Abstract
Surface immobilization and characterization of the functional activity of fibronectin (Fn) type-III domains are reported. The domains FnIII9-10 or FnIII10 containing the RGD loop and PHSRN synergy site were recombinantly produced and covalently bound to chemically activated PEG methacrylate (MA) hydrogel coatings by microcontact printing. Such fabricated biochip surfaces were 6 mm in diameter and consisted of 190 µm wide protein stripes separated by 200 µm spacing. They were analyzed by imaging null ellipsometry, atomic force microscopy and fluorescence microscopy. Also, the coatings were tested in human foreskin fibroblast and HeLa cultures for at least 96 h, thus evaluating their suitability for controlled cell adhesion and proliferation. However, while HeLa cultures were equally well responsive to the FnIII9-10, FnIII10 and Fn surfaces, the fibroblasts displayed lower cell and lower focal adhesion areas, as well as lower proliferation rates on the Fn fragment surfaces as compared to Fn. Nevertheless, full functional activity of the fibroblasts was confirmed by immunostaining of Fn produced by the cells adherent on the biochip surfaces. The observed interaction differences that were either cell type or surface composition-dependent demonstrate the potential use of specifically engineered Fn and other ECM protein-derived domains in biochip architectures.
1. Introduction
The extracellular matrix (ECM) is a complex cellular environment and integral mechanical and structural part of the tissues. It provides pathways for cellular signaling, directing cell adhesion, function, differentiation, migration, proliferation and tissue formation [1]. Fibronectin (Fn) is a ubiquitous ECM protein, which is responsible for cytoskeletal assembly, cell attachment and migration. The globule of Fn displays several exposed molecular recognition sites for other ECM macromolecules and for cell receptors, including the well-studied Arginine–Glycine–Aspartic acid (RGD) loop and Pro–His–Ser–Arg–Asn (PHSRN) synergy site located at the Fn type-III domain repeats 10 (FnIII10) and 9 (FnIII9), respectively [2]. Controlling the interactions between the Fn molecule and cellsoffers an interesting strategy to stimulate cellular responses [3]. Specific fragments of Fn can be deposited on artificial surfaces and employed, for example, in analyzing phenotypes of adherent cells [4]. On the other hand, further dissection of Fn into smaller peptide fragments, e.g., RGD or RGD-PHSRN, leads to the loss of the correct structural context of the interacting cells and significantly lowers the overall adhesion strength [5].
Conformation of FnIII9-10 and/or FnIII10 at artificial interfaces has been analyzed by different modeling and experimental techniques. Molecular dynamics (MD) studies have been carried out on the physisorption of FnIII9-10 [6] and FnIII8-10 [7], describing the biological activity of the physisorbed Fn fragments. Additionally, MD simulations of FnIII7-10 indicated transient interactions with self-assembled monolayers [8]. A force spectroscopy study [9] reported on FnIII8-14 transition from “side-on” to “end-on” orientation during surface adsorption. However, non-covalent protein immobilization on surfaces does not provide a stable interface, leading to substantial protein desorption in cell culture medium [10].
There are numerous different approaches for surface functionalization by covalently attaching the entire Fn molecules (recently reviewed by Palomino et al. [11]). Some previous reports have indicated that coupling chemistry and/or choice of specific pining positions on the polypeptide chain are important for the functionality of the bound protein. For example, Vallieres et al. [12] covalently anchored Fn on amino-functionalized silica via primary amine groups, finding that such an immobilization was more favorable for cell adhesion than immobilization via the thiol moiety.
Furthermore, a few reports have suggested controlled immobilization of the Fn fragments as a strategy for investigation of cellular adhesion: covalent docking via primary amines [5], thiol [13,14] and fusion protein [15] have been reported. Markowski et al. [13] analyzed interactions of epithelial cells with FnIII9-10 variants covalently immobilized on polyacrylamide hydrogels, taking into account the substrate stiffness. They found that Fn fragments and the substrate mechanics act synergistically to direct the epithelial-to-mesenchymal transition of the cells. Hui et al. [14] covalently bound FnIII9-10 variants to hyaluronic acid hydrogels, mimicking the mechanics of healthy and fibrotic lung tissue to study the adhesion of lung fibroblasts. They found that the viscoelastic and adhesive cues together regulate fibroblast mechanobiology. However, the above-referred studies lack the details on the surface characterization of the coatings consisting of the Fn fragments.
Precise tailoring of the cellular environment in terms of the selection and orientation of specific ECM building blocks could be combined with the promising technologies of cell patterning and networking [16]. Cell chips comprising micro/nanometer-scale features consisting of ECM fragments could open new possibilities for miniaturized and highly parallelized analysis of the fundamental mechanisms involved in cellular adhesion, proliferation, differentiation and migration.
Recently, we introduced an efficient platform for cell chip fabrication taking advantage of initiator-free synthesis of polyethylene glycol methacrylate (PEG MA) hydrogels on glass substrates [17]. We demonstrated that such substrates can be efficiently used for the covalent immobilization and patterning of different proteins. Covalent immobilization of Fn in micropatterns with a high density of molecules in a hydrogel-like state has proved useful, for example, in studies of human primary cardiac stem cell differentiation [18]. Therefore, in the present study, we report on the possibility of fabricating biochip surfaces consisting of FnIII9-10 or FnIII10 covalently immobilized on a chemically activated PEG MA surface by microcontact printing [19].
Contrary to the above-cited studies on various Fn fragment immobilization, we performed characterization of our biochip surfaces by a set of surface analysis techniques. Having ensured that the Fn fragments can be docked to the bioinert PEG hydrogel substrates with a controlled surface density, homogeneity and stability, we further compared cell adhesion. We tested Fn fragment-decorated biochip surfaces in human foreskin fibroblast and epithelial HeLa cell cultures. Our study suggests different responses of the selected cell types to the Fn fragment vs. full-length Fn surfaces, as judged from the cell proliferation assay.
2. Materials and Methods
2.1. DNA Constructs, Recombinant Protein Production and Purification
Human fibronectin 1 (Fn1) (NG_012196.1) fragments corresponding to FnIII9-10 and FnIII10 included segments 4338-4910 and 4608-4910 of the Fn1 transcript mRNA (NM_212474.3) sequence. The fragment FnIII9-10 was PCR cloned from FN207 plasmid [20] (No. 50497; Addgene, Watertown, MA, USA); for FnIII10, the codon usage was optimized for E. coli and the nucleotide sequence was produced by gene synthesis (Thermo Fisher Scientific, Waltham, MA, USA) (Supplemental Materials, Figure S1).
The Impact (New England BioLabs, Ipswich, MA, USA) protein purification system utilizing a C-terminus-fused, self-cleavable intein tag with a chitin-binding domain was used for protein production. All steps were carried out according to manufacturer instructions with several modifications. Restriction enzymes NdeI and LguI (Thermo Fisher Scientific Baltics, Vilnius, Lithuania) were used to clone the constructs into a pTXB1 vector (New England BioLabs, Ipswich, MA, USA) and electrocompetent E. coli DH5 alpha (McLab, South San Francisco, CA, USA) cells were transformed for clone selection and E. coli BL21 (DE3) (McLab, South San Francisco, CA, USA) was used for protein production. The overnight bacterial culture was diluted in Lysogeny Broth (LB) medium [21] containing 100 µg·mL−1 of ampicillin, incubated to reach the OD600 0.4–0.6 and protein production, was induced with 1 mM of isopropyl-β-D-thiogalactoside for 4 h at 37 °C. The cells were centrifuged, and the pellet was resuspended in ~1/10 volume of ice-cold 20 mM Tris-HCl, pH 8.8, supplemented with 0.5 mM of phenylmethylsulfonyl fluoride and 1 mM of tris (2-carboxyethyl) phosphine. The cells were lysed, cellular debris was removed by centrifugation and the supernatant was loaded on the column, packed with 10 mL of chitin beads (New England BioLabs, Ipswich, MA, USA). Intein cleavage was induced by 40 mM dithiothreitol (DTT) at 4 °C overnight. The eluted protein was desalted using PD-10 columns (GE Healthcare, Chicago, IL, USA).
2.2. Recombinant Protein Characterization
The recombinant protein samples were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS PAGE) on 12% polyacrylamide gel using the Mini Protean system (BioRad, Hercules, CA, USA). After staining with Expedeon Instant Blue dye (Abcam, Cambridge, UK), the gels were scanned with an FLA 9000 scanner (GE Healthcare, Chicago, IL, USA), and the target protein purity was assessed using ImageQuant TL v.7.0 software (GE Healthcare, Chicago, IL, USA). The protein bands were excised from the gel, and in-gel digestion was carried out using MS-grade trypsin (Pierce, Appleton, WI, USA) according to Kondo and Hirohashi [22] and Shevchenko et al. [23]. Tryptic peptides were loaded and desalted on a 100 μm × 20 mm Acclaim PepMap C18 trap column and separated on a 75 μm × 150 mm Acclaim PepMap C18 column using an Ultimate3000 RSLC system (Thermo Fisher Scientific, Waltham, MA, USA), coupled to a Maxis G4 Q-TOF mass spectrometer detector with a Captive Spray nanoelectrospray ionization source (Bruker, Billerica, MA, USA). Peptide identification was performed using the MASCOT server (Matrix Science, London, UK) against the corresponding Fn1 fragment sequence and the SwissProt database (The UniProt Consortium, 2021). The data were visualized using the ProteinScape software (Bruker, Billerica, MA, USA).
2.3. Hydrogel-Coated Substrates
The detailed procedures for hydrogel synthesis are described elsewhere [17]. Briefly, commercial glass (AG Schott, Perai, Malaysia) or silica (Accurion GmbH, Göttingen, Germany) substrates were rinsed in ethanol and dried under a stream of nitrogen gas (99.999% purity). Then, they were washed for 10 min using the SC-1-cleaning procedure, in a 5:1:1 mixture of ultrapure water, 30% H2O2 (Carl Roth GmbH, Karlsruhe, Germany) and 25% NH4OH (Carl Roth GmbH) solutions, at 85 °C temperature for 10 min. Subsequently, they were treated in a plasma dry cleaner for 3 min at a 20 W power (Femto, Diener Electronic GmbH, Ebhausen, Germany). The plasma-treated glass or silica substrates were modified with (3-aminopropyl)trimethoxysilane (APTMS) (Sigma, St. Louis, MO, USA) in a custom-made vacuum chamber for 20 min at a 50 mm bar pressure and baked for 2 h at 120 °C temperature. Just before hydrogel synthesis by self-initiated photografting and photopolymerization (SIPGP) reaction, the samples were rinsed in water for 20 s and dried in a stream of nitrogen gas. Then, a drop of 5% of glutaraldehyde solution (GA) (Sigma) in 0.1 M phosphate buffer (PB) pH 8.0 was placed on the sample surface for 10 min to convert the amine groups into aldehyde groups. After this step, the samples were rinsed in ultrapure water and gently blown dry in nitrogen gas. Monomer solutions of 360 mM of 2-hydroxyethyl methacrylate (HEMA), poly(ethylene glycol) methacrylate (PEGMA) and methacrylic acid (MA) (all from Sigma) were all prepared in ultrapure water (taken directly from a Synergy 185 UV unit, Millipore). The monomer solutions were mixed in a 1:1:1 ratio using a regular shaker. Drops of 8 μL of the mixed monomers were used to coat 400 mm2 size samples for the subsequent SIPGP procedure, which was performed under UV light with the main emission peak at 254 nm. The glass and silica substrates with the synthesized hydrogel coatings were rinsed in ultrapure water for 1 min, dried under a nitrogen gas stream and kept dry at 4 °C till further use.
2.4. Protein Immobilization
The polypeptides were immobilized on the hydrogel substrate by means of random covalent coupling through the primary amines (lysines and N-terminus) to activated carboxyls of the PEG-based hydrogel coating. Briefly, substrates coated with the hydrogel containing functional carboxyl groups were incubated in ethanol solution of N-hydroxysuccinimide (NHS) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) (both from Sigma) for 25 min. After incubation, the chip substrates were cleaned in an ultrasonic bath in ethanol and ultrapure water, respectively. Cell-adhesive zones consisting of the Fn protein or Fnfragments were fabricated by microcontact printing [19]. Briefly, circular 6 mm diameter polydimethylsiloxane PDMS (Dow Corning) elastomeric stamps containing a surface topography of protruding 190 µm wide stripes with 200 µm spacing were employed. They were treated with oxygen plasma for 30 s, then coated with solutions of 1 mg·mL−1 FnIII9-10, FnIII10 or 0.1 mg·mL−1 Fn (Yo Proteins AB, Huddinge, Sweden) and incubated for 5 min. After incubation, the excess protein solution was removed with a pipette, and the elastomeric stamp was dried under the nitrogen gas flow and transferred into contact with the activated hydrogel surface. After 5 min, the elastomeric stamp was removed from the hydrogel surface, which was chemically quenched in 0.1 M ethanolamine solution in phosphate buffer for 40 min. After the passivation step, the fabricated chips were rinsed in ultrapure water and blown dry in the nitrogen gas stream. For inspection of the FnIII9-10 and FnIII10 RGD fragments, we chose to use antibodies that recognize the cell attachment fragments of human fibronectin molecules. We obtained biochip surfaces containing the FnIII9-10, FnIII10 and Fn zones, which were visualized with anti-fibronectin cell attachment fragment antibody (Merck, MAB88916-C), diluted 1:1000 with PBS buffer and incubated on substrates for 1 h at room temperature (RT). The secondary antibody Alexa Fluor goat anti-mouse IgG (H&L) was used at 2 μg·mL−1 for 30 min in the dark.
2.5. Imaging Ellipsometry
A single-wavelength imaging null-ellipsometer Nanofilm EP3, equipped with a laser emitting at the 658 nm wavelength (Accurion GmbH, Germany), was employed for the hydrogel and protein thickness mapping on silica-coated substrates (Accurion GmbH, Germany). Measurements were performed at a 60° angle of incidence. The thickness of the protein layer was modeled using the Accurion data analysis program, EP4 Model. The refractive index values were taken from the inbuilt databases. The analyzed hydrogel layers and protein zones were modeled as an ‘organic’ layer with a refractive index of n = 1.5 and k = 0. At least nine FnIII9-10 and FnIII10 surfaces that were prepared in three batches were measured, and at least six height maps were acquired from each sample for analysis. Four Fn surfaces were prepared, and at least six height maps were acquired for analysis.
2.6. Atomic Force Microscopy
Surface topography and thickness of the Fn/Fn fragment layers were analyzed using a NanoWizard 3 (Bruker) atomic force microscope (AFM). To evaluate the layer changes as the hydrogel and Fn/Fn fragments swell, the samples were scanned both in air and in 1× phosphate-buffered saline (PBS) (pH 7.4). All measurements were carried out in the quantitative imaging (QI) mode with MLCT-A (Bruker AFM) probes, at a set point of 1 nN. The scanned area ranged from 10 µm × 10 µm to 5 µm × 5 µm with a set resolution of 256 × 256 pixels. The QI mode is based on measuring force–displacement curves for each pixel. Therefore, for the scans of a swelled sample, a contact point height (CPH) was calculated to eliminate any surface deformations eventually occurring during the measurements. The CPH was determined by finding a height from the approach part of the force–displacement curve at which deflection was detected. The AFM scan data were processed employing the JPK Data Processing (v.6.1.120) and Gwyddion (v.2.53) [24] software. All the samples mapped by imaging ellipsometry samples were additionally analyzed by AFM, and at least three zones were imaged on each sample.
2.7. Cell Adhesion and Proliferation
Human foreskin fibroblasts transfected with a green fluorescent protein (HF-GFPs) and HeLa cells were maintained in a humidified 37 °C incubator (Binder C150, Tuttlingen, Germany) purged with 5% CO2. HF-GFPs were cultivated in the Medium106 media (Gibco, ThermoFisher Scientific, Waltham, MA, USA) with a low serum growth supplement (LSGS) (Gibco, ThermoFisher Scientific), 0.1 mg·mL−1 streptomycin/penicillin. The HeLa cells were cultivated in the Advance DMEM medium (Gibco, ThermoFisher Scientific) with 10% fetal bovine serum (FBS) and 200 mM L-glutamine, 0.1 mg·mL−1 streptomycin/penicillin. All samples were fixed in 4% paraformaldehyde in PBS for 10 min, washed three times with PBS, and examined under an Olympus BX51 upright microscope and an Olympus IX81 inverted microscope.
The biochips consisting of the hydrogel coatings with the immobilized FnIII9-10, FnIII10 or Fn proteins were used for single-cell shape analysis. They were placed into a tissue culture plate (TCP) (Techno Plastic Products AG, Trasadingen, Switzerland), and then approx. 2500 HF-GFP or HeLa cell were seeded. The biochip samples were kept in the cell culture for 24 h and then fixed. For permeabilization and blocking of nonspecific binding, the samples were incubated in PBS containing 0.1% Triton X-100 for 5 min and in 1% bovine serum albumin (BSA) for 30 min. The primary antibodies to vinculin (ab18058) were diluted 1/250 and incubated with samples for 2 h at RT. The secondary antibodies ab150113 AlexaFluor 488 goat anti-mouse IgG (H&L) were used at 2 μg·mL−1 for 30 min. Seven samples of each FnIII9-10, FnIII10 and Fn-modified biochips were prepared for HF-GFP single-cell shape analysis and 178, 198 and 83 single cells, respectively, were selected for analysis. Four of each FnIII9-10, FnIII10 and Fn chip, samples were prepared for HeLa single-cell shape analysis, and at least 265 cells from each surface type were selected for analysis. For controlled cell culture experiments, the HF-GFP and HeLa cells were seeded (approx. 2500 cells) both on the chips placed into the TCPs, as well as on empty TCPs, to facilitate cell counting. After 96 h of culturing, the chips were washed with 10 mM PBS twice and fixed. For cell proliferation, the CCK8 assay (Abcam, ab228554) was used. Cell proliferation experiments were performed on Days 1, 2 and 4, according to the manufacturer’s instructions. Fixed cell blockings were performed as described above. The primary antibodies to the full length of Fn protein (ab268020) were diluted 1/500 and incubated with samples for 1 h 30 min at RT. The secondary antibodies were ab6719 TexasRed goat anti-rabbit IgG (H&L), used at 2 μg·mL−1 for 30 min in the dark. Proliferation experiments on FnIII9-10, FnIII10, Fn and TCP surfaces were repeated three times, and four samples (chips) were used in each experiment.
2.8. Statistical Analysis
The imaging ellipsometry, AFM and CCK8 data were analyzed in Microsoft Office Excel 2010, and the quantitative results are presented as mean ± standard errors.
In the single-cell adhesion analysis, the cell borders, area, perimeter and focal adhesion areas were calculated with the ImageJ v.1.49h [25] software. The cell shape index was calculated applying the equation: shape index = 4πA/P2, where A is the cell area, and P is the cell perimeter. The quantitative results are presented as mean ± standard errors. The statistical data analysis was performed by applying the Analysis of variance (ANOVA) with the LSD post hoc test. Differences were considered statistically significant when p < 0.05. The data were processed using the Microsoft Office Excel 2010 and SPSS 20 (IBM) software.
3. Results
The fibronectin fragments corresponding to the type-III repeats 9-10 (FnIII9-10) and repeat 10 (FnIII10) include either combination of the cell adhesion synergy site with the RGD motif (FnIII9-10) or the RGD motif only (FnIII10). Recombinant protein production using a C-terminus fused self-cleavable intein tag with a chitin-binding domain, followed by chitin affinity matrix purification, yielded purities of FnIII9-10 and FnIII10 of 98% and 93%, respectively (see Supplementary Materials, Figure S2). This was comparable to the purity of the commercial Fn, 95%. To assess the potential effect of post-translational modifications resulting from expression in the bacterial cells or protein purification procedure, tryptic digestion of the SDS PAGE band corresponding to the FnIII9-10 fragment was analyzed by mass spectrometry (Supplementary Materials, Figure S3). The analysis resulted in the 97.4% coverage of the protein sequence and revealed partial oxidation of the two methionine residues (Met1 and Met192). However, we assume that the type and location of this modification would not be likely to affect the efficiency of immobilization and the cell-binding properties of the fragments.
3.1. Characterization of Biochip Surfaces
We characterized the fabricated biochip surfaces that consisted of the cell-adhesive (Fn fragment or Fn) zones and bioinert (cell- and protein-repellent) zones by a set of complementary surface analysis techniques: imaging ellipsometry, fluorescence microscopy and atomic force microscopy (AFM). Figure 1a–c show representative examples of label-free visualization of the surfaces: imaging ellipsometry height maps of FnIII9-10, FnIII10 and Fn covalently immobilized onto the PEG MA hydrogel on silica, respectively. We could confirm that the microcontact printing process yielded homogeneous zones consisting of both the Fn fragment and full Fn protein. The ellipsometric height of the obtained layers ranged from 1.4 ± 0.4 nm for FnIII9-10 and FnIII10 to 2.4 ± 0.5 nm for Fn. Such thickness values can be compared to the average size of the Fn globule, which is 2 nm [26], indicating formation of a dense layer of the selected biomolecules.
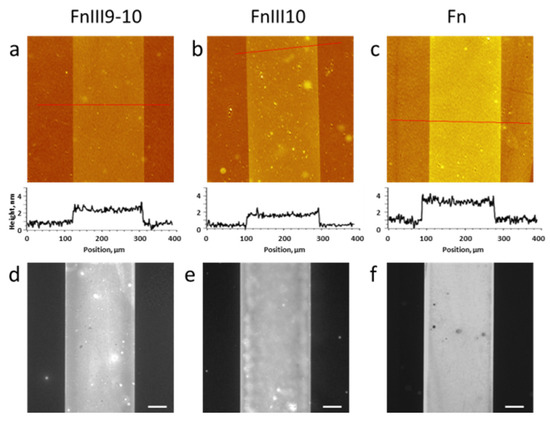
Figure 1.
Visualization of FnIII9-10, FnIII10 and Fn zones formed on ultrathin PEG methacrylate hydrogel coatings. (a–c) Representative imaging ellipsometry thickness maps recorded in air, shown together with their respective height cross-sections. The biochip substrate was silica. (d–f) Fluorescence microscopy images of the same type of biochip surfaces fabricated on glass substrates, after antibody binding against the cell attachment fragment in PBS buffer. The scale bar is 50 µm. (see Materials and Methods (Section 2) for details).
We tested the functionality of the two immobilized Fn fragments and Fn by antibody binding against the cell attachment fragment. As shown in Figure 1d–f, in all three cases, antifibronectin (cell attachment fragment antibody) binding suggested the RGD motive being exposed to the ambient. Note that for fluorescence visualization and for the cell studies presented below, we fabricated samples containing the Fn fragments/Fn on PEG MA hydrogels synthesized on glass substrates. The efficient antibody binding to the fragment/protein zones confirms the completeness of the immobilized fragment/protein layers, despite the exposure of the molecules to air during the microcontact printing process and covalent attachment to the PEG MA surface via multiple primary amine groups of the polypeptide chain. Additionally, this experiment confirms the excellent resistance of the PEG MA coating to nonspecific protein binding [17].
To verify the Fn protein/fragment layer thickness independently and to obtain nanometer-scale structural information, we carried out AFM analysis of the surfaces both in air and in buffer. Figure 2a–c shows representative AFM topography images recorded in air. The AFM topography height was 2.4 ± 0.2 nm, 1.8 ± 0.2 nm and 3.9 ± 0.4 nm for FnIII9-10, FnIII10 and Fn, respectively. The formed protein/fragment zones were complete, smooth and resembled the topography of the hydrogel surface (within the range of standard deviation), and they had well-defined borders.
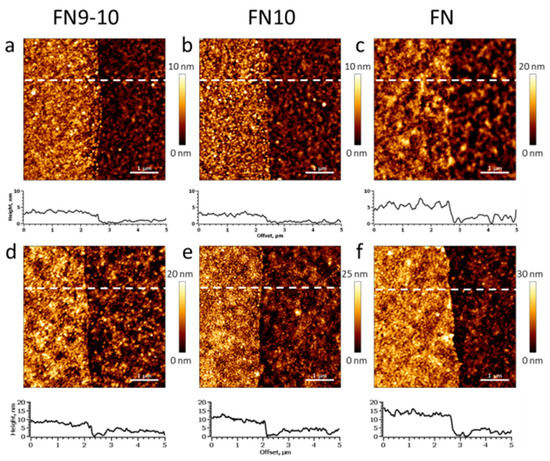
Figure 2.
Atomic force microscopy (AFM) analysis of FnIII9-10, FnIII10 and Fn zones formed on ultrathin PEG methacrylate hydrogel coating on silica. The topography images were recorded in dry (a–c) and hydrated (d,e) states. The surface height was recalculated from the acquired AFM topography data (d–f) for elimination of any potential surface deformations by the scanning probe (see Materials and Methods (Section 2) for details). The scale bar is 1 µm.
Upon submerging the biochip samples in PBS buffer (pH 7.4), the AFM height of the protein layer increased to 6.2 ± 0.7 nm, 6.5 ± 0.7 nm and 10.9 ± 1.1 nm for FnIII9-10, FnIII10 and Fn, respectively. The RMS surface roughness also increased. Despite the observed swelling, all FnIII9-10, FnIII10 and Fn zones remained stable and preserved their structural integrity (see Table 1 for details).

Table 1.
Comparison of atomic force microscopy topographic data for FnIII9-10, FnIII10 and Fn layers formed on ultrathin PEG methacrylate coatings on silica (represented as mean and standard error values).
3.2. Single-Cell Analysis
We estimated single-cell adhesion behavior by carrying out fluorescence microscopy of individual HF-GFP and HeLa cells, respectively. We fixed the cells after 24 h and immunostained them for vinculin (the red signal channel). Figure 3 shows representative epifluorescence microscopy images of selected Fn fragment/protein zones containing adherent cells. We used the same images also for the statistical analysis of the mean values of the cell area, cell shape index and focal adhesion area (see text below).
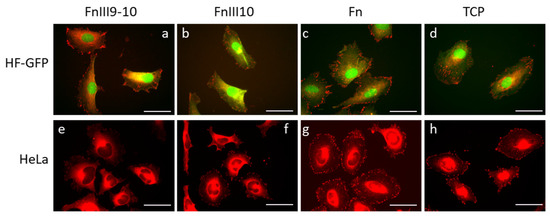
Figure 3.
Fluorescence micrographs showing GFP-transfected human fibroblast (HF-GFP, a–d) and HeLa (e–h) cell adhesion on FnIII9-10, FnIII10 and Fn -modified biochip surfaces. A tissue culture plate (TCP) was used for control. The cells were immunostained for vinculin (red filter). The scale bar is 50 µm in all images.
We determined the cell shape and focal adhesion area from the fluorescence microscopy images and analyzed the data by standard image processing. For this purpose, we repositioned the cell images placing a cell in the center of the image and rotating it to align the cell axis horizontally. Figure 4 shows the overlays of the contours of 30 randomly selected HF-GFP and HeLa cells on the biochip surface and tissue culture plate (TCP).
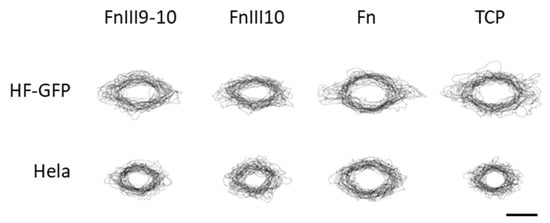
Figure 4.
Cell shape analysis of GFP-transfected human fibroblasts (HF-GFP) and HeLa cells on FnIII9-10, FnIII10 and Fn -modified biochip surfaces and on tissue culture plate (TCP). The images show 30 randomly selected single-cell contours that were centered and aligned on the cell axis horizontally prior to the overlay. The scale bar is 50 µm.
Further on, Figure 5 shows the summary of the performed statistical analysis. The adhesion behavior of HF-GFP on the FnIII9-10 and FnIII10 surfaces was rather similar and did not present any statistically significant differences. The values for the area of the HF-GFP, cell shape index and area of the cell adhesion were around 3000 µm2, 0.4 and 300 µm2, respectively. We could state that HF-GFP cells on the Fn surface and TCP plate presented similar adhesion behavior in terms of the cell area and adhesion zone area, respectively, with the cell area being around 3700 µm2 and a focal adhesion area of around 550 µm2. The cell shape index was similar for the cells on all protein/fragment surfaces, but statistically different from TCP.

Figure 5.
Statistical analysis of single GFP-transfected human fibroblast (HF-GFP) and HeLa cells adherent onto FnIII9-10, FnIII10 and Fn-modified biochip surfaces and tissue culture plate (TCP). The cells were immunostained for vinculin. The focal adhesion area per cell was calculated according to the number of vinculin points. The data are presented as mean ± standard error of between 83 and 335 cells from 4–7 samples of each type. * marks a statistically significant difference compared to the samples of the same cell type grown on Fn and TCP, ∧—compared to the samples on TCP, #—compared to Fn (projected cell area), #—compared to Fn domains (focal adhesions area per cell). ANOVA with post hoc LSD test, p < 0.05.
Similar to fibroblasts, the adhesion behavior of the HeLa cells on FnIII9-10 and FnIII10 was statistically similar. HeLa on the Fn surfaces had more than 1.5 times larger cell and adhesion areas, respectively. The cell shape index increased from 0.37 and 0.34 for FnIII9-10 and FnIII10, respectively, and to 0.43 for Fn. The HeLa cells on TCP displayed an intermediate behavior, and the cell area was closer to cells on FnIII9-10 and FnIII10. However, the cell shape index and area of the adhesion were closer to the cells on Fn.
3.3. Proliferation
To estimate cell proliferation, we employed the CCK-8 assay (Figure 6). Again, HF-GFP proliferation on the FnIII9-10 and FnIII10 surfaces was similar, at least with the selected period of 96 h. Notably, the HF-GFP cells on the Fn surfaces proliferated faster, and at 96 h, the optical density (OD) value was 2–2.5 times higher than in the case of FnIII9-10 and FnIII10. The cells on the TCP plate had the largest area to proliferate, and this could explain the observed highest OD value at 96 h.
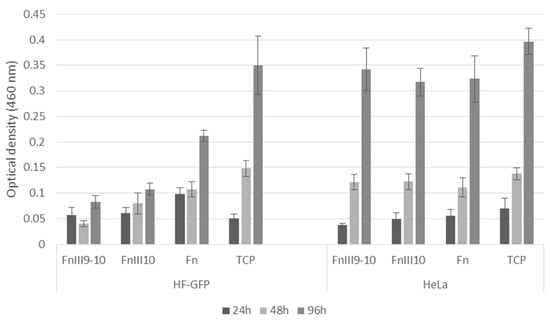
Figure 6.
CCK-8 proliferation assay of GFP-transfected human fibroblast (HF-GFP) and HeLa cells cultured on FnIII9-10, FnIII10 and Fn -modified biochip surfaces and tissue culture plate (TCP). The error bars represent the standard error between all counts for each sample type.
Contrary to the HF-GFP culture, the HeLa cells within the error range showed a similar proliferation rate on all types of surfaces and TCP plates within the course of the experiment. This result suggests that cytotoxicity effects due to the potential presence of the pyrogenic lipopolysaccharide (LPS) impurities of the FnIII9-10 and FnIII10 surfaces were unlikely. Previously, Hao et al. reported LPS-mediated stimulation of HeLa cell proliferation [27]. However, in our cell cultures, we observed statistically similar proliferation behavior on both the commercial Fn-made and recombinant FnIII9-10 and FnIII10 surfaces.
3.4. Secretion of Fibronectin
Finally, we verified the functional activity of the adherent cells by analyzing the secretion of fibronectin during proliferation on the different biochip surfaces. After one week, despite the above-mentioned differences in proliferation, confluent cell layers formed on all protein/fragment surfaces. We fixed the HF-GFP cells and immunostained them for the full-length fibronectin. We could detect fibronectin secretion by HF-GFP cells on all biochip surfaces (Figure 7), which indicates that the cells preserved their basic function. At the same time, we could observe no fibronectin bound to the bare PEG MA zones of the biochip surfaces, again confirming the excellent cell-/protein-repellent properties of this hydrogel coating serving as a substrate for protein/fragment immobilization [17].
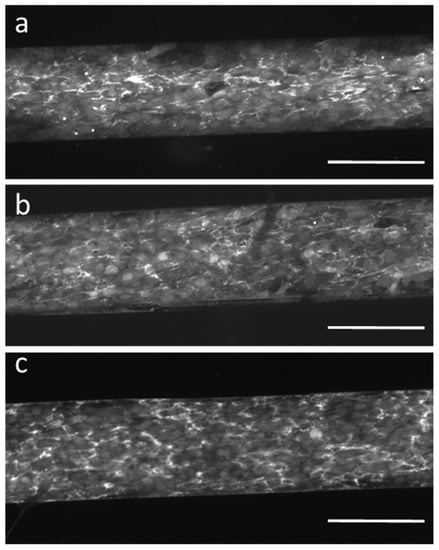
Figure 7.
Immunocytochemical staining visualizing secretion of full-length fibronectin by GFP-transfected human fibroblasts (HF-GFP) proliferating on (a) FnIII9-10-, (b) FnIII10- and (c) Fn-modified biochip surfaces, respectively. The scale bar is 200 µm.
4. Discussion
4.1. Orientation of Covalently Immobilized Fibronectin Fragments
The results of the present study show that both FnIII9-10 and FnIII10 surfaces are homogeneously coated as estimated on the micro- and nanometer scales. However, the ellipsometric height of both Fn fragment coatings (1.4 nm) was significantly lower than the estimated size of the fragments packed in their native state (approx. 8 nm and 4 nm for FnIII9-10 and FnIII10, respectively). Thus, we suggest that the randomly coupled (nonselective) covalent immobilization of the Fn fragments to the PEG MA hydrogel via the available five amino acids leads to the preferential “side-on” orientation of the polypeptide on the surface.
Figure 8 shows a simulated structure of FnIII9-10 with the marked amino acids bearing the primary amines (four lysines and the N-terminus) that are available for covalent immobilization to the carboxyl groups of the PEG MA hydrogel. Notably, three out of four lysines are positioned on the opposite side of the RGD region suggesting a probability that the latter motif can be exposed on the surface for available for cell adhesion. Indeed, we could confirm a certain degree of the RGD exposure to the ambient by performing the antibody binding experiments on our biochip surfaces. Still, one should bear in mind that due to the random coupling via the primary amines present at different locations at both ends of the protein, different degree of surface presentation of the RGD motifs is possible.
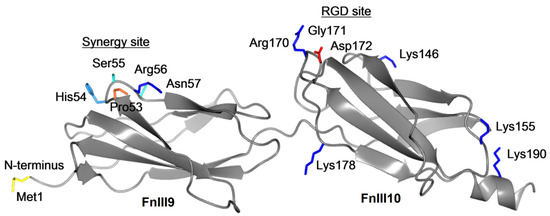
Figure 8.
Ribbon diagram of the human fibronectin 1 type-III repeat 9-10 fragment. The structure was modeled based on the fibronectin fragment PDB id 1FNF [29] and visualized with the CCP4mg 2.10.11 software [30].
Further on, random coupling of the FnIII9-10 fragment via the N-terminal amine can substantially limit the interdomain tilt angle. Altroff et al. showed [28] that the tilt angle between the FnIII9 and FnIII10 domains has an integrin-dependent function, which can be compromised by the conformational constraints due to the covalent coupling to the surface.
4.2. Cell Adhesion and Proliferation Behavior
We analyzed the perimeter, cell area and cell focal adhesion areas and proliferation in the HF-GFP and HeLa cultures on the FnIII9-10, FnIII10, Fn and TCP surfaces. The measured parameters, including the calculated cell shape index, were statistically similar for both cell types on the FnIII9-10 and FnIII10 surfaces. The differences might appear at the integrin engagement level since integrins α5β1 and αIIbβ3 in addition to RGD binding require interaction with the synergy site of the FnIII9 domain to reinforce binding [31]. In general, recognition of RGD by integrins could be highly sensitive to changes in the Fn conformation [3]; however, the detailed characterization of integrin binding was beyond the scope of this study.
Based on the molecular masses of protein/fragments and ellipsometric heights of the formed coatings, we estimate that the RGD binding site density on the FnIII9-10 and FnIII10 surfaces is 5 and 9 times higher in comparison to the Fn-modified biochip surface. This might have an effect on the cell adhesion process, as for both HF-GFP and HeLa cells, the cell area and focal adhesion areas were statistically lower for the FnIII9-10 and FnIII10 than for the Fn surfaces. Different results were described in a previous study [32], where gingival fibroblasts exhibited an equivalent biological activity FnIII9-10 and Fn physisorbed on surfaces on nontissue culture plates. Therefore, we explain the differences observed in our study by covalent pinning of the polypeptides at a certain orientation and conformation to the surface, thus significantly restricting the conformational flexibility, which likely would be more possible for physisorbed proteins/fragments. Additional conformational differences in the formed Fn fragment layers could be caused by the employed microcontact printing process. One cannot exclude that such a treatment could not affect the conformation and surface presentation of the cell-interacting motifs of the Fn fragments, leading to different effects on the interaction with the cell surface receptors (integrins). However, despite the above-mentioned differences, the cell shape index on all examined biochip surface types was statistically similar for both cell lines. The size and number of focal adhesions depend on the strength of cell-substrate adhesion, meanwhile, the cell shape depends on the particular cell function or cell–cell contact status. So far, we can state that cells on the different biochip surfaces in general maintain their function.
Cell spreading facilitates its division and proliferation. Therefore, it is generally accepted that a smaller cell area results in a lower proliferation rate. For our experiments, we chose fibroblasts and epithelial cells because of their well-documented differences in the morphogenetic behavior. Different cells might have distinct intracellular signaling pathways activated in response to the same ECM stimuli [33]. In our case, the HF-GFP and HeLa cells indeed showed different proliferation results. Since the HeLa cells were equally responsive to FnIII9, FnIII9-10 and Fn surfaces, this suggests that the surface stimuli for this cell line are the same. Meanwhile, fibroblast cell proliferation is stimulated by the RGD motif on the full-length fibronectin surface, suggesting a better integrin–protein interaction, as discussed above. Furthermore, the FnIII9-10, FnIII10 and Fn coatings promoted different cell adhesion for both cell lines. Therefore, the immobilized Fn fragments can serve as a convenient model platform to study the role of fibronectin in cell biology, tissue engineering, tumorigenesis and mechanobiology.
5. Conclusions
To conclude, we have shown that engineered Fn fragments can be used in prototyping biochip surfaces by microcontact printing, a process that yields densely packed, homogenous and stable layers, as proved by several surface analysis techniques. We found that the FnIII9-10- and FnIII10-modified PEG MA hydrogel surfaces were stable in HF-GFP and HeLa cultures for at least 96 h. The HF-GFP cells displayed lower cell and focal adhesion areas, respectively, as well as lower proliferation rates on the Fn fragment surfaces as compared to the immobilized full-length Fn. The observed interaction differences could be either related to the differences in the preferential integrin engagement or to the molecular conformation differences of the surface-pinned fragments. Despite of that, we could confirm the secretion of Fn by the adherent HF-GFP cells both on the Fn fragment and full-length Fn coatings.
Future studies could employ amino acid residue replacement and different chemical conjugation approaches to obtain biochip surfaces with oriented immobilization of FnIII9-10 and FnIII10 proteins. Additionally, it would be useful to introduce post-translational modifications to FnIII9-10 and FnIII10 by selecting a different protein expression system. Finally, we plan to explore the Fn, FnIII9-10 and FnIII10 biochip surfaces as a strategy to stimulate specific cellular responses by differential engagement of integrins.
Our present study is a step toward the development of stable biochip surface architectures with well-controlled biomolecular composition and physicochemical characteristics. More complex cell–cell and cell–ECM interaction analysis in the high-throughput, high-content mode can be designed in the future by employing the combined biochip and engineered protein fragment platform, e.g., by designing combinations of the fragments, printing fragment gradients and introducing geometrical/mechanobiological cues [16].
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/coatings12070880/s1, Figure S1: DNA sequences used for cloning of human fibronectin 1 (Fn1) type III domain fragments corresponding to domains 9-10 (A) and 10 (B). Restriction enzyme NdeI and LguI recognition sequences are marked in yellow and green, respectively; Figure S2: SDS PAGE separation of purified recombinant fibronectin fragments. Molecular weight (MW) marker (lane 1), and 5 μg of type-III repeat 9-10 (FnIII9-10) (lane 2) and 10 (FnIII10) (lane 3) fragments were separated on 12% polyacrylamide gel; Figure S3: Characterization of recombinant fibronectin fragment Fn9-10 using mass spectrometry. The purified fragment was separated by SDS PAGE, the corresponding molecular weight band was excised and in-gel digested with trypsin. Tryptic peptide mass fingerprinting was carried out using MASCOT server (Matrix science, London, UK) and corresponding peptide sequences. Data processed and visualized using ProteinScape software (Bruker, Billerica, MA, USA). (A): Sequence map. The residues matching and not matching database sequence are shown in red and grey font colour, respectively. Gray bars indicate the identified peptides and the relative TIC peak intensity is indicated by the darkness of the shading (the darker the shading, the higher the intensity value). Red boxes indicate observed fragment ions that correspond to the sequence. The potential site of N-glycosylation is marked in yellow. (B): Peptide results table. m/z-measured mass-to-charge ratio, Δm/z-difference in mass-to-charge ratio, RMS90-root mean square 90% confidence value, Scores–Mascot protein score, Range-position in protein (aa number to aa number), Sequence-peptide sequence, Modifications-chemical modifications present.
Author Contributions
Conceptualization, M.G., D.B. and R.V.; Methodology, G.S., A.M.-G., T.J., J.V. and P.H., Validation, M.G., A.M.-G. and P.H.; Formal analysis, M.G., A.M.-G., T.J., J.V. and P.H.; Investigation, M.G., G.S., A.M.-G., T.J., J.V. and P.H.; Resources, M.G. and D.B.; Data curation, M.G., G.S., A.M.-G., T.J., J.V. and P.H.; Writing—original draft preparation, M.G., D.B. and R.V.; Writing—review and editing, M.G., D.B. and R.V.; Visualization, M.G., G.S., A.M.-G., T.J., J.V. and P.H.; Supervision, M.G., D.B. and R.V.; Project administration, M.G., D.B. and R.V.; Funding acquisition, M.G., D.B. and R.V.; All authors have read and agreed to the published version of the manuscript.
Funding
This project has received funding from European Regional Development Fund (Project No. 01.2.2-LMT-K-718-01-0037) under grant agreement with the Research Council of Lithuania (LMTLT).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The raw data can be obtained from the corresponding author upon reasonable request.
Acknowledgments
The authors would like to thank Agnė Vailionytė, Šarūnas Vaitekonis and Karolis Gimbutis for laboratory assistance and discussion.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Doyle, A.D.; Nazari, S.S.; Yamada, K.M. Cell–Extracellular Matrix Dynamics. Phys. Biol. 2022, 19, 021002. [Google Scholar] [CrossRef] [PubMed]
- Patten, J.; Wang, K. Fibronectin in Development and Wound Healing. Adv. Drug Deliv. Rev. 2021, 170, 353–368. [Google Scholar] [CrossRef] [PubMed]
- Bachman, H.; Nicosia, J.; Dysart, M.; Barker, T.H. Utilizing Fibronectin Integrin-Binding Specificity to Control Cellular Responses. Adv. Wound Care 2015, 4, 501–511. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.C.; Rowe, J.A.; Barker, T.H. Guiding Epithelial Cell Phenotypes with Engineered Integrin-Specific Recombinant Fibronectin Fragments. Tissue Eng. Part A 2011, 17, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Petrie, T.A.; Capadona, J.R.; Reyes, C.D.; García, A.J. Integrin Specificity and Enhanced Cellular Activities Associated with Surfaces Presenting a Recombinant Fibronectin Fragment Compared to RGD Supports. Biomaterials 2006, 27, 5459–5470. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Hao, L.; Li, J.; Du, C.; Wang, Y. Role of Ninth Type-III Domain of Fibronectin in the Mediation of Cell-Binding Domain Adsorption on Surfaces with Different Chemistries. Langmuir 2018, 34, 9847–9855. [Google Scholar] [CrossRef] [PubMed]
- Liamas, E.; Kubiak-Ossowska, K.; Black, R.A.; Thomas, O.R.T.; Zhang, Z.J.; Mulheran, P.A. Adsorption of Fibronectin Fragment on Surfaces Using Fully Atomistic Molecular Dynamics Simulations. Int. J. Mol. Sci. 2018, 19, 3321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, L.; Li, T.; Wang, L.; Shi, X.; Fan, Y.; Du, C.; Wang, Y. Mechanistic Insights into the Adsorption and Bioactivity of Fibronectin on Surfaces with Varying Chemistries by a Combination of Experimental Strategies and Molecular Simulations. Bioact. Mater. 2021, 6, 3125–3135. [Google Scholar] [CrossRef] [PubMed]
- Liamas, E.; Black, R.A.; Mulheran, P.A.; Tampé, R.; Wieneke, R.; Thomas, O.R.T.; Zhang, Z.J. Probing Fibronectin Adsorption on Chemically Defined Surfaces by Means of Single Molecule Force Microscopy. Sci. Rep. 2020, 10, 15662. [Google Scholar] [CrossRef]
- Custódio, C.A.; Alves, C.M.; Reis, R.L.; Mano, J.F. Immobilization of Fibronectin in Chitosan Substrates Improves Cell Adhesion and Proliferation. J. Tissue Eng. Regen. Med. 2010, 4, 316–323. [Google Scholar] [CrossRef] [Green Version]
- Durand, C.P.; Pauthe, E.; Gand, A. Fibronectin-Enriched Biomaterials, Biofunctionalization, and Proactivity: A Review. Appl. Sci. 2021, 11, 12111. [Google Scholar] [CrossRef]
- Vallières, K.; Chevallier, P.; Sarra-Bournet, C.; Turgeon, S.; Laroche, G. AFM Imaging of Immobilized Fibronectin: Does the Surface Conjugation Scheme Affect the Protein Orientation/Conformation? Langmuir 2007, 23, 9745–9751. [Google Scholar] [CrossRef] [PubMed]
- Markowski, M.C.; Brown, A.C.; Barker, T.H. Directing Epithelial to Mesenchymal Transition through Engineered Microenvironments Displaying Orthogonal Adhesive and Mechanical Cues. J. Biomed. Mater. Res. Part A 2012, 100A, 2119–2127. [Google Scholar] [CrossRef] [PubMed]
- Hui, E.; Moretti, L.; Barker, T.H.; Caliari, S.R. The Combined Influence of Viscoelastic and Adhesive Cues on Fibroblast Spreading and Focal Adhesion Organization. Cell. Mol. Bioeng. 2021, 14, 427–440. [Google Scholar] [CrossRef]
- Eisenberg, J.L.; Piper, J.L.; Mrksich, M. Using Self-Assembled Monolayers to Model Cell Adhesion to the 9th and 10th Type III Domains of Fibronectin. Langmuir 2009, 25, 13942–13951. [Google Scholar] [CrossRef] [PubMed]
- Blanchoin, L.; Letort, G.; Ennomani, H.; Gressin, L.; Théry, M. Dynamic Reorganization of the Actin Cytoskeleton. F1000Research 2015, 4, 940. [Google Scholar] [CrossRef] [Green Version]
- Cėpla, V.; Rakickas, T.; Stankevičienė, G.; Mazėtytė-Godienė, A.; Baradokė, A.; Ruželė, Ž.; Valiokas, R. nas Photografting and Patterning of Poly(Ethylene Glycol) Methacrylate Hydrogel on Glass for Biochip Applications. ACS Appl. Mater. Interfaces 2020, 12, 32233–32246. [Google Scholar] [CrossRef] [PubMed]
- Miksiunas, R.; Aldonyte, R.; Vailionyte, A.; Jelinskas, T.; Eimont, R.; Stankeviciene, G.; Cepla, V.; Valiokas, R.; Rucinskas, K.; Janusauskas, V.; et al. Cardiomyogenic Differentiation Potential of Human Dilated Myocardium-derived Mesenchymal Stem/Stromal Cells: The Impact of HDAC Inhibitor SAHA and Biomimetic Matrices. Int. J. Mol. Sci. 2021, 22, 12702. [Google Scholar] [CrossRef]
- Qin, D.; Xia, Y.; Whitesides, G.M. Soft Lithography for Micro-and Nanoscale Patterning. Nat. Protoc. 2010, 5, 491–502. [Google Scholar] [CrossRef] [Green Version]
- Danen, E.H.J.; Aota, S.I.; Van Kraats, A.A.; Yamada, K.M.; Ruiter, D.J.; Van Muijen, G.N.P. Requirement for the Synergy Site for Cell Adhesion to Fibronectin Depends on the Activation State of Integrin A5β1. J. Biol. Chem. 1995, 270, 21612–21618. [Google Scholar] [CrossRef] [Green Version]
- Bertani, G. Studies on Lysogenesis. I. The Mode of Phage Liberation by Lysogenic Escherichia Coli. J. Bacteriol. 1951, 62, 293–300. [Google Scholar] [CrossRef] [Green Version]
- Kondo, T.; Hirohashi, S. Application of Highly Sensitive Fluorescent Dyes (CyDye DIGE Fluor Saturation Dyes) to Laser Microdissection and Two-Dimensional Difference Gel Electrophoresis (2D-DIGE) for Cancer Proteomics. Nat. Protoc. 2006, 1, 2940–2956. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Tomas, H.; Havliš, J.; Olsen, J.V.; Mann, M. In-Gel Digestion for Mass Spectrometric Characterization of Proteins and Proteomes. Nat. Protoc. 2006, 1, 2856–2860. [Google Scholar] [CrossRef] [PubMed]
- Nečas, D.; Klapetek, P. Gwyddion: An Open-Source Software for SPM Data Analysis. Open Phys. 2012, 10, 181–188. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Meth. 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Erikson, H.P.; Carrell, N.; McDonagh, J. Fibronectin Molecule Visualized in Electron Microscopy: A Long, Thin, Flexible Strand. J. Cell Biol. 1981, 91, 673–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, H.; Xu, F.; Hao, J.; He, Y.Q.; Zhou, X.Y.; Dai, H.; Wu, L.Q.; Liu, F.R. Lipoxin A4 Suppresses Lipopolysaccharide-Induced Hela Cell Proliferation and Migration via NF-ΚB Pathway. Inflammation 2015, 38, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Altroff, H.; Schlinkert, R.; Van Der Walle, C.F.; Bernini, A.; Campbell, I.D.; Werner, J.M.; Mardon, H.J. Interdomain Tilt Angle Determines Integrin-Dependent Function of the Ninth and Tenth FIII Domains of Human Fibronectin. J. Biol. Chem. 2004, 279, 55995–56003. [Google Scholar] [CrossRef] [Green Version]
- Leahy, D.J.; Aukhil, I.; Erickson, H.P. 2.0 Å Crystal Structure of a Four-Domain Segment of Human Fibronectin Encompassing the RGD Loop and Synergy Region. Cell 1996, 84, 155–164. [Google Scholar] [CrossRef] [Green Version]
- McNicholas, S.; Potterton, E.; Wilson, K.S.; Noble, M.E.M. Presenting Your Structures: The CCP4mg Molecular-Graphics Software. Acta Crystallogr. Sect. D 2011, 67, 386–394. [Google Scholar] [CrossRef] [Green Version]
- Benito-Jardón, M.; Klapproth, S.; Gimeno-LLuch, I.; Petzold, T.; Bharadwaj, M.; Müller, D.J.; Zuchtriegel, G.; Reichel, C.A.; Costell, M. The Fibronectin Synergy Site Re-Enforces Cell Adhesion and Mediates a Crosstalk between Integrin Classes. elife 2017, 6, e22264. [Google Scholar] [CrossRef] [PubMed]
- Abla, A.B.; Boeuf, G.; Elmarjou, A.; Dridi, C.; Poirier, F.; Changotade, S.; Lutomski, D.; Elm’selmi, A. Engineering of Bio-Adhesive Ligand Containing Recombinant RGD and PHSRN Fibronectin Cell-Binding Domains in Fusion with a Colored Multi Affinity Tag: Simple Approach for Fragment Study from Expression to Adsorption. Int. J. Mol. Sci. 2021, 22, 7362. [Google Scholar] [CrossRef] [PubMed]
- Balion, Z.; Cėpla, V.; Svirskiene, N.; Svirskis, G.; Druceikaitė, K.; Inokaitis, H.; Rusteikaitė, J.; Masilionis, I.; Stankevičienė, G.; Jelinskas, T.; et al. Cerebellar Cells Self-Assemble into Functional Organoids on Synthetic, Chemically Crosslinked ECM-Mimicking Peptide Hydrogels. Biomolecules 2020, 10, 754. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).