
Preparation and Self-Healing Application of Isocyanate Prepolymer Microcapsules

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
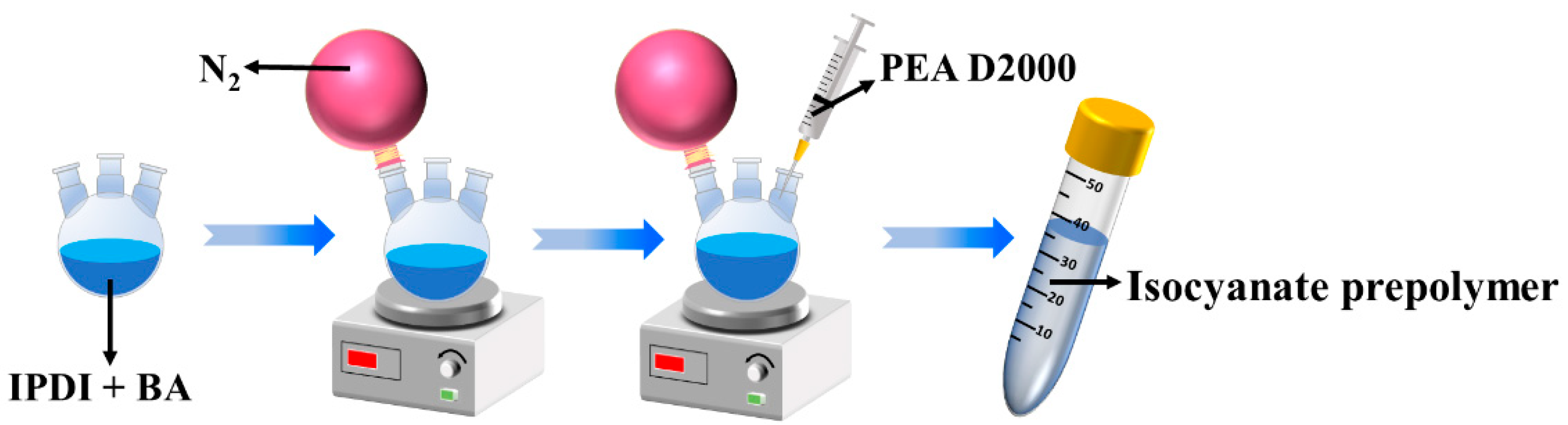
2.2. Preparation of Isocyanate Prepolymer
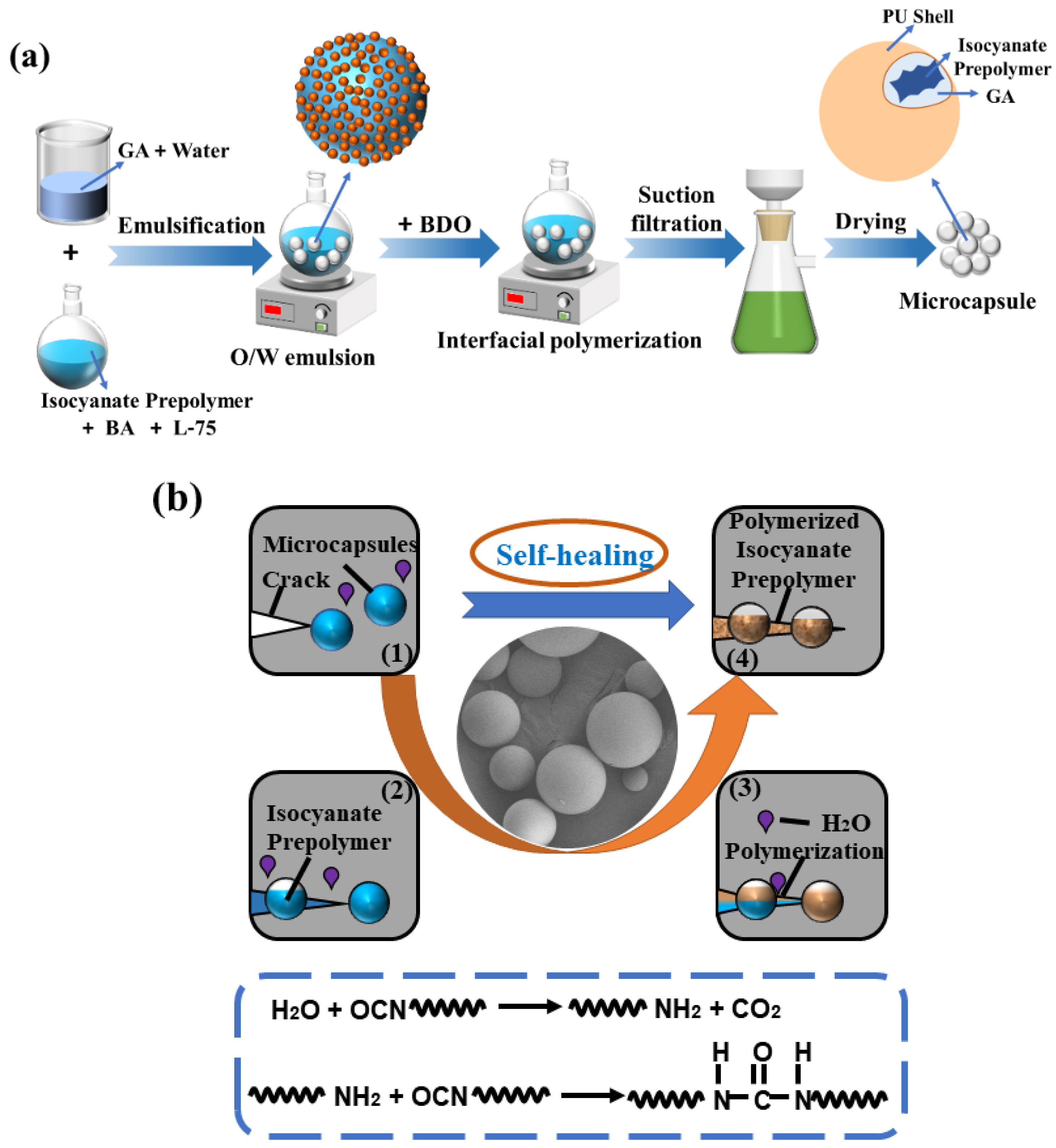
2.3. Preparation of Microcapsules
2.4. Core Content Determination
2.5. Characterizations
2.6. Evaluation of Self-Healing Performance
2.7. Evaluation of Anti-Corrosion Performance
3. Results and Discussion
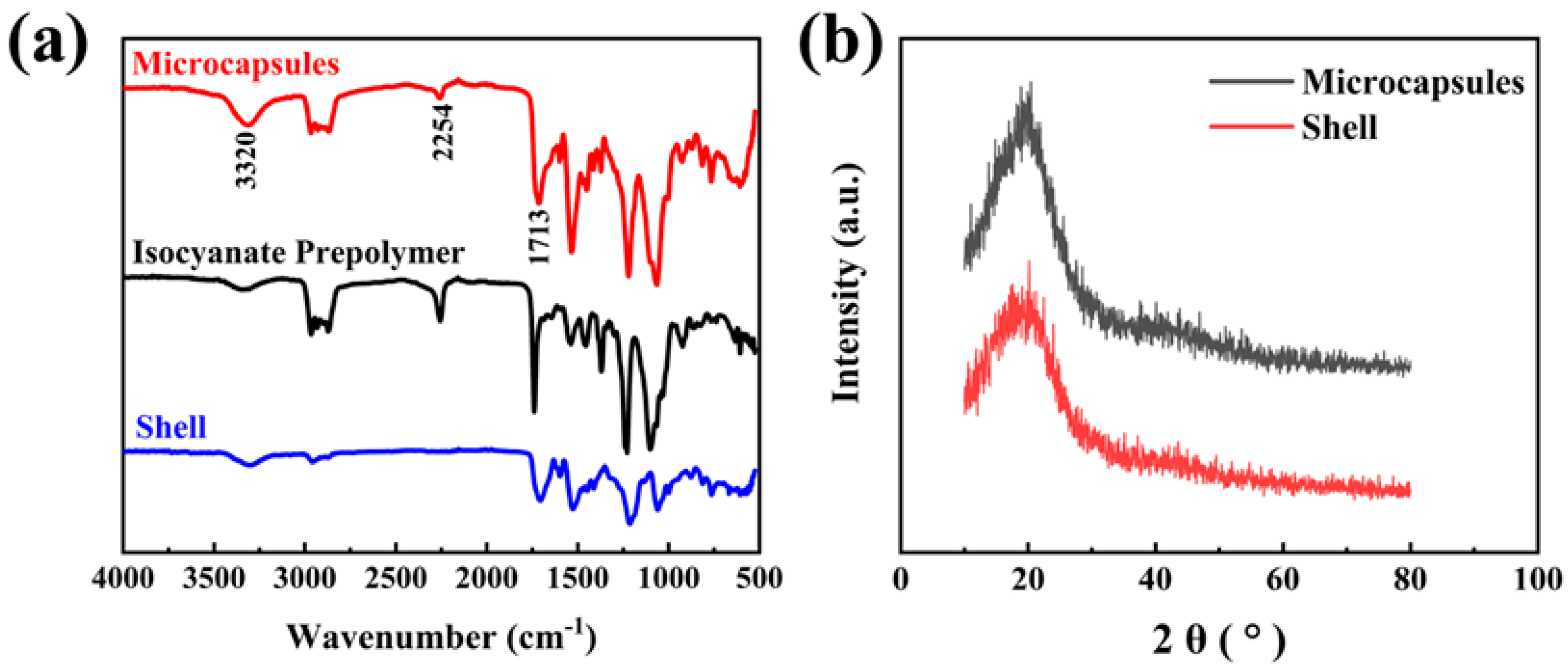
3.1. Analysis of the Chemical Structure of Microcapsules
3.2. Effect of the Concentration of Gum Arabic on Microcapsules
3.3. Effect of the Concentration of BDO on Microcapsules
3.4. Core Content
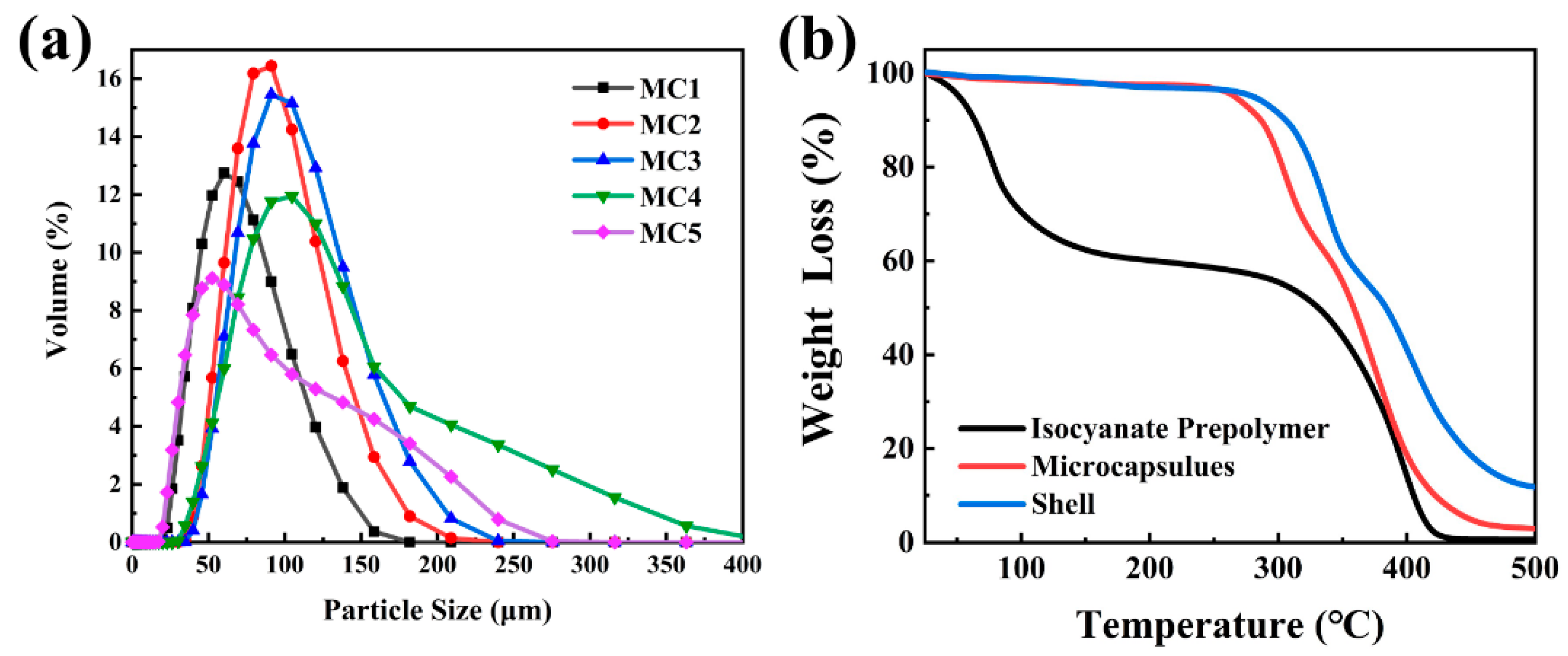
3.5. Particle Size and Thermal Analysis of Microcapsules
3.6. Preliminary Self-Healing and Anti-Corrosion Performance of Epoxy Coatings
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wilson, G.O.; Moore, J.S.; White, S.R.; Sottos, N.R.; Andersson, H.M. Autonomic healing of epoxy vinyl esters via ring opening metathesis polymerization. Adv. Funct. Mater. 2008, 18, 44–52. [Google Scholar] [CrossRef]
- Jin, H.; Mangun, C.L.; Stradley, D.S.; Moore, J.S.; Sottos, N.R.; White, S.R. Self-healing thermoset using encapsulated epoxy-amine healing chemistry. Polymer 2012, 53, 581–587. [Google Scholar] [CrossRef]
- Zhu, D.Y.; Wetzel, B.; Noll, A.; Rong, M.Z.; Zhang, M.Q. Thermo-molded self-healing thermoplastics containing multilayer microreactors. J. Mater. Chem. A 2013, 1, 7191–7198. [Google Scholar] [CrossRef]
- Jin, H.; Mangun, C.L.; Griffin, A.S.; Moore, J.S.; Sottos, N.R.; White, S.R. Thermally stable autonomic healing in epoxy using a dual-microcapsule system. Adv. Mater. 2014, 26, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, R.F.A.; van den Berg, O.; Nguyen, L.T.T.; Fehér, K.; Du Prez, F.E. Microencapsulation of active ingredients using PDMS as shell material. Macromolecules 2014, 47, 8231–8237. [Google Scholar] [CrossRef]
- White, S.R.; Sottos, N.R.; Geubelle, P.H.; Moore, J.S.; Kessler, M.R.; Sriram, S.R.; Brown, E.N.; Viswanathan, S. Autonomic healing of polymer composites. Nature 2001, 409, 794–797. [Google Scholar] [CrossRef]
- Alizadegan, F.; Mirabedini, S.M.; Pazokifard, S.; Moghadam, S.G.; Farnood, R. Improving self-healing performance of polyurethane coatings using PU microcapsules containing bulky-IPDI-BA and nano-clay. Prog. Org. Coat. 2018, 123, 350–361. [Google Scholar] [CrossRef]
- Cao, C.F.; Liu, W.J.; Xu, H.; Yu, K.X.; Gong, L.X.; Guo, B.F.; Li, Y.T.; Feng, X.L.; Lv, L.Y.; Pan, H.T.; et al. Temperature-induced resistance transition behaviors of melamine sponge composites wrapped with different graphene oxide derivatives. J. Mater. Sci. Technol. 2021, 85, 194–204. [Google Scholar] [CrossRef]
- Cao, B.; Souza, L.; Xu, J.; Mao, W.; Wang, F.; Al-Tabbaa, A. Soil mix cutoff wall materials with microcapsule-based self-Healing grout. J. Geotech. Geoenviron. Eng. 2021, 147, 04021124. [Google Scholar] [CrossRef]
- Nguyen, L.T.T.; Hillewaere, X.K.D.; Teixeira, R.F.A.; van den Berg, O.; Du Prez, F.E. Efficient microencapsulation of a liquid isocyanate with in situ shell functionalization. Polym. Chem. 2015, 6, 1159–1170. [Google Scholar] [CrossRef]
- Song, Y.; Chen, K.F.; Wang, J.J.; Liu, Y.; Qi, T.; Li, G.L. Synthesis of polyurethane/poly (urea-formaldehyde) double-shelled microcapsules for self-healing anticorrosion coatings. Chin. J. Polym. Sci. 2020, 38, 45–52. [Google Scholar] [CrossRef]
- Jeoung, H.J.; Kim, K.W.; Chang, Y.J.; Jung, Y.C.; Ku, H.; Oh, K.W.; Choi, H.M.; Chung, J.W. Self-healing EPDM rubbers with highly stable and mechanically-enhanced urea-formaldehyde (UF) microcapsules prepared by multi-step in situ polymerization. Polymers 2020, 12, 1918. [Google Scholar] [CrossRef]
- Rodriguez, R.; Bekas, D.G.; Flórez, S.; Kosarli, M.; Paipetis, A.S. Development of self-contained microcapsules for optimised catalyst position in self-healing materials. Polymer 2020, 187, 122084. [Google Scholar] [CrossRef]
- Fan, W.; Zhang, Y.; Li, W.; Wang, W.; Zhao, X.; Song, L. Multilevel self-healing ability of shape memory polyurethane coating with microcapsules by induction heating. Chem. Eng. J. 2019, 368, 1033–1044. [Google Scholar] [CrossRef]
- Chen, L.; Yu, H.; Li, W.; Dirican, M.; Liu, Y.; Zhang, X. Interlayer design based on carbon materials for lithium–sulfur batteries: A review. J. Mater. Chem. A 2020, 8, 10709–10735. [Google Scholar] [CrossRef]
- Zhou, S.; Jia, Y.; Wang, C. Global sensitivity analysis for the polymeric microcapsules in self-healing cementitious composites. Polymers 2020, 12, 2990. [Google Scholar] [CrossRef]
- Shchukin, D.G. Container-based multifunctional self-healing polymer coatings. Polym. Chem. 2013, 4, 4871–4877. [Google Scholar] [CrossRef] [Green Version]
- Blaiszik, B.J.; Jones, A.R.; Sottos, N.R.; White, S.R. Microencapsulation of gallium–indium (Ga–In) liquid metal for self-healing applications. J. Microencapsul. 2014, 31, 350–354. [Google Scholar] [CrossRef]
- Wu, G.; An, J.; Sun, D.; Tang, X.; Xiang, Y.; Yang, J. Robust microcapsules with polyurea/silica hybrid shell for one-part self-healing anticorrosion coatings. J. Mater. Chem. A 2014, 2, 11614–11620. [Google Scholar] [CrossRef]
- Habib, S.; Khan, A.; Nawaz, M.; Sliem, M.H.R.; Shakoor, R.A.; Kahraman, R.; Abdullah, A.M.; Zekri, A. Self-healing performance of multifunctional polymeric smart coatings. Polymers 2019, 11, 1519. [Google Scholar] [CrossRef] [Green Version]
- An, S.; Yoon, S.S.; Lee, M.W. Self-healing structural materials. Polymers 2021, 13, 2297. [Google Scholar] [CrossRef]
- Yan, X.; Peng, W. Preparation of microcapsules of urea formaldehyde resin coated waterborne coatings and their effect on properties of wood crackle coating. Coatings 2020, 10, 764. [Google Scholar] [CrossRef]
- Tao, Z.; Cui, J.; Qiu, H.; Yang, J.; Gao, S.; Li, J. Microcapsule/silica dual-fillers for self-healing, self-reporting and corrosion protection properties of waterborne epoxy coatings. Prog. Org. Coat. 2021, 159, 106394. [Google Scholar] [CrossRef]
- Katoueizadeh, E.; Zebarjad, S.M.; Janghorban, K. Mechanical properties of epoxy composites embedded with functionalized urea-formaldehyde microcapsules containing an oxidizable oil. Mater. Chem. Phys. 2021, 260, 124106. [Google Scholar] [CrossRef]
- Attaei, M.; Loureiro, M.V.; Do Vale, M.; Condeço, J.A.; Pinho, I.; Bordado, J.C.; Marques, A.C. Isophorone diisocyanate (IPDI) microencapsulation for mono-component adhesives: Effect of the active H and NCO sources. Polymers 2018, 10, 825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attaei, M.; Calado, L.M.; Taryba, M.G.; Morozov, Y.; Shakoor, R.A.; Kahramna, R.; Marques, A.C.; Monmor, M.F. Autonomous self-healing in epoxy coatings provided by high efficiency isophorone diisocyanate (IPDI) microcapsules for protection of carbon steel. Prog. Org. Coat. 2020, 139, 105445. [Google Scholar] [CrossRef]
- Fathabadi, H.F.; Javidi, M. Self-Healing and Corrosion Performance of Polyurethane Coating Containing Polyurethane Microcapsules. Preprint. 2021. Available online: https://doi.org/10.21203/rs.3.rs-348120/v1 (accessed on 30 March 2021).
- Haghayegh, M.; Mirabedini, S.M.; Yeganeh, H. Preparation of microcapsules containing multi-functional reactive isocyanate-terminated-polyurethane-prepolymer as healing agent, part II: Corrosion performance and mechanical properties of a self-healing coating. RSC Adv. 2016, 6, 50874–50886. [Google Scholar] [CrossRef]
- Nik Md Noordin Kahar, N.N.F.; Osman, A.F.; Alosime, E.; Arsat, N.; Azman, N.A.M.; Syamsir, A.; Itam, Z.; Hamid, Z.A.A. The versatility of polymeric materials as self-healing agents for various types of applications: A review. Polymers 2021, 13, 1194. [Google Scholar] [CrossRef]
- Koh, E.; Kim, N.K.; Shin, J.; Kim, Y.W. Polyurethane microcapsules for self-healing paint coatings. RSC Adv. 2014, 4, 16214–16223. [Google Scholar] [CrossRef]
- Dry, C. Procedures developed for self-repair of polymer matrix composite materials. Compos. Struct. 1996, 35, 263–269. [Google Scholar] [CrossRef]
- Li, J.; Shi, H.; Liu, F.; Han, E.H. Self-healing epoxy coating based on tung oil-containing microcapsules for corrosion protection. Prog. Org. Coat. 2021, 156, 106236. [Google Scholar] [CrossRef]
- Sima, W.X.; Shao, Q.Q.; Sun, P.T.; Liang, C.; Yang, M.; Yin, Z.; Deng, Q. Magnetically gradient-distributed microcapsule/epoxy composites: Low capsule load and highly targeted self-healing performance. Chem. Eng. J. 2021, 405, 126908. [Google Scholar] [CrossRef]
- Yang, J.; Keller, M.W.; Moore, J.S.; White, S.R.; Sottos, N.R. Microencapsulation of isocyanates for self-healing polymers. Macromolecules 2008, 41, 9650–9655. [Google Scholar] [CrossRef]
- Wang, W.; Xu, L.; Li, X.; Lin, Z.; Yang, Y.; An, E. Self-healing mechanisms of water triggered smart coating in seawater. J. Mater. Chem. A 2014, 2, 1914–1921. [Google Scholar] [CrossRef]
- Di Credico, B.; Levi, M.; Turri, S. An efficient method for the output of new self-repairing materials through a reactive isocyanate encapsulation. Eur. Polym. J. 2013, 49, 2467–2476. [Google Scholar] [CrossRef]
- Woo, J.H.; Kim, N.H.; Kim, S.I.; Park, O.K.; Lee, J.H. Effects of the addition of boric acid on the physical properties of mxene/polyvinyl alcohol (PVA) nanocomposite. Compos. Part B Eng. 2020, 199, 108205. [Google Scholar] [CrossRef]
- Liang, F.; Chen, W.; Ma, A.; Zhou, H.W.; Jin, X.L. Preparation and properties of isocyanate microcapsules by interfacial polymerization method. Polym. Mater. Sci. Eng. 2018, 34, 150–154. [Google Scholar]
- Yi, H.; Yang, Y.; Gu, X.; Huang, J.; Wang, C. Multilayer composite microcapsules synthesized by Pickering emulsion templates and their application in self-healing coating. J. Mater. Chem. A 2015, 3, 13749–13757. [Google Scholar] [CrossRef]
- Santos, A.N.B.; Santos, D.J.; Carastan, D.J. Microencapsulation of reactive isocyanates for application in self-healing materials: A review. J. Microencapsul. 2021, 38, 338–356. [Google Scholar] [CrossRef]
- Tripathi, M.; Dwivedi, R.; Kumar, D.; Roy, P.K. Thermal activation of mendable epoxy through inclusion of microcapsules and imidazole complexes. Polym. Plast. Technol. Eng. 2016, 55, 129–137. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | m(GA)/g | m(L-75)/g | m(Isocyanate Prepolymer)/g | m(BDO)/g |
|---|---|---|---|---|
| MC1 | 2 | 3 | 6 | 2 |
| MC2 | 3 | 3 | 6 | 2 |
| MC3 | 5 | 3 | 6 | 2 |
| MC4 | 3 | 3 | 6 | 2.5 |
| MC5 | 3 | 3 | 6 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiang, G.; Tu, J.; Xu, H.; Ji, J.; Liang, L.; Li, H.; Chen, H.; Tian, J.; Guo, X. Preparation and Self-Healing Application of Isocyanate Prepolymer Microcapsules. Coatings 2022, 12, 166. https://doi.org/10.3390/coatings12020166
Xiang G, Tu J, Xu H, Ji J, Liang L, Li H, Chen H, Tian J, Guo X. Preparation and Self-Healing Application of Isocyanate Prepolymer Microcapsules. Coatings. 2022; 12(2):166. https://doi.org/10.3390/coatings12020166
Chicago/Turabian StyleXiang, Guifeng, Jing Tu, Heng Xu, Jie Ji, Li Liang, Haozhe Li, Haoran Chen, Jingqing Tian, and Xiaode Guo. 2022. "Preparation and Self-Healing Application of Isocyanate Prepolymer Microcapsules" Coatings 12, no. 2: 166. https://doi.org/10.3390/coatings12020166
APA StyleXiang, G., Tu, J., Xu, H., Ji, J., Liang, L., Li, H., Chen, H., Tian, J., & Guo, X. (2022). Preparation and Self-Healing Application of Isocyanate Prepolymer Microcapsules. Coatings, 12(2), 166. https://doi.org/10.3390/coatings12020166