
First-Principles Investigation of the Structural, Magnetic, and Electronic Properties of Janus MXene Material CrScCO2

 ,
,
Abstract
1. Introduction
2. Computational Details
3. Results and Discussion
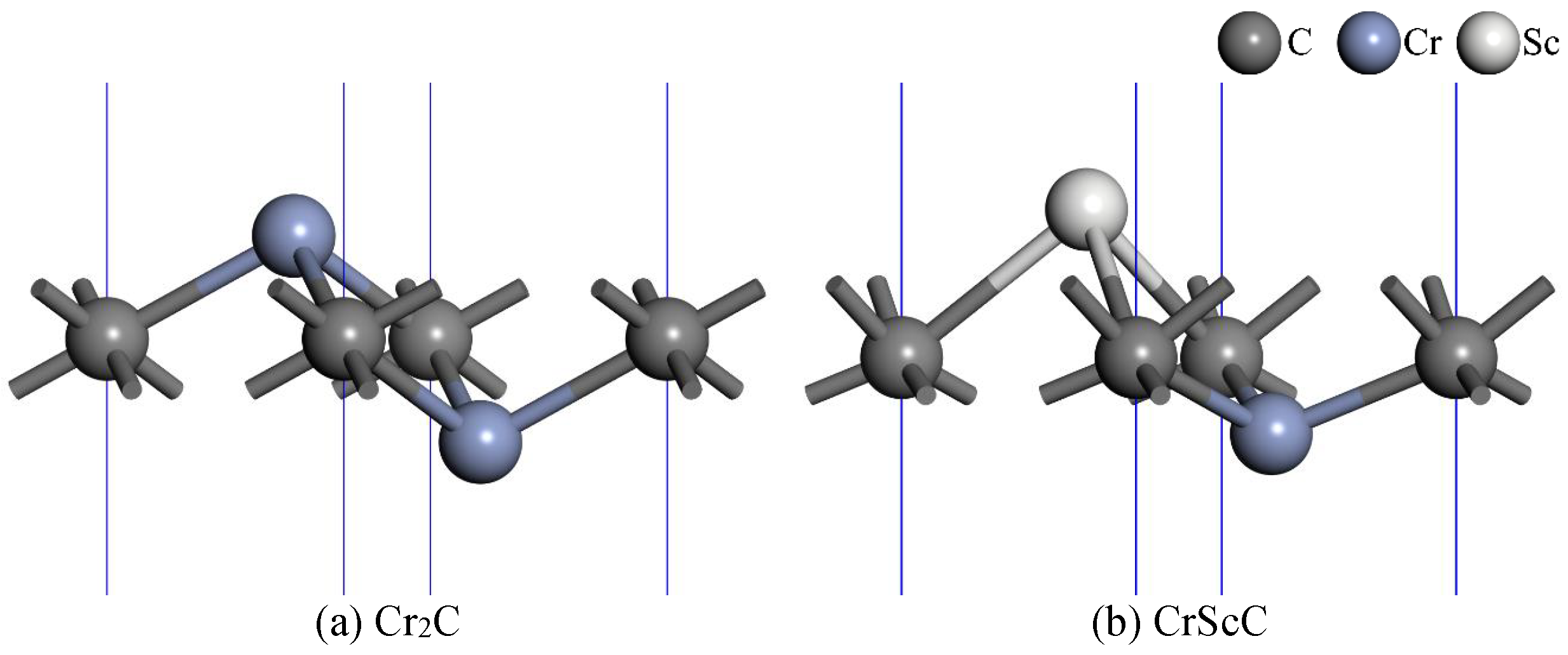
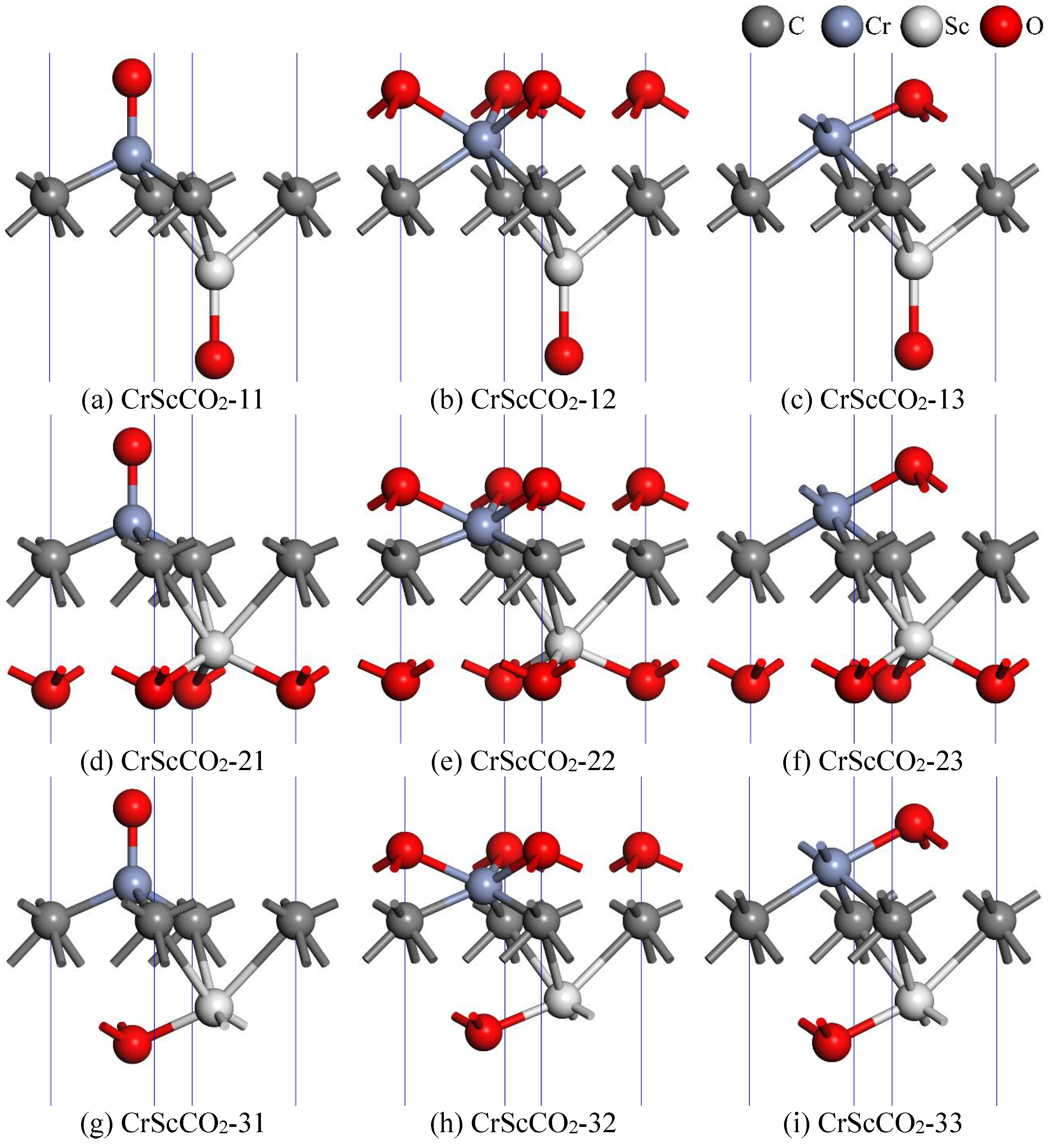
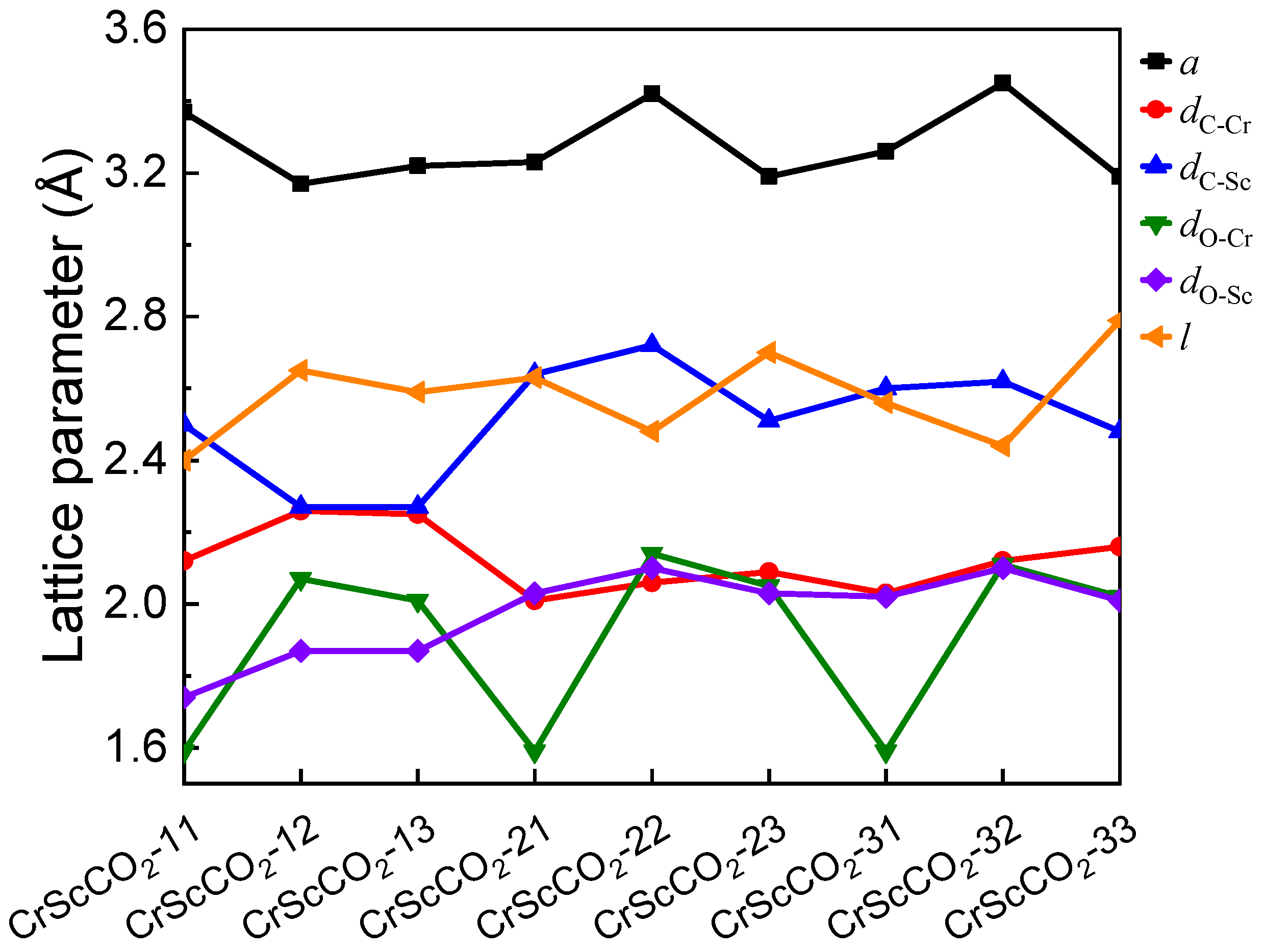
3.1. The Geometry and Stability
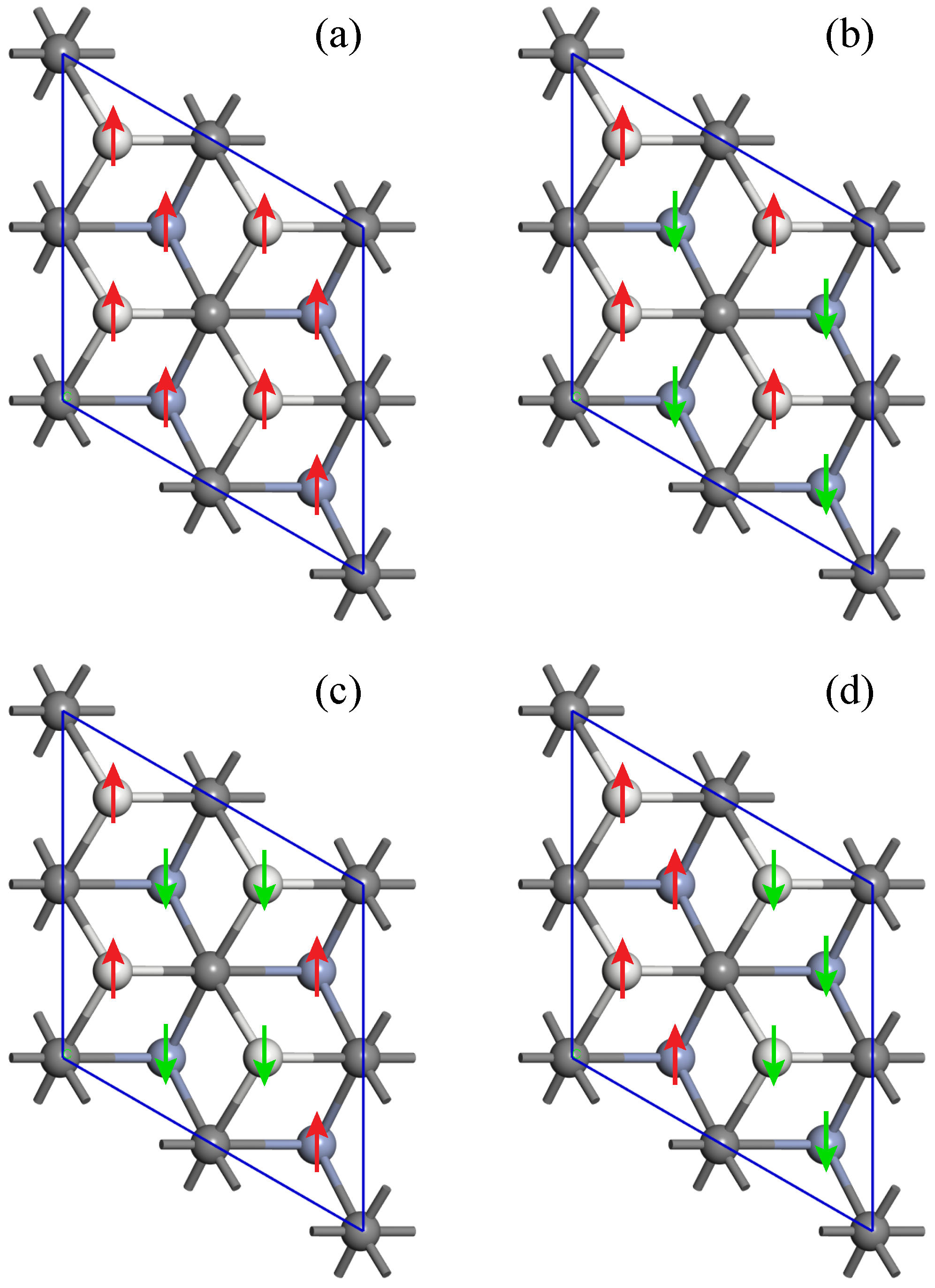
3.2. Magnetic and Electronic Structure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Naguib, M.; Kurtoglu, M.; Presser, V.; Lu, J.; Niu, J.; Heon, M.; Hultman, L.; Gogotsi, Y.; Barsoum, M. Two-Dimensional Nanocrystals Produced by Exfoliation of Ti3AlC2. Adv. Mater. 2011, 23, 4248–4253. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Bao, W.; Jaumaux, P.; Zhang, S.; Wang, C.; Wang, G. MXene-Based Composites: Synthesis and Applications in Rechargeable Batteries and Supercapacitors. Adv. Mater. Interfaces 2019, 6, 1802004. [Google Scholar] [CrossRef]
- Pang, J.; Mendes, R.G.; Bachmatiuk, A.; Zhao, L.; Ta, H.; Gemming, T.; Liu, H.; Liu, Z.; Rümmeli, M. Applications of 2D MXenes in energy conversion and storage systems. Chem. Soc. Rev. 2018, 48, 72–133. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.; Anwer, S.; Bhat, K.; Infas, M.; Mohideen, H.; Liao, K.; Qurashi, A. Prospects challenges and stability of 2D MXenes for clean energy conversion and storage applications. NPJ 2D Mater. Appl. 2021, 5, 61. [Google Scholar] [CrossRef]
- Naguib, M.; Mochalin, V.N.; Barsoum, M.W.; Gogotsi, Y. 25th anniversary article: MXenes: A new family of two-dimensional materials. Adv. Mater. 2014, 26, 992–1005. [Google Scholar] [CrossRef]
- Khazaei, M.; Ranjbar, A.; Arai, M.; Sasaki, T.; Yunoki, S. Electronic properties and applications of MXenes: A theoretical review. J Mater. Chem. C 2017, 5, 2488–2503. [Google Scholar] [CrossRef]
- Naguib, M.; Mashtalir, O.; Carle, J.; Presser, V.; Lu, J.; Hultman, L.; Gogotsi, Y.; Barsoum, M.W. Two-dimensional transition metal carbides. ACS Nano 2012, 6, 1322–1331. [Google Scholar] [CrossRef]
- Gomaa, I.; Hosny, N.M.; Elhaes, H.; Ezzat, H.A.; Elmahgary, M.G.; Ibrahim, M.A. Two-Dimensional MXene as a Promising Adsorbent for Trihalomethanes Removal: A Density-Functional Theory Study. Nanomaterials 2024, 14, 454. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Krist.-Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Bučko, T.; Hafner, J.; Lebègue, S.; Ángyán, J.G. Improved description of the structure of molecular and layered crystals: Ab initio DFT calculations with van der Waals corrections. J. Phys. Chem. C 2010, 114, 11814. [Google Scholar] [CrossRef] [PubMed]
- Si, C.; Zhou, J.; Sun, Z. Half-metallic ferromagnetism and surface functionalization-induced metal–insulator transition in graphene-like two-dimensional Cr2C crystals. ACS Appl. Mater. Inter. 2015, 7, 17510. [Google Scholar] [CrossRef] [PubMed]
- Akgenc, B.; Vatansever, E.; Ersan, F. Tuning of electronic structure, magnetic phase, and transition temperature in two-dimensional Cr-based Janus MXenes. Phys. Rev. Mater. 2021, 5, 083403. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Togo, A.; Chaput, L.; Tadano, T.; Tanaka, I. Implementation strategies in phonopy and phono3py. J. Phys. Condens. Matter 2023, 35, 353001. [Google Scholar] [CrossRef]
- Zhou, Y.; Wo, D.; Cho, Y.; He, Q.; Yang, X.; Herrera, K.; Chu, Z.; Han, Y.; Downer, M.C.; Hailin, P.; et al. Out-of-Plane Piezoelectricity and Ferroelectricity in Layered α-In2Se3 Nanoflakes. Nano Lett. 2017, 17, 5508–5513. [Google Scholar] [CrossRef]
- Zhang, K.; Bao, C.; Gu, Q.; Ren, X.; Zhang, H.; Deng, K.; Wu, Y.; Li, Y.; Feng, J.; Zhou, S. Raman signatures of inversion symmetry breaking and structural phase transition in type-II Weyl semimetal MoTe2. Nat. Commun. 2016, 7, 13552. [Google Scholar] [CrossRef]
- Lu, S.; Ren, W.; He, J.; Yu, C.; Jiang, P.; Chen, J. Enhancement of the lattice thermal conductivity of two-dimensional functionalized MXenes by inversion symmetry breaking. Phys. Rev. B 2022, 105, 165301. [Google Scholar] [CrossRef]
- Wolverton, C.; Ozoliṇš, V. First-principles aluminum database: Energetics of binary Al alloys and compounds. Phys. Rev. B 2006, 73, 144104. [Google Scholar] [CrossRef]
- Lv, P.; Li, Y.L.; Wang, J.F. Monolayer Ti2C MXene: Manipulating magnetic properties and electronic structures by an electric field. Phys. Chem. Chem. Phys. 2020, 22, 11266. [Google Scholar] [CrossRef]
- Arias-Camacho, I.M. Influence of the Hubbard U Parameter on the Structural, Electronic, Magnetic, and Transport Properties of Cr/Fe/Zr-Based MBenes. ACS Omega 2023, 8, 45003–45012. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Ai, L.; Chen, M.; Lü, Y. The mechanism of hydrogen-accelerated melting of polycrystalline copper. Phys. Chem. Chem. Phys. 2021, 23, 3942. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Akinola, O.; Chakraborty, I.; Celio, H.; Akinwande, D.; Incorvia, J.A.C. Synthesis and characterization of Cr2C MXenes. J. Mater. Res. 2021, 36, 1980–1989. [Google Scholar] [CrossRef]
- Wu, H.; Guo, Z.; Zhou, J.; Sun, Z. Vacancy-mediated lithium adsorption and diffusion on MXene. Appl. Surf. Sci. 2019, 488, 578–585. [Google Scholar] [CrossRef]
- García-Romeral, N.; Morales-García, Á.; Viñes, F.; de P., R.; de P. R. Moreira, I.; Illas, F. How does thickness affect magnetic coupling in Ti-based MXenes. Phys. Chem. Chem. Phys. 2023, 25, 17116. [Google Scholar] [CrossRef]
- You, H.; Zhang, Y.; Chen, J.; Ding, N.; An, M.; Miao, L.M.; Shuai, D. Peierls transition driven ferroelasticity in the two-dimensional d-f hybrid magnets. Phys. Rev. B 2021, 103, L161408. [Google Scholar] [CrossRef]
- Liu, X.; Huang, H.; Sun, L.; Pan, M.; Shang, Z. First-principles Study on the Electronic and Magnetic Properties of CrVCF2 MXene. J. Synth. Cryst. 2024, 53, 1386–1393. [Google Scholar] [CrossRef]
- García-Romeral, N.; Morales-García, A.; Viñes, F.; Moreira, I.d.P.R.; Illas, F. Theoretical Analysis of Magnetic Coupling in the Ti2C Bare MXene. J. Phys. Chem. C 2023, 127, 3706. [Google Scholar] [CrossRef]
- Yu, H.; Chen, M.; Shao, Z.; Tao, Y.; Jiang, X.; Dong, Y.; Zhang, J.; Yang, X.; Liu, Y. Giant tunneling magnetoresistance in in-plane double-barrier magnetic tunnel junctions based on MXene Cr2C. Phys. Chem. Chem. Phys. 2023, 25, 10991. [Google Scholar] [CrossRef]
- Lee, J.; Seko, A.; Shitara, K.; Nakayama, K.; Tanaka, I. Prediction model of band gap for inorganic compounds by combination of density functional theory calculations and machine learning techniques. Phys. Rev. B 2016, 93, 115104. [Google Scholar] [CrossRef]
- Makkar, P.; Ghosh, N.N. A review on the use of DFT for the prediction of the properties of nanomaterials. RSC Adv. 2021, 11, 27897. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Fang, Z.; Li, B.; Yang, Y. First-Principles Study of the Ferromagnetic Properties of Cr2CO2 and Cr2NO2 MXenes. ACS Omega 2020, 5, 25848. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zhang, Z.; Wang, Z.; Liu, B.; Zhou, D.; Zhou, X.; Zhang, P.; Zhao, X. Novel magneto-electrocatalyst Cr2CO2-MXene for boosting nitrogen reduction to ammonia. Mater. Horiz. 2024, 11, 1769. [Google Scholar] [CrossRef]
- Khazaei, M.; Arai, M.; Sasaki, T.; Chung, C.; Venkataramanan, N.S.; Estili, M.; Sakka, Y.; Kawazoe, Y. Novel Electronic and Magnetic Properties of Two-Dimensional Transition Metal Carbides and Nitrides. Adv. Funct. Mater. 2012, 23, 2185. [Google Scholar] [CrossRef]
- Zhou, T.; He, J.; Yang, K.; Wu, B.; Shen, G.; Huang, H.; Gu, Y.; Wen, L.; Zhang, Q. Ultrahigh tunneling magnetoresistance ratios in the sandwich-structured full MXene Cr2NO2/Ti2CO2/Cr2NO2 van der Waals heterojunction. Results Phys. 2024, 61, 107753. [Google Scholar] [CrossRef]
- Huang, H.; Yang, K.; Zhao, W.; Zhou, T.; Yang, X.; Wu, B. High-Pressure-Induced Transition from Ferromagnetic Semiconductor to Spin Gapless Semiconductor in Quaternary Heusler Alloy VFeScZ (Z = Sb, As, P). Appl. Sci. 2019, 9, 2859. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a | dC–Cr | dC–Sc | l | ||
|---|---|---|---|---|---|
| Cr2C | Current work | 3.27 | 2.18 | – | 2.17 |
| Ref. [13] | 3.21 | 2.13 | – | 2.11 | |
| Ref. [12] | 3.13 | 2.10 | – | 2.12 | |
| Ref. [25] | 2.81 | 2.09 | – | 1.93 | |
| CrScC | Current work | 3.36 | 2.21 | 2.32 | 2.33 |
| Ref. [13] | 3.29 | 2.09 | 2.39 | 2.34 |
| ΔE | Eb | Ef | Mtotal | MCr | MSc | |
|---|---|---|---|---|---|---|
| CrScCO2-11 | 5.26 | 4.76 | −0.03 | 1.00 | 2.62 | 0.14 |
| CrScCO2-12 | 5.32 | 4.75 | −0.04 | 1.00 | 3.58 | 0.03 |
| CrScCO2-13 | 4.53 | 4.91 | 0.11 | 1.00 | 3.43 | 0.04 |
| CrScCO2-21 | 1.50 | 5.51 | 0.72 | 1.00 | 2.19 | 0.00 |
| CrScCO2-22 | 0.31 | 5.75 | 0.96 | 3.00 | 3.88 | −0.10 |
| CrScCO2-23 | 1.30 | 5.55 | 0.76 | 1.00 | 3.25 | −0.17 |
| CrScCO2-31 | 1.02 | 5.61 | 0.81 | 1.00 | 2.23 | −0.06 |
| CrScCO2-32 | 0.00 | 5.81 | 1.02 | 3.00 | 4.03 | −0.13 |
| CrScCO2-33 | 0.84 | 5.64 | 0.85 | 1.20 | 3.30 | −0.12 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, H.; Liu, X.; Sun, L.; Shang, Z.; Zhou, T.; Li, P.; Wu, B. First-Principles Investigation of the Structural, Magnetic, and Electronic Properties of Janus MXene Material CrScCO2. Coatings 2025, 15, 580. https://doi.org/10.3390/coatings15050580
Huang H, Liu X, Sun L, Shang Z, Zhou T, Li P, Wu B. First-Principles Investigation of the Structural, Magnetic, and Electronic Properties of Janus MXene Material CrScCO2. Coatings. 2025; 15(5):580. https://doi.org/10.3390/coatings15050580
Chicago/Turabian StyleHuang, Haishen, Xiaoying Liu, Li Sun, Zhenzhen Shang, Tingyan Zhou, Ping Li, and Bo Wu. 2025. "First-Principles Investigation of the Structural, Magnetic, and Electronic Properties of Janus MXene Material CrScCO2" Coatings 15, no. 5: 580. https://doi.org/10.3390/coatings15050580
APA StyleHuang, H., Liu, X., Sun, L., Shang, Z., Zhou, T., Li, P., & Wu, B. (2025). First-Principles Investigation of the Structural, Magnetic, and Electronic Properties of Janus MXene Material CrScCO2. Coatings, 15(5), 580. https://doi.org/10.3390/coatings15050580