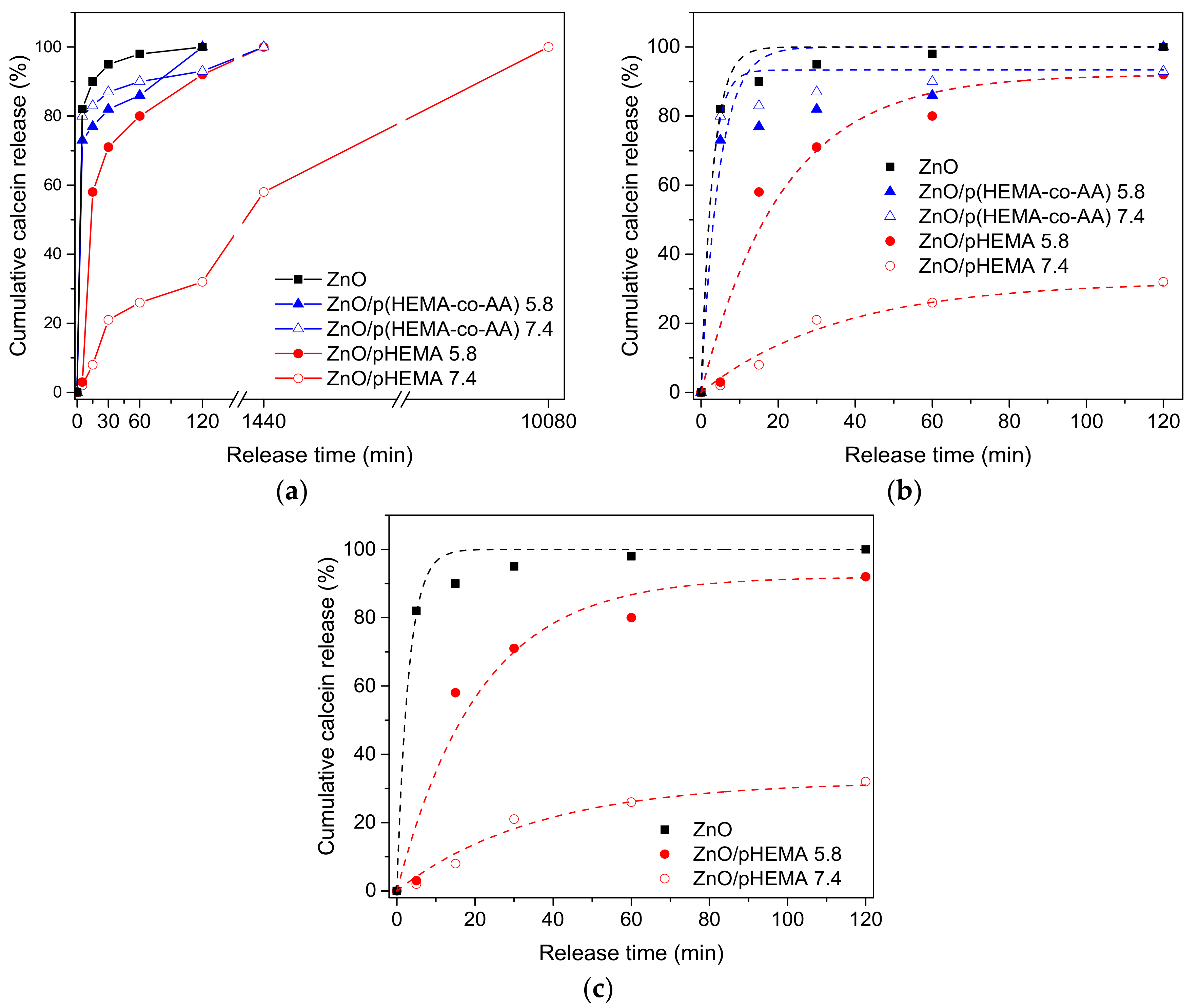
3.3. pH-Triggered Release of Calcein from Drop-Casted ZnO/pHEMA and ZnO/p(HEMA-co-AA) Bilayer Coatings
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gultepe, E.; Nagesha, D.; Sridhar, S.; Amiji, M. Nanoporous inorganic membranes or coatings for sustained drug delivery in implantable devices. Adv. Drug Deliv. Rev. 2010, 62, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Felton, L.A.; Porter, S.C. An update on pharmaceutical film coating for drug delivery. Expert Opin. Drug Deliv. 2013, 10, 421–435. [Google Scholar] [CrossRef] [PubMed]
- La, W.G.; Park, S.; Yoon, H.H.; Jeong, G.J.; Lee, T.J.; Bhang, S.H.; Han, J.Y.; Char, K.; Kim, B.S. Delivery of a therapeutic protein for bone regeneration from a substrate coated with graphene oxide. Small 2013, 9, 4051–4060. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Kim, D.J.; Park, S.J.; Yoo, J.B.; Ryu, Y.S. Controlled drug release using nanoporous anodic aluminum oxide on stent. Thin Solid Films 2007, 515, 5184–5187. [Google Scholar] [CrossRef]
- Gulati, K.; Ramakrishnan, S.; Aw, M.S.; Atkins, G.J.; Findlay, D.M.; Losic, D. Biocompatible polymer coating of titania nanotube arrays for improved drug elution and osteoblast adhesion. Acta Biomater. 2012, 8, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Salonen, J.; Kaukonen, A.M.; Hirvonen, J.; Lehto, V.P. Mesoporous silicon in drug delivery applications. J. Pharm. Sci. 2008, 97, 632–653. [Google Scholar] [CrossRef] [PubMed]
- Leung, J.W.C.; Lau, G.T.C.; Sung, J.J.Y.; Costerton, J.W. Decreased bacterial adherence to silver-coated stent material: An in vitro study. Gastrointest. Endosc. 1992, 38, 338–340. [Google Scholar] [CrossRef]
- Cauda, V.; Chiodoni, A.; Laurenti, M.; Canavese, G.; Tommasi, T. Ureteral double-J stents performances toward encrustation after long-term indwelling in a dynamic in vitro model. J. Biomed. Mater. Res. B 2017, 105, 2244–2253. [Google Scholar] [CrossRef] [PubMed]
- Cauda, F.; Cauda, V.; Fiori, C.; Onida, B.; Garrone, E. Heparin coating on ureteral double J stents prevents encrustations: An in vivo case study. J. Endourol. 2008, 22, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Laube, N.; Kleinen, L.; Bradenahl, J.; Meissner, A. Diamond-like carbon coatings on ureteral stents—A new strategy for decreasing the formation of crystalline bacterial biofilms? J. Urol. 2007, 177, 1923–1927. [Google Scholar] [CrossRef] [PubMed]
- John, T.; Rajpurkar, A.; Smith, G.; Fairfax, M.; Triest, J. Antibiotic pretreatment of hydrogel ureteral stent. J. Endourol. 2007, 21, 1211–1215. [Google Scholar] [CrossRef] [PubMed]
- Leung, J.W.; Liu, Y.L.; Cheung, S.W.; Chan, R.C.Y.; Inciardi, J.F.; Cheng, A.F. Effect of antibiotic-loaded hydrophilic stent in the prevention of bacterial adherence: A study of the charge, discharge, and recharge concept using ciprofloxacin. Gastrointest. Endosc. 2001, 53, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Cormio, L.; La Forgia, P.; La Forgia, D.; Siitonen, A.; Ruutu, M. Bacterial adhesion to urethral catheters: Role of coating materials and immersion in antibiotic solution. Eur. Urol. 2001, 40, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Thierry, B.; Merhi, Y.; Silver, J.; Tabrizian, M. Biodegradable membrane-covered stent from chitosan-based polymers. J. Biomed. Mater. Res. A 2005, 75A, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Wang, Z.; Li, G.; Cui, F.; Zhang, Y.; Zhang, X. Comparison of a biodegradable ureteral stent versus the traditional double-J stent for the treatment of ureteral injury: An experimental study. Biomed. Mater. 2012, 7, 065002. [Google Scholar] [CrossRef] [PubMed]
- Chew, B.H.; Paterson, R.F.; Clinkscales, K.W.; Levine, B.S.; Shalaby, S.W.; Lange, D. In vivo evaluation of the third generation biodegradable stent: A novel approach to avoiding the forgotten stent syndrome. J. Urol. 2013, 189, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Lo, J.; Lange, D.; Chew, B.H. Ureteral stents and foley catheters-Associated Urinary tract infections: The role of coatings and materials in infection prevention. Antibiotics 2014, 3, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hu, C.; Shao, L. The antimicrobial activity of nanoparticles: Present situation and prospects for the future. Int. J. Nanomed. 2017, 12, 1227–1249. [Google Scholar] [CrossRef] [PubMed]
- Huh, A.J.; Kwon, Y.J. “Nanoantibiotics”: A new paradigm for treating infectious diseases using nanomaterials in the antibiotics resistant era. J. Control. Release 2011, 156, 128–145. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Weng, Z.; Li, X.; Liu, X.; Wu, S.; Yeung, K.W.K.; Wang, X.; Cui, Z.; Yang, X.; Chu, P. Biomedical applications of functionalized ZnO nanomaterials: From biosensors to bioimaging. Adv. Mater. Interfaces 2016, 3, 150049. [Google Scholar] [CrossRef]
- Racca, L.; Canta, M.; Dumontel, B.; Ancona, A.; Limongi, T.; Garino, N.; Laurenti, M.; Canavese, G.; Cauda, V. Zinc oxide nanostrucutres in biomedicine. In Smart Nanoparticles for Biomedicine; Ciofani, G., Ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2018; pp. 171–187. [Google Scholar]
- Sirelkhatim, A.; Mahmud, S.; Seeni, A.; Kaus, N.H.M.; Ann, L.C.; Bakhori, S.K.M.; Hasan, H.; Mohamad, D. Review on zinc oxide nanoparticles: Antibacterial activity and toxicity mechanism. Nano-Micro Lett. 2015, 7, 219–242. [Google Scholar] [CrossRef]
- Cauda, V.; Stassi, S.; Lamberti, A.; Morello, M.; Pirri, C.F.; Canavese, G. Leveraging ZnO morphologies in piezoelectric composites for mechanical energy harvesting. Nano Energy 2015, 18, 212–221. [Google Scholar] [CrossRef]
- Chiolerio, A.; Roppolo, I.; Cauda, V.; Crepaldi, M.; Bocchini, S.; Bejtka, K.; Verna, A.; Pirri, C.F. Ultraviolet mem-sensors: Flexible anisotropic composites featuring giant photocurrent enhancement. Nano Res. 2015, 8, 1956–1963. [Google Scholar] [CrossRef]
- Hernandez, S.; Hidalgo, D.; Sacco, A.; Chiodoni, A.; Lamberti, A.; Cauda, V.; Tresso, E.; Saracco, G. Comparison of photocatalytic and transport properties of TiO2 and ZnO nanostructures for solar-driven water splitting. Phys. Chem. Chem. Phys. 2015, 17, 7775–7786. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Available online: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=182.8991 (accessed on 12 September 2018).
- Laurenti, M.; Verna, A.; Fontana, M.; Stassi, S.; Canavese, G.; Marasso, S.L.; Cauda, V. How micropatterning and surface functionalization affect the wetting behavior of ZnO nanostructured surfaces. Adv. Mater. Interfaces 2016, 3, 1600110. [Google Scholar] [CrossRef]
- Laurenti, M.; Cauda, V. Porous zinc oxide thin films: Synthesis approaches and applications. Coatings 2018, 8, 67. [Google Scholar] [CrossRef]
- Rivera, V.F.; Auras, F.; Motto, P.; Stassi, S.; Canavese, G.; Celasco, E.; Bein, T.; Onida, B.; Cauda, V. Length-dependent charge generation from vertical arrays of high-aspect-ratio ZnO nanowires. Chem. A Eur. J. 2013, 19, 14665–14674. [Google Scholar] [CrossRef] [PubMed]
- Dumontel, B.; Canta, M.; Engelke, H.; Chiodoni, A.; Racca, L.; Ancona, A.; Limongi, T.; Canavese, G.; Cauda, V. Enhanced biostability and cellular uptake of zinc oxide nanocrystals shielded with a phospholipid bilayer. J. Mater. Chem. B 2017, 5, 8799–8813. [Google Scholar] [CrossRef] [PubMed]
- Cauda, V.; Pugliese, D.; Garino, N.; Sacco, A.; Bianco, S.; Bella, F.; Lamberti, A.; Gerbaldi, C. Multi-functional energy conversion and storage electrodes using flower-like Zinc oxide nanostructures. Energy 2014, 65, 639–646. [Google Scholar] [CrossRef]
- Miccoli, B.; Cauda, V.; Bonanno, A.; Sanginario, A.; Bejtka, K.; Bella, F.; Fontana, M.; Demarchi, D. One-dimensional ZnO/gold junction for simultaneous and versatile multisensing measurements. Sci. Rep. 2016, 6, 29763. [Google Scholar] [CrossRef] [PubMed]
- Laurenti, M.; Cauda, V. ZnO nanostructures for tissue engineering applications. Nanomaterials 2017, 7, 374. [Google Scholar] [CrossRef] [PubMed]
- Laurenti, M.; Cauda, V. Gentamicin-releasing mesoporous ZnO structures. Materials 2018, 11, 314. [Google Scholar] [CrossRef] [PubMed]
- Ancona, A.; Dumontel, B.; Garino, N.; Demarco, B.; Chatzitheodoridou, D.; Fazzini, W.; Engelke, H.; Cauda, V. Lipid-coated zinc oxide nanoparticles as innovative ROS-generators for photodynamic therapy in cancer cells. Nanomaterials 2018, 8, 143. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Sasidharan, A.; Rani, V.V.D.; Menon, D.; Nair, S.; Manzoor, K.; Raina, S. Role of size scale of ZnO nanoparticles and microparticles on toxicity toward bacteria and osteoblast cancer cells. J. Mater. Sci. Mater. Med. 2009, 20, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Montheard, J.P.; Chatzopoulos, M.; Chappard, D. 2-Hydroxyethyl methacrylate (HEMA)—Chemical-properties and applications in biomedical fields. J. Macromol. Sci. Part C Polym. Rev. 1992, C32, 1–34. [Google Scholar] [CrossRef]
- Yarimkaya, S.; Basan, H. Swelling behavior of poly(2-hydroxyethyl methacrylateco-acrylic acid-co-ammonium acrylate) hydrogels. J. Macromol. Sci. A 2007, 44, 939–946. [Google Scholar] [CrossRef]
- Omidian, H.; Park, K.; Kandalam, U.; Rocca, J.G. Swelling and mechanical properties of modified HEMA-based superporous hydrogels. J. Bioact. Compat. Pol. 2010, 25, 483–497. [Google Scholar] [CrossRef]
- Zhang, Y.; Chan, H.F.; Leong, K.W. Advanced materials and processing for drug delivery: The past and the future. Adv. Drug Deliver Rev. 2013, 65, 104–120. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.; Wang, T.; Li, Y.; Tu, I. Synthesis, characterization and ocular biocompatibility of potential keratoprosthetic hydrogels based on photopolymerized poly(2-hydroxyethyl methacrylate)-co-poly(acrylic acid). J. Mater. Chem. 2012, 22, 1812–1823. [Google Scholar] [CrossRef]
- Kokubo, T.; Takadama, H. How useful is SBF in predicting in vivo bone bioactivity? Biomaterials 2006, 27, 2907–2915. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Zhang, Y.; Zhou, T.; Zhou, S. Stability of hydrogen-bonded hydroxypropylcellulose/poly(acrylic acid) microcapsules in aqueous solutions. Soft Matter 2009, 5, 842–849. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, Y.; Wang, L.; Hong, S.; Xu, J.; Chen, Y. Composite thin film by hydrogen-bonding assembly of polymer brush and poly(vinylpyrrolidone). Langmuir 2006, 22, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Sacco, A.; Lamberti, A.; Gazia, R.; Bianco, S.; Manfredi, D.; Shahzad, N.; Cappelluti, F.; Ma, S.; Tresso, E. High efficiency dye-sensitized solar cells exploiting sponge-like ZnO nanostructures. Phys. Chem. Chem. Phys. 2012, 14, 16203–16208. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, W.A.; Carrasquer, G. Mechanisms of acidification of the urine. Prog. Cardiovasc. Dis. 1961, 4, 105–133. [Google Scholar] [CrossRef]
- Barrett, K.E.; Barman, S.M.; Boitano, S.; Brooks, H.L. Acidification of the urine & bicarbonate excretion. In Ganong’s Review of Medical Physiology, 25th ed.; Weitz, M., Kearns, B., Eds.; McGraw-Hill Education: New York, NY, USA, 2016. [Google Scholar]
- Moe, O.W.; Preisig, P.A. Dual role of citrate in mammalian urine. Curr. Opin. Nephrol. Hypertens. 2006, 15, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Fatehah, M.O.; Aziz, H.A.; Stoll, S. Stability of ZnO nanoparticles in solution. Influence of pH, dissolution, aggregation and disaggregation effects. J. Coll. Sci. Biotechnol. 2014, 3, 75–84. [Google Scholar] [CrossRef]
- Moe, O.W. Uric acid nephrolithiasis: Proton titration of an essential molecule? Curr. Opin. Nephrol. Hypertens. 2006, 15, 366–373. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | pH | (w120min/w0), % | K (min−1) | Χ2 |
|---|---|---|---|---|
| ZnO | 7.4 | 100 | 0.33 ± 0.05 | 0.98 |
| ZnO/pHEMA | 7.4 | 32 | 0.03 ± 0.01 | 0.98 |
| 5.8 | 92 | 0.05 ± 0.01 | 0.95 | |
| ZnO/p(HEMA-co-AA) | 7.4 | 93 | 0.38 ± 0.08 | 0.98 |
| 5.8 | 100 | 0.21 ± 0.07 | 0.85 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laurenti, M.; Grochowicz, M.; Cauda, V. Porous ZnO/2–Hydroxyethyl Methacrylate Eluting Coatings for Ureteral Stent Applications. Coatings 2018, 8, 376. https://doi.org/10.3390/coatings8110376
Laurenti M, Grochowicz M, Cauda V. Porous ZnO/2–Hydroxyethyl Methacrylate Eluting Coatings for Ureteral Stent Applications. Coatings. 2018; 8(11):376. https://doi.org/10.3390/coatings8110376
Chicago/Turabian StyleLaurenti, Marco, Marta Grochowicz, and Valentina Cauda. 2018. "Porous ZnO/2–Hydroxyethyl Methacrylate Eluting Coatings for Ureteral Stent Applications" Coatings 8, no. 11: 376. https://doi.org/10.3390/coatings8110376
APA StyleLaurenti, M., Grochowicz, M., & Cauda, V. (2018). Porous ZnO/2–Hydroxyethyl Methacrylate Eluting Coatings for Ureteral Stent Applications. Coatings, 8(11), 376. https://doi.org/10.3390/coatings8110376