Design and Synthesis of Small Molecules as Potent Staphylococcus aureus Sortase A Inhibitors


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
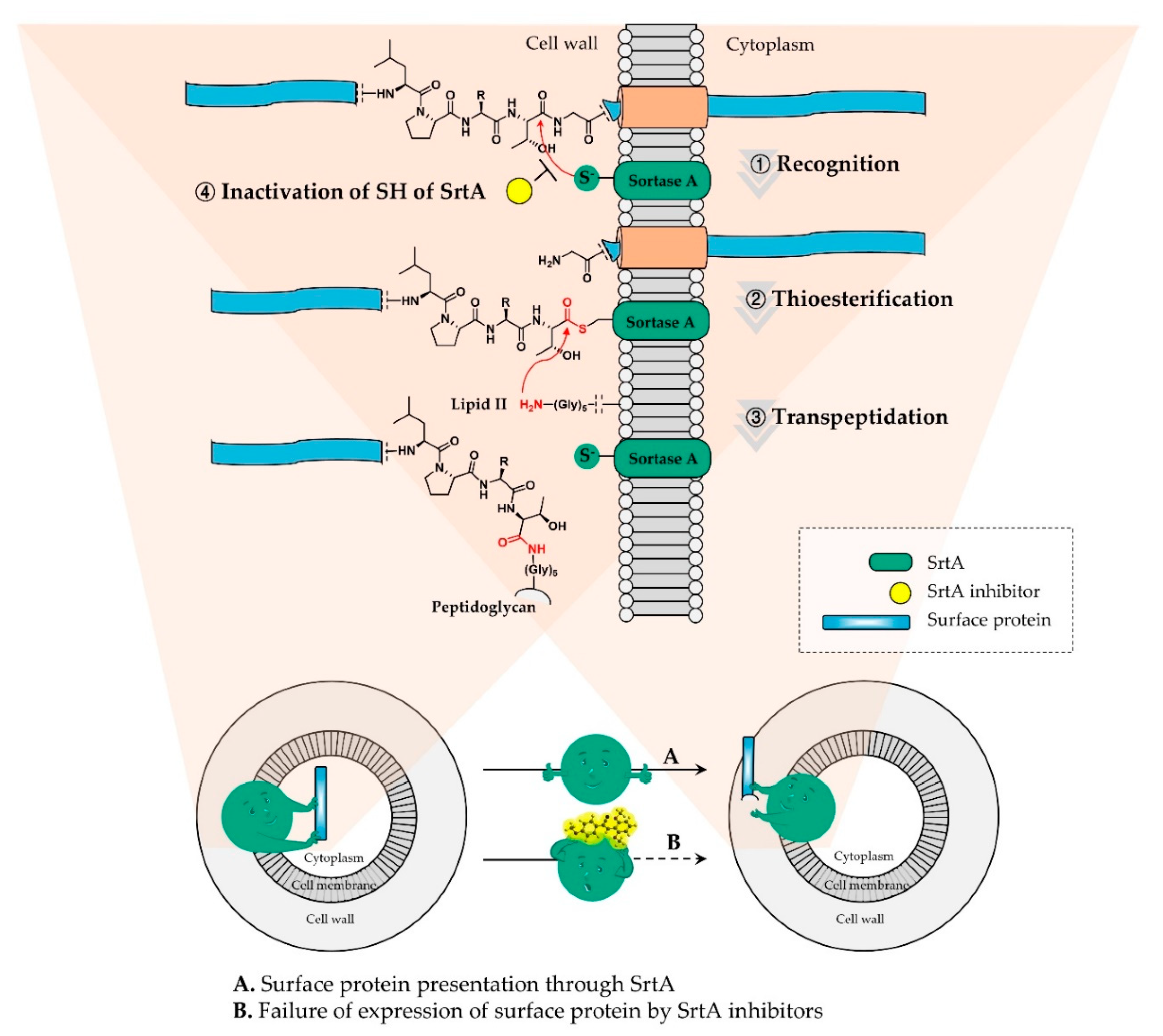
1. Introduction
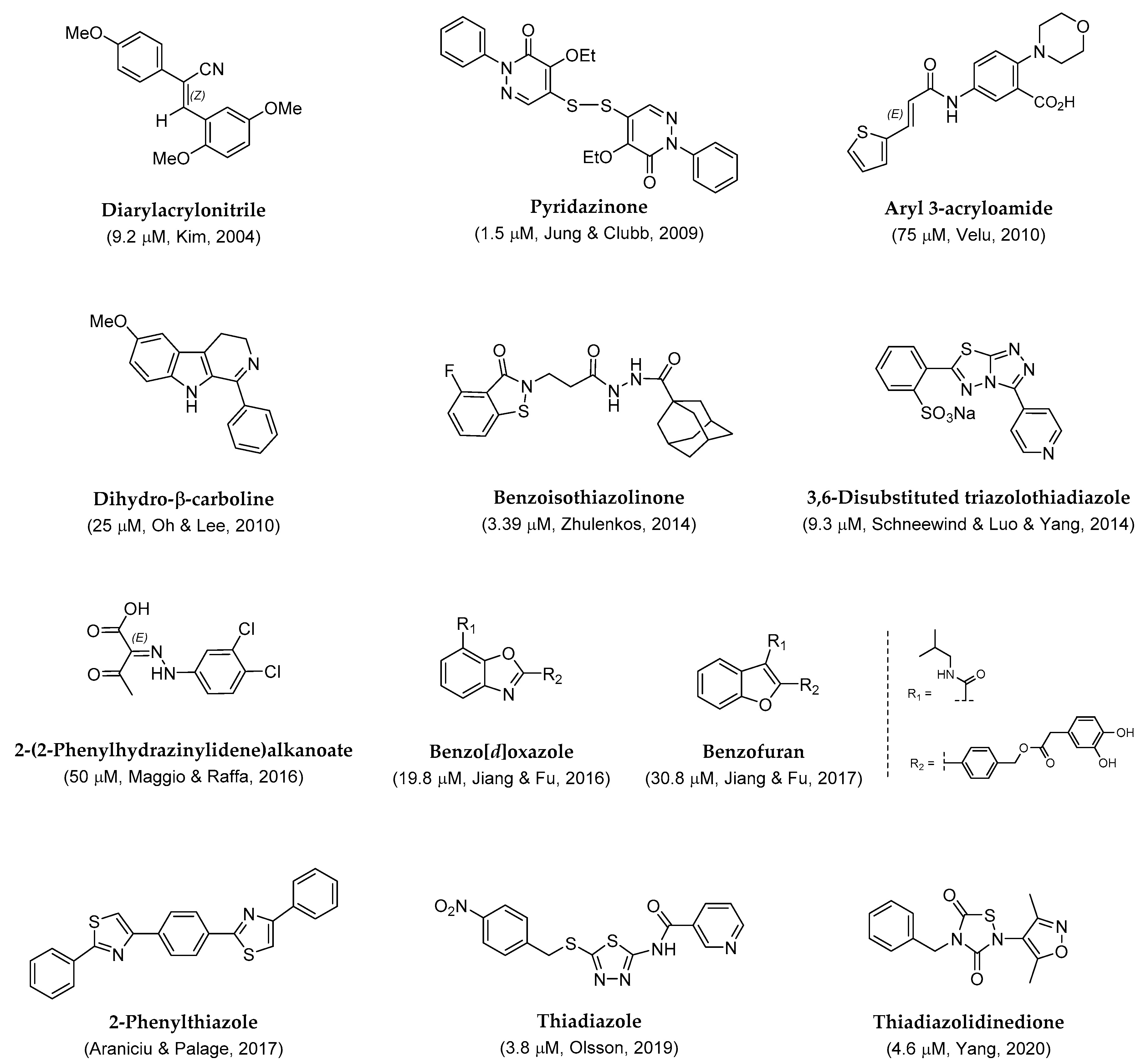
2. Results and Discussion
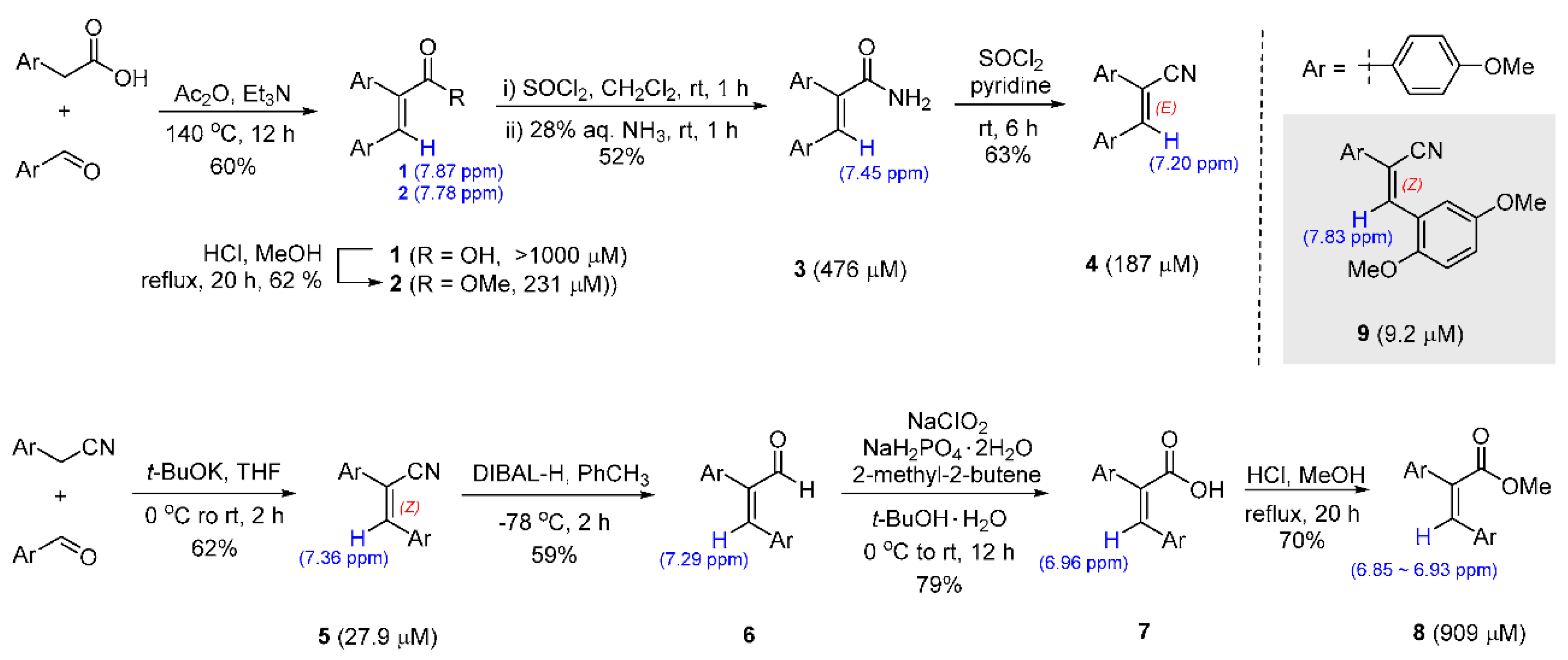
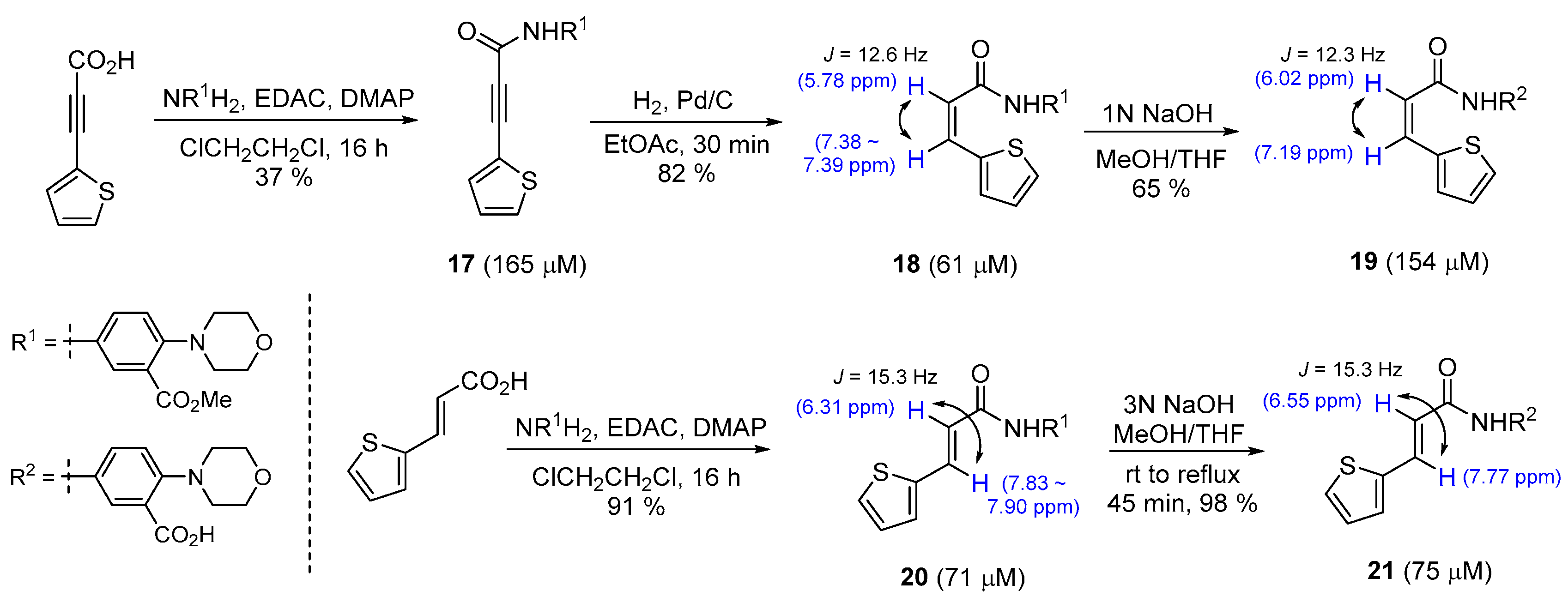
2.1. Preparation of Diarylacrylonitriles
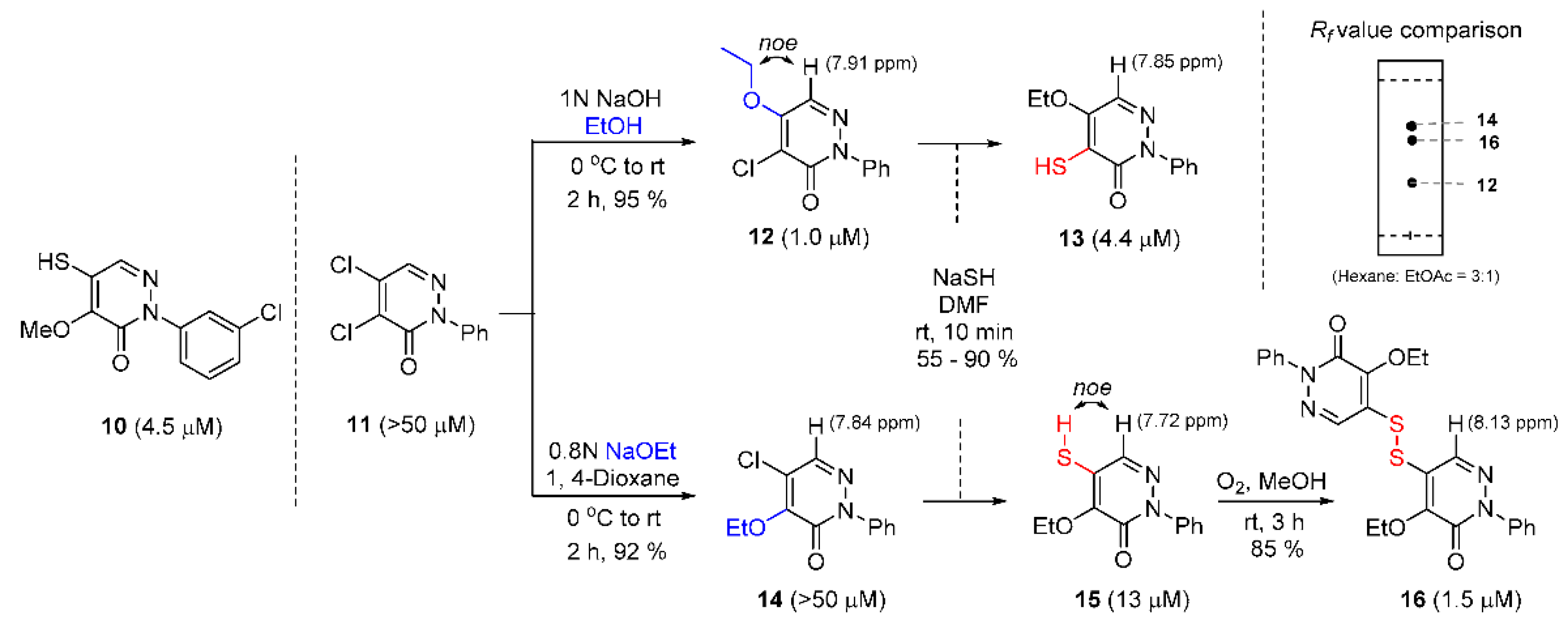
2.2. Preparation of Pyridazinones
2.3. Preparation of Aryl 3-Acryloamide
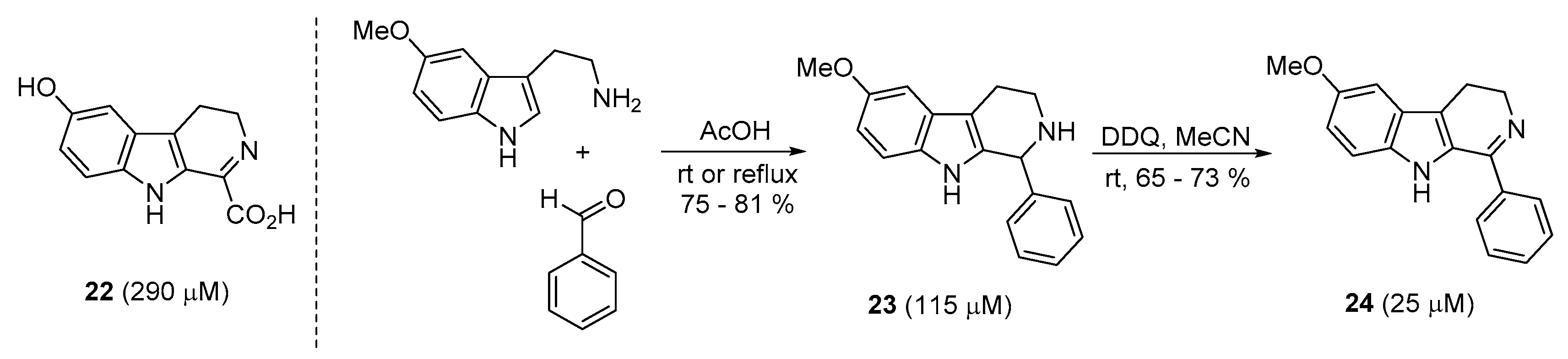
2.4. Preparation of Dihydro-β-Carboline
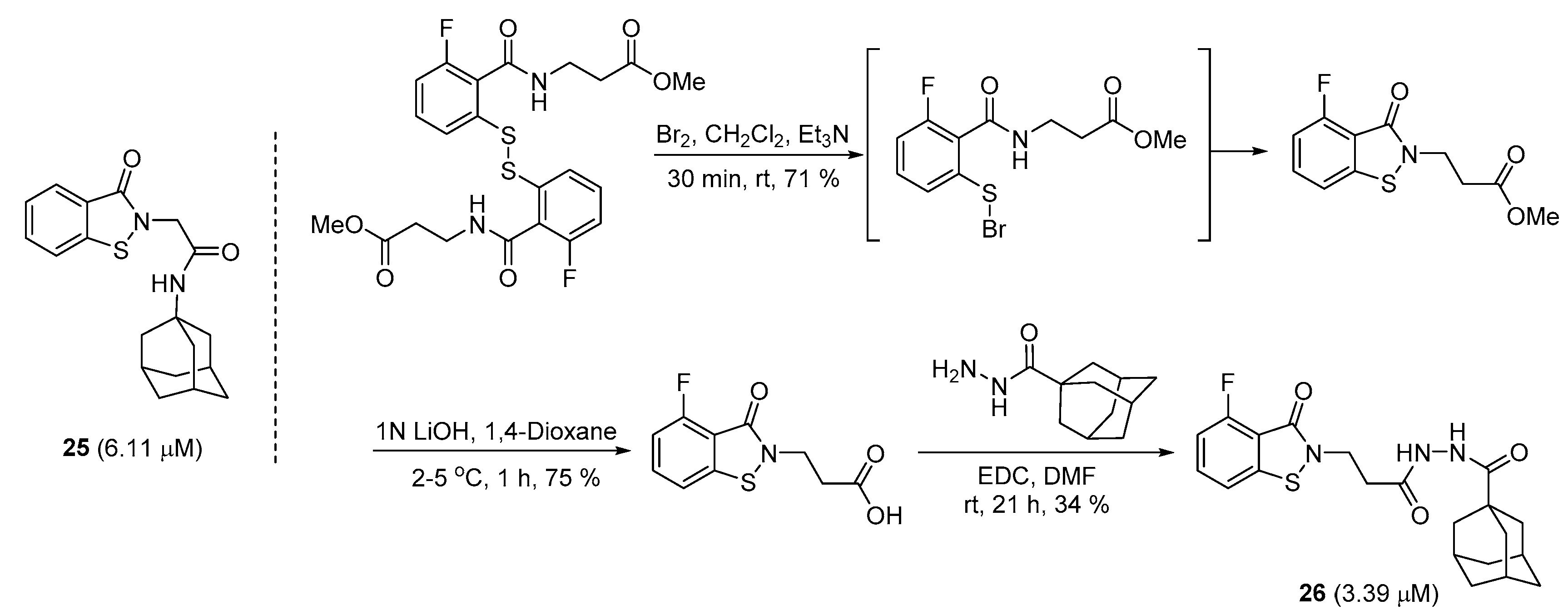
2.5. Preparation of Benzoisothiazolinones
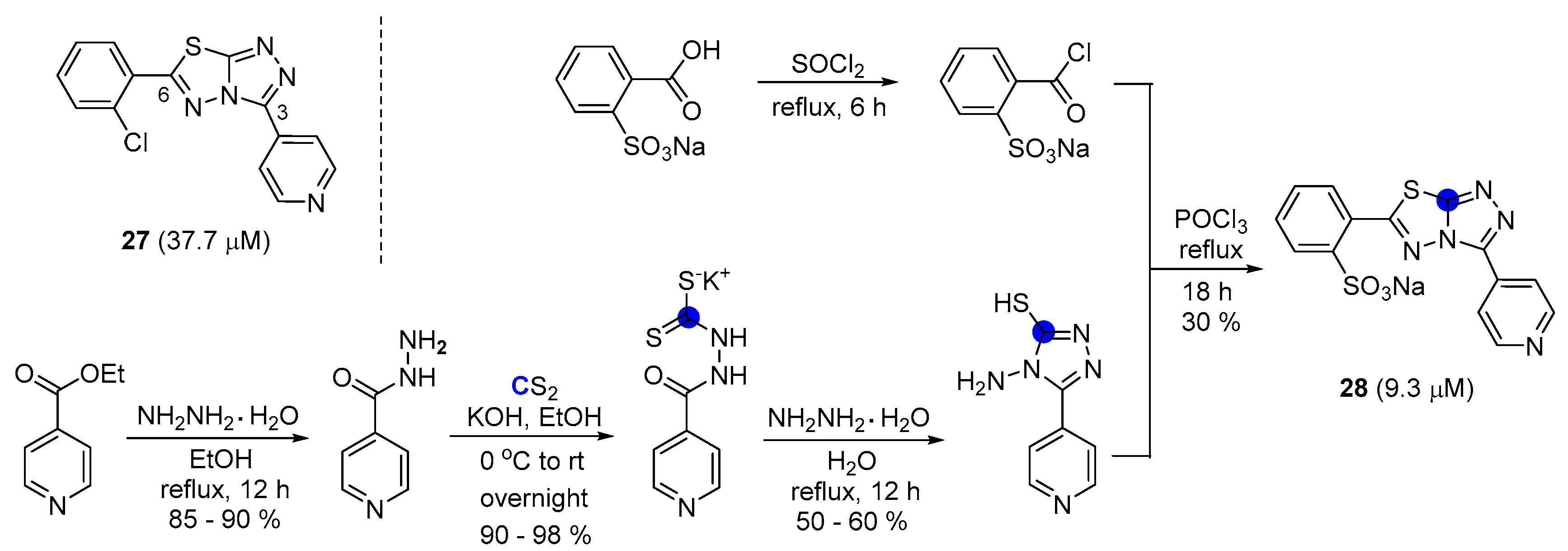
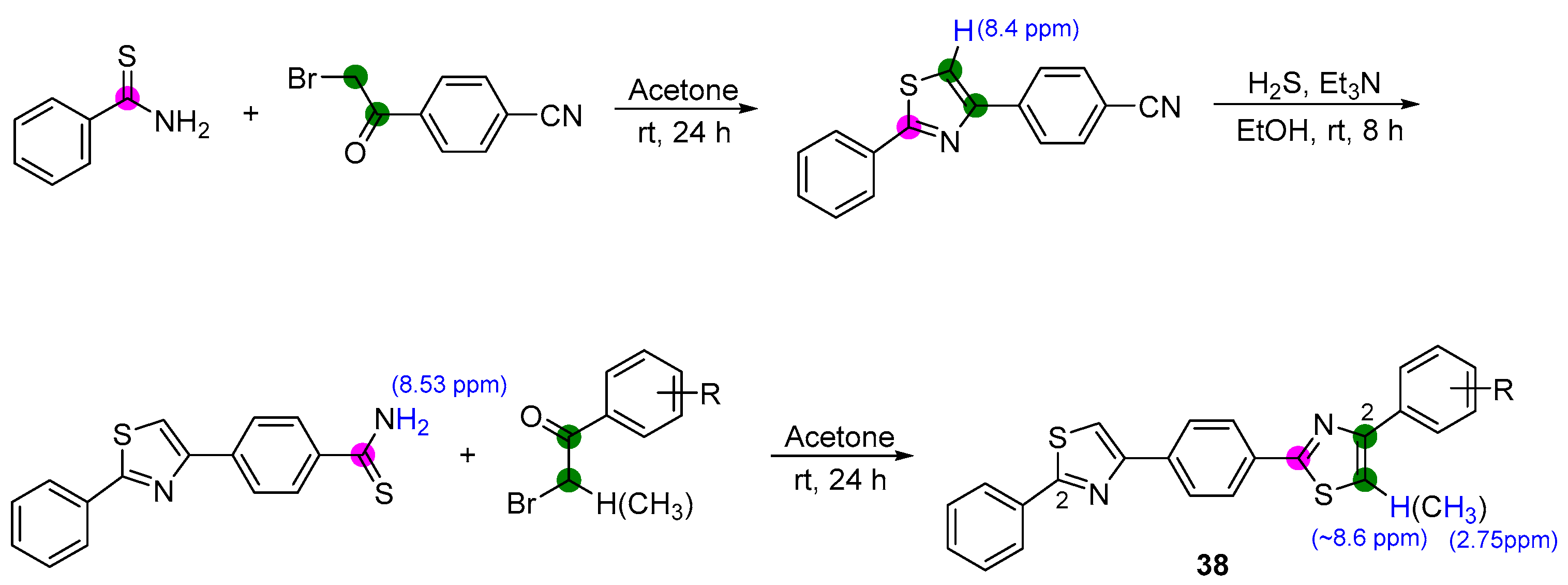
2.6. Preparation of Triazolothiadiazoles
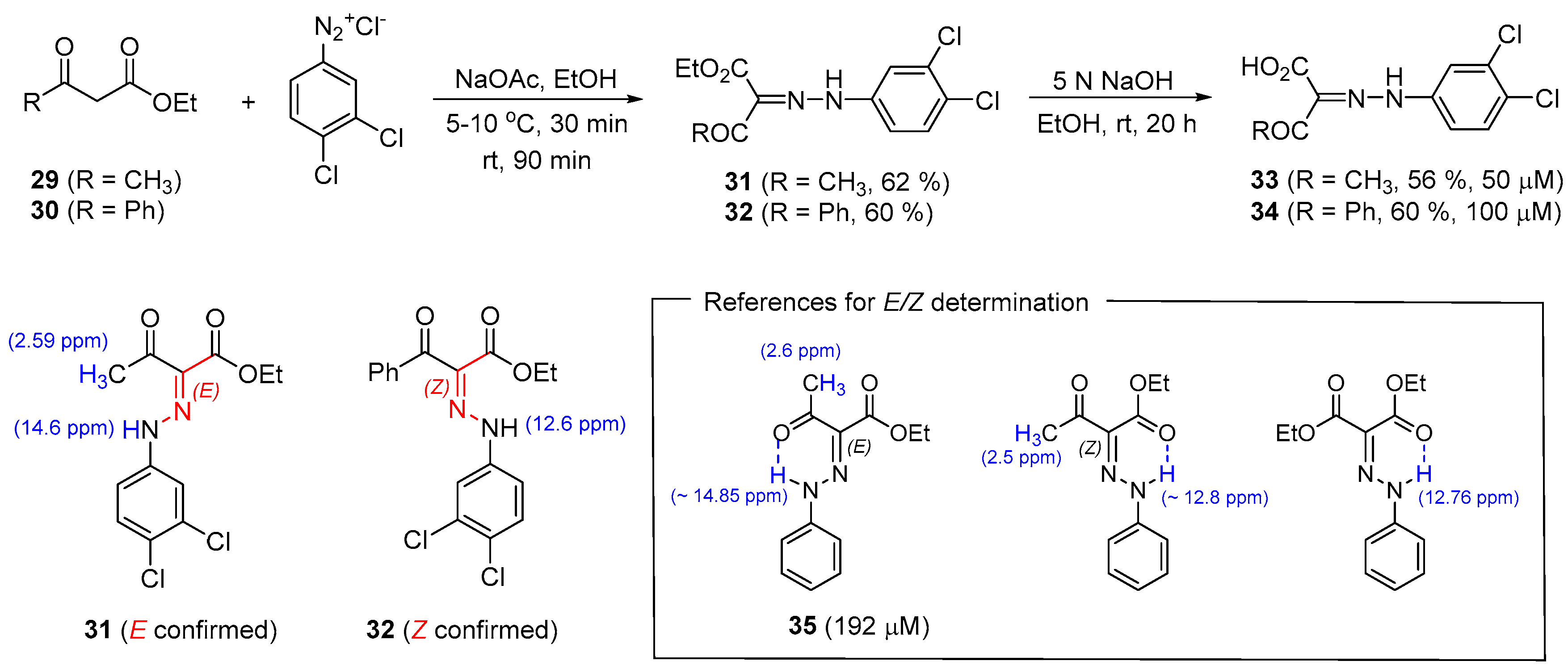
2.7. Preparation of 2-(2-Phenylhydrazinylidene)Alkanoates
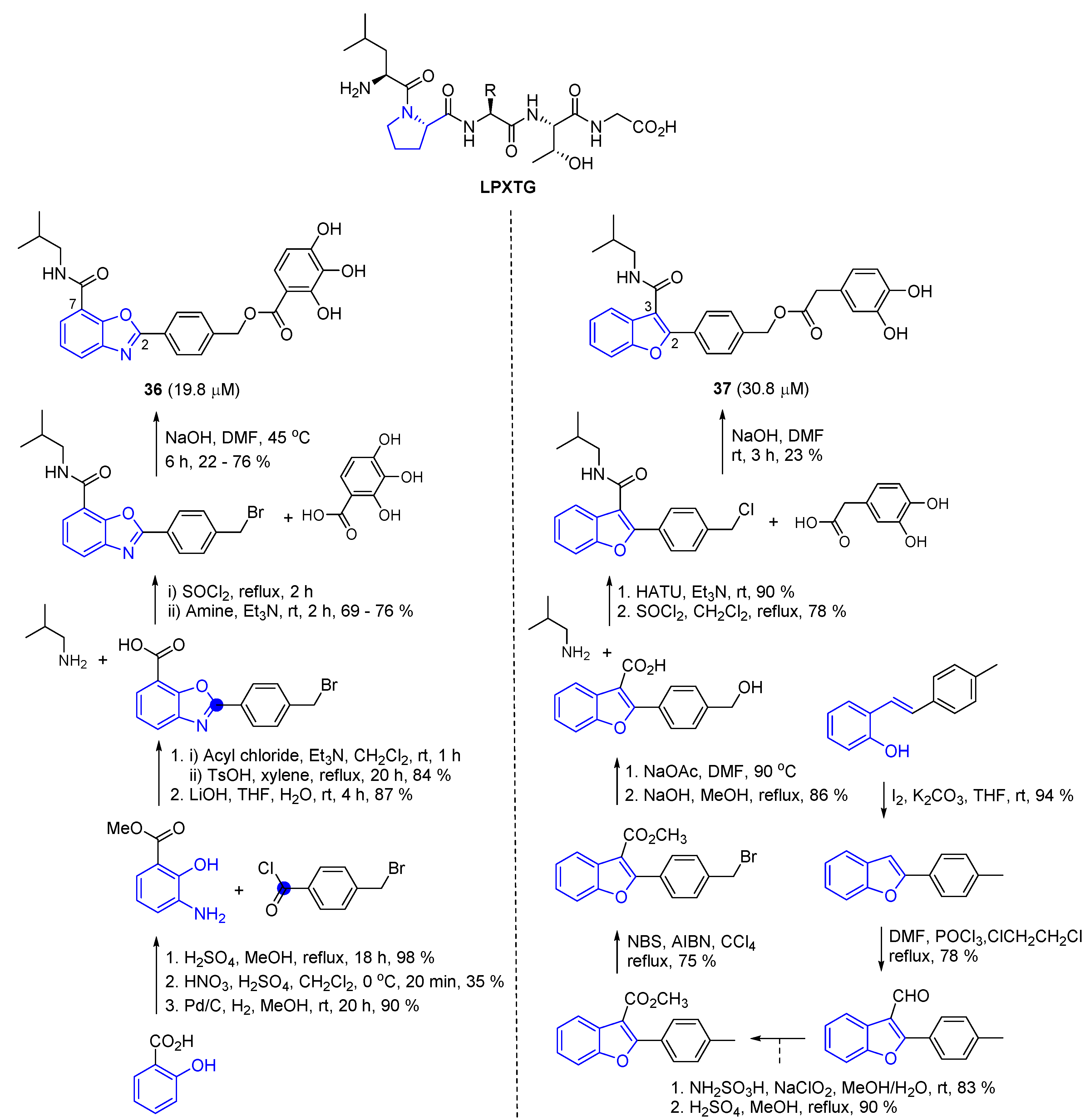
2.8. Preparation of Benzo[d]Oxazoles and Benzofuran
2.9. Preparation of Phenylthiazoles
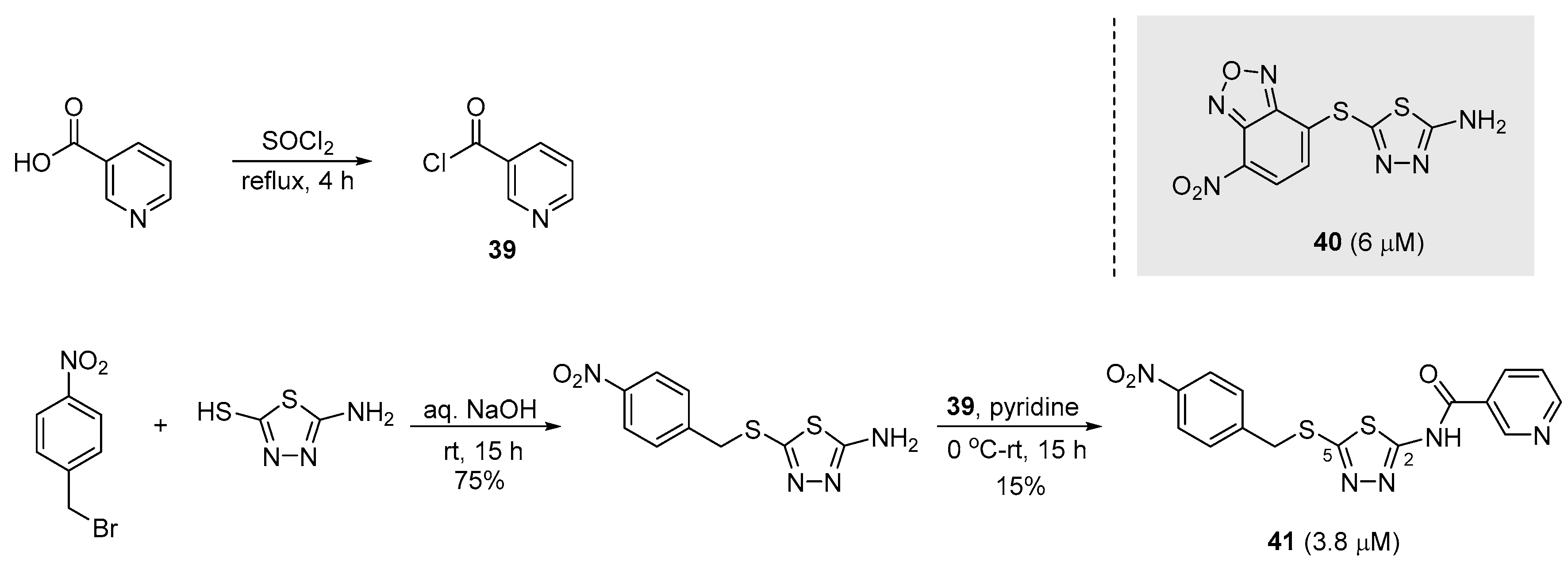
2.10. Preparation of Thiadiazole
2.11. Preparation of Thiadiazolidinedione
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chaudhary, A.S. A review of global initiatives to fight antibiotic resistance and recent antibiotics’ discovery. Acta Pharm. Sin. B 2016, 6, 552–556. [Google Scholar] [CrossRef]
- Blair, J.M.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef]
- Aslam, B.; Wang, W.; Arshad, M.I.; Khurshid, M.; Muzammil, S.; Rasool, M.H.; Nisar, N.A.; Alvi, R.F.; Aslam, M.A.; Qamar, M.U.; et al. Antibiotic resistance: A rundown of a global crisis. Infect. Drug. Resist. 2018, 11, 1645–1658. [Google Scholar] [CrossRef]
- Kohanski, M.A.; Dwyer, D.J.; Hayete, B.; Lawrence, C.A.; Collins, J.J. A common mechanism of cellular death induced by bactericidal antibiotics. Cell 2007, 130, 797–810. [Google Scholar] [CrossRef]
- DeLeo, F.R.; Chambers, H.F. Reemergence of antibiotic-resistant Staphylococcus aureus in the genomics era. J. Clin. Investig. 2009, 11, 2464–2474. [Google Scholar] [CrossRef]
- Chambers, H.J.; DeLeo, F.R. Waves of resistance: Staphylococcus aureus in the antibiotics. Nat. Rev. Microbiol. 2009, 7, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Neu, H.C. The crisis in antibiotic resistance. Science 1992, 257, 1064–1073. [Google Scholar] [CrossRef]
- Baquero, F. Gram-positive resistance: Challenge for the development of new antibiotics. J. Antimicrob. Chemother. 1997, 39, 1–6. [Google Scholar] [CrossRef]
- Andersson, H.; Lindholm, C.; Fossum, B. MRSA–global threat and personal disaster: patients’ experiences. Int. Nurs. Rev. 2001, 58, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Grema, H.A.; Geidam, Y.A.; Gadzama, G.B.; Ameh, J.A.; Suleiman, A. Methicillin resistant Staphyloccus aureus (MRSA): A review. Adv. Anim. Vet. Sci. 2015, 3, 79–98. [Google Scholar] [CrossRef]
- Kurosu, M.; Siricilla, S.; Mitachi, K. Advances in MRSA drug discovery: Where are we and where do we need to be? Expert Opin. Drug Discov. 2013, 8, 1095–1116. [Google Scholar] [CrossRef]
- Wright, G.D.; Sutherland, A.D. New strategies for combating multidrug-resistant bacteria. Trends Mol. Med. 2007, 13, 260–267. [Google Scholar] [CrossRef]
- Long, D.D.; Aggen, J.B.; Christensen, B.G.; Judice, J.K.; Hegde, S.S.; Kaniga, K.; Krause, K.M.; Linsell, M.S.; Moran, E.J.; Pace, J.L. A multivalent approach to drug discovery for novel antibiotics. J. Antibiot. Res. 2008, 61, 595–602. [Google Scholar] [CrossRef]
- Ton-That, H.; Liu, G.; Mazmanian, S.K.; Faull, K.F.; Schneewind, O. Purification and characterization of sortase, the transpeptidase that cleaves surface proteins of Staphylococcus aureus at the LPXTG motif. Proc. Natl. Acad. Sci. USA 1999, 96, 12424–12429. [Google Scholar] [CrossRef]
- Mazmanian, S.K.; Liu, G.; Ton-That, H.; Schneewind, O. Staphylococcus aureus sortase, an enzyme that anchors surface proteins to the cell wall. Science 1999, 285, 760–763. [Google Scholar] [CrossRef] [PubMed]
- Ilangovan, U.; Ton-That, H.; Iwahara, J.; Schneewind, O.; Clubb, R.T. Structure of sortase, the transpeptidase that anchors proteins to the cell wall of Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2001, 98, 6056–6061. [Google Scholar] [CrossRef] [PubMed]
- Frankel, B.A.; Kruger, R.G.; Robinson, D.E.; Kelleher, N.L.; McCafferty, D.G. Staphylococcus aureus sortase transpeptidase SrtA: Insight into the kinetic mechanism and evidence for a reverse protonation catalytic mechanism. Biochemistry 2005, 44, 11188–11200. [Google Scholar] [CrossRef]
- Suree, N.; Liew, C.K.; Villareal, V.A.; Thieu, W.; Fadeev, E.A.; Clemens, J.J.; Jung, M.E.; Clubb, R.T. The structure of the Staphylococcus aureus sortase-substrate complex reveals how the universally conserved LPXTG sorting signal is recognized. J. Biol. Chem. 2009, 284, 24465–24477. [Google Scholar] [CrossRef] [PubMed]
- Cascioferro, S.; Totsika, M.; Schillaci, D. Sortase A: An ideal target for anti-virulence drug development. Microb. Pathog. 2014, 77, 105–112. [Google Scholar] [CrossRef]
- Maresso, A.W.; Schneewind, O. Sortase as a target of anti-infective therapy. Pharmacol. Rev. 2008, 60, 128–141. [Google Scholar] [CrossRef]
- Connolly, K.M.; Smith, B.M.; Pilpa, R.; Ilangovan, U.; Jung, M.E.; Clubb, R.T. Sortase from S. aureus does not contain a thiolate-imidazolium ion pair in its active site. J. Biol. Chem. 2003, 278, 34061–34065. [Google Scholar] [CrossRef]
- Liew, C.K.; Smith, B.T.; Pilpa, R.; Ilangovan, U.; Connolly, K.M.; Jung, M.E.; Clubb, R.T. Localization and mutagenesis of the sorting signal binding site on sortase A from Staphylococcus aureus. FEBS Lett. 2004, 571, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.E.; Clemens, J.J.; Suttee, N.; Liew, C.K.; Clubb, R.T. Synthesis of (2R, 3S) 3-amino-4-mercapto-2-butanol, a threonine analogue for covalent inhibition of sortases. Bioorg. Med. Chem. Letts. 2005, 15, 5076–5079. [Google Scholar] [CrossRef] [PubMed]
- Naik, M.T.; Suree, N.; Ilangovan, N.U.; Liew, C.K.; Clemens, J.; Jung, M.E.; Clubb, R.T. A calcium modulated loop closure mechanism activates cell surface protein anchoring by the Staphylococcus aureus sortase A transpeptidase. J. Biol. Chem. 2006, 281, 1817–182642. [Google Scholar] [CrossRef] [PubMed]
- Ton-That, H.; Marraffini, L.A.; Schneewind, O. Protein sorting to the cell wall envelope of Gram-positive bacteria. Biochim. Biophys. Acta 2004, 1694, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.H.; Wereszczynski, J.; Amer, B.R.; Yi, S.W.; Jung, M.E.; Clubb, R.T.; McCammon, J.A. Discovery of Staphylococcus aureus sortase A inhibitors using virtual screening and the relaxed complex scheme. Chem. Biol. Drug Des. 2013, 82, 418–428. [Google Scholar] [CrossRef]
- Chan, A.H.; Yi, S.W.; Weiner, E.M.; Amer, B.R.; Sue, C.; Wereszczynski, J.; Dillen, C.; Senese, S.; Torres, J.Z.; McCammon, J.A.; et al. NMR structure-based optimization of Staphylococcus aureus sortase A pyridazinone inhibitors. Chem. Biol. Drug Des. 2017, 90, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.; Bice, T.W.; Ton-That, H.; Schneewind, O.; Narayana, S.V. Crystal structures of Staphylococcus aureus sortase A and its substrate complex. J. Biol. Chem. 2004, 279, 31383–31389. [Google Scholar] [CrossRef]
- Kruger, R.G.; Otvos, B.; Frankel, B.A.; Bentley, M.; Dostal, P.; McCafferty, D.G. Analysis of the substrate specificity of the Staphylococcus aureus sortase transpeptidase SrtA. Biochemistry 2004, 43, 1541–1551. [Google Scholar] [CrossRef]
- Ton-That, H.; Mazmanian, S.K.; Faull, K.F.; Schneewind, O. Anchoring of surface proteins to the cell wall of Staphylococcus aureus sortase catalyzed in vitro transpeptidation reaction using LPXTG peptide and NH2-GLY3SUBSTRATES. J. Biol. Chem. 2000, 275, 9876–9881. [Google Scholar] [CrossRef]
- Mazmanian, S.K.; Ton-That, H.; Schneewind, O. Sortase-catalysed anchoring of surface proteins to the cell wall of Staphylococcus aureus. Mol. Microbiol. 2001, 40, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Kruger, R.G.; Barkallah, S.; Frankel, B.A.; McCafferty, D.G. Inhibition of the Staphylococcus aureus sortase transpeptidase SrtA by phosphinic peptidomimetics. Bioorg. Med. Chem. 2004, 12, 3723–3729. [Google Scholar] [CrossRef] [PubMed]
- Perry, A.M.; Ton-That, H.; Mazmanian, S.K.; Schneewind, O. Anchoring of surface proteins to the cell wall of Staphylococcus aureus III. Lipid II is an in vivo peptidoglycan substrate for sortase-catalyzed surface protein anchoring. J. Biol. Chem. 2002, 277, 16241–16248. [Google Scholar] [CrossRef] [PubMed]
- Marraffini, L.A.; Ton-That, H.; Zong, Y.; Narayana, S.V.; Schneewind, O. Anchoring of surface proteins to the cell wall of Staphylococcus aureus a conserved arginine residue is required for efficient catalysis of sortase A. J. Biol. Chem. 2004, 279, 37763–37770. [Google Scholar] [CrossRef]
- Kappel, K.; Wereszczynski, J.; Clubb, R.T.; McCammon, J.A. The binding mechanism, multiple binding modes, and allosteric regulation of Staphylococcus aureus Sortase A probed by molecular dynamics simulations. Protein Sci. 2012, 21, 1858–1871. [Google Scholar] [CrossRef]
- Cascioferro, S.; Raffa, D.; Maggio, B.; Raimondi, M.V.; Schillaci, D.; Daidone, G. Sortase A inhibitors: Recent advances and future perspectives. J. Med. Chem. 2015, 58, 9108–9123. [Google Scholar] [CrossRef]
- Guo, Y.; Cai, S.; Gu, G.; Guo, Z.; Long, Z. Recent progress in the development of sortase A inhibitors as novel anti-bacterial virulence agents. RSC Adv. 2015, 5, 49880–49889. [Google Scholar] [CrossRef]
- Oh, K.-B.; Kim, S.-H.; Lee, J.; Cho, W.-J.; Lee, T.; Kim, S. Discovery of diarylacrylonitriles as a novel series of small molecule sortase A inhibitors. J. Med. Chem. 2004, 47, 2418–2421. [Google Scholar] [CrossRef]
- Suree, N.; Yi, S.W.; Thieu, W.; Marohn, M.; Damoiseaux, R.; Chan, A.; Jung, M.E.; Clubb, R.T. Discovery and structure-activity relationship analysis of Staphylococcus aureus sortase A inhibitors. Bioorg. Med. Chem. 2009, 17, 7174–7185. [Google Scholar] [CrossRef]
- Chenna, B.C.; King, J.R.; Shinkre, B.A.; Glover, A.L.; Lucius, A.L.; Velu, S.E. Synthesis and structure activity relationship studies of novel Staphylococcus aureus sortase A inhibitors. Eur. J. Med. Chem. 2010, 45, 3752–3761. [Google Scholar] [CrossRef]
- Lee, Y.-J.; Han, Y.-R.; Park, W.; Nam, S.-H.; Oh, K.-B.; Lee, H.-S. Synthetic analogs of indole-containing natural products as inhibitors of sortase A and isocitrate lyase. Bioorg. Med. Chem. Lett. 2010, 20, 6882–6885. [Google Scholar] [CrossRef] [PubMed]
- Zhulenkovs, D.; Rudevica, Z.; Jaudzems, K.; Turks, M.; Leonchiks, A. Discovery and structure-activity relationship studies of irreversible benzisothiazolinone-based inhibitors against Staphylococcus aureus sortase A transpeptidase. Bioorg. Med. Chem. 2014, 22, 5988–6003. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, H.; Zhu, K.; Gong, S.; Dramsi, S.; Wang, Y.-T.; Li, J.; Chen, F.; Zhang, R.; Zhou, L.; et al. Antiinfective therapy with a small molecule inhibitor of Staphylococcus aureus sortase. Proc. Natl. Acad. Sci. USA 2014, 111, 13517–13522. [Google Scholar] [CrossRef]
- Maggio, B.; Raffa, D.; Raimondi, M.V.; Cascioferro, S.; Plescia, F.; Schillaci, D.; Cusimano, M.G.; Leonchiks, A.; Zhulenkovs, D.; Basile, L.; et al. Discovery of a new class of sortase A transpeptidase inhibitors to tackle Gram-positive pathogens: 2-(2-phenylhydrazinylidene)alkanoic acids and related derivatives. Molecules 2016, 21, 241. [Google Scholar] [CrossRef]
- Zhang, Y.; Bao, J.; Deng, X.-X.; He, W.; Fan, J.-J.; Jiang, F.-Q.; Fu, L. Synthesis, biological evaluation and molecular docking of 2-phenyl-benzo[d]oxazole-7-carboxamide derivatives as potential Staphylococcus aureus Sortase A inhibitors. Bioorganic Med. Chem. Lett. 2016, 26, 4081–4085. [Google Scholar] [CrossRef]
- He, W.; Zhang, Y.; Bao, J.; Deng, X.; Batara, J.; Casey, S.; Guo, Q.; Jiang, F.; Fu, L. Synthesis, biological evaluation and molecular docking analysis of 2-phenyl-benzofuran-3-carboxamide derivatives as potential inhibitors of Staphylococcus aureus Sortase A. Bioorg. Med. Chem. 2017, 25, 1341–1351. [Google Scholar] [CrossRef]
- Oniga, S.D.; Araniciu, C.; Palage, M.D.; Popa, M.; Chifiriuc, M.C.; Marc, G.; Pirnau, A.; Stoica, C.I.; Lagoudis, I.; Dragoumis, T.; et al. New 2-phenylthiazoles as potential sortase A inhibitors: Synthesis, biological evaluation and molecular docking. Molecules 2017, 22, 1827. [Google Scholar] [CrossRef] [PubMed]
- Wehrli, P.M.; Uzelac, I.; Olsson, T.; Jacso, T.; Tietze, D.; Gottfries, J. Discovery and development of substituted thiadiazoles as inhibitors of Staphylococcus aureus sortase A. Bioorg. Med. Chem. 2019, 27, 115043–115053. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Zhang, T.; Guan, X.N.; Dong, Z.; Lan, L.; Yang, S.; Yang, C.G. Tideglusib and its analogues as inhibitors of Staphylococcus aureus SrtA. J. Med. Chem. 2020, 63, 8442–8457. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, H.C.; Craig, P.N.; Groves, W.G. Spasmolytics. III. 3-Tropanyl 2, 3-diarylacrylates. J. Med. Chem. 1970, 13, 1079–1081. [Google Scholar] [CrossRef]
- Ohsumi, K.; Nakagawa, R.; Fukuda, Y.; Hatanaka, T.; Morinaga, Y.; Nihei, Y.; Ohishi, K.; Suga, Y.; Akiyama, Y.; Tsuji, T. Novel combretastatin analogues effective against murine solid tumors: Design and structure-activity relationships. J. Med. Chem. 1998, 41, 3022–3032. [Google Scholar] [CrossRef]
- Meyers, M.J.; Sun, J.; Carlson, K.E.; Marriner, G.A.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A. Estrogen receptor-β potency-selective ligands: Structure-activity relationship studies of diarylpropionitriles and their acetylene and polar analogues. J. Med. Chem. 2001, 44, 4230–4251. [Google Scholar] [CrossRef]
- Rummens, F.H.A.; De Haan, J.W. Spectroscopic studies on olefins—III: NMR of cis-and trans-disubstituted olefins. Org. Magn. Reson. 1970, 2, 351–355. [Google Scholar] [CrossRef]
- Zhang, J.; Morton, H.E.; Ji, J. Confirmation and prevention of halogen exchange: Practical and highly efficient one-pot synthesis of dibromo-and dichloropyridazinones. Tetrahedron Lett. 2006, 47, 8733–8735. [Google Scholar] [CrossRef]
- Lyga, J.W. The reaction of 2-substituted-4,5-dichloro-3(2H)-pyridazinones with alkoxides and alkylthiolates. Heterocycl. Chem. 1988, 25, 1757–1760. [Google Scholar] [CrossRef]
- Skotnicki, J.S.; Kearney, R.M.; Smith, A.L. Synthesis of secorapamycin esters and amides. Tetrahedron Lett. 1994, 35, 197–200. [Google Scholar] [CrossRef]
- Wadsworth, D.H.; Geer, S.M.; Detty, M.R. Preparation of arylpropiolate esters from trichlorocyclopropenium cation and elaboration of the esters to unsymmetrical 1, 4-pentadiyn-3-ones and unsymmetrical tellurapyranones. J. Org. Chem. 1987, 52, 3662–3668. [Google Scholar] [CrossRef]
- Cox, E.D.; Cook, J. The Pictet-Spengler condensation: A new direction for an old reaction. Chem. Rev. 1995, 95, 1797–1842. [Google Scholar] [CrossRef]
- Trujillo, J.I.; Meyers, M.J.; Anderson, D.R.; Hegde, S.; Mahoney, M.W.; Vernier, W.F.; Buchler, I.P.; Wu, K.W.; Yang, S.; Hartman, S.J.; et al. Novel tetrahydro-β-carboline-1-carboxylic acids as inhibitors of mitogen activated protein kinase-activated protein kinase 2 (MK-2). Bioorg. Med. Chem. Lett. 2007, 17, 4657–4663. [Google Scholar] [CrossRef]
- Kawashima, Y.; Horiguchi, A.; Taguchi, M.; Tuyuki, Y.; Karasawa, Y.; Araki, H.; Hatayama, K. Synthesis and pharmacological evaluation of 1, 2, 3, 4-tetrahydro-β-carboline derivatives. Chem. Pharm. Bull. 1995, 43, 783–787. [Google Scholar] [CrossRef]
- Shen, Y.-C.; Chen, C.-Y.; Hsieh, P.W.; Duh, C.-Y.; Lin, Y.-M.; Ko, C.-L. The preparation and evaluation of 1-substituted 1, 2, 3, 4-tetrahydro-and 3, 4-dihydro-β-carboline derivatives as potential antitumor agents. Chem. Pharm. Bull. 2005, 53, 32–36. [Google Scholar] [CrossRef]
- Sławik, T. Aminomethyl derivatives of (benzisothiazolin-3-one-2-yl) acetic acid amides and 2-(1, 2-benzisothiazoline-3-one-2-yl) propionic acid amides. Pharmazie 1991, 46, 777–780. [Google Scholar]
- Lai, H.; Dou, D.; Aravapalli, S.; Teramoto, T.; Lushington, G.H.; Mwania, T.M.; Alliston, K.R.; Eichhorn, D.M.; Padmanabhan, R.; Groutas, W.C. Design, synthesis and characterization of novel 1, 2-benzisothiazol-3 (2H)-one and 1, 3, 4-oxadiazole hybrid derivatives: Potent inhibitors of Dengue and West Nile virus NS2B/NS3 proteases. Bioorg. Med. Chem. 2013, 21, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Dou, D.; Alex, D.; Du, B.; Tiew, K.C.; Aravapalli, S.; Mandadapu, S.R.; Calderone, R.; Groutas, W.C. Antifungal activity of a series of 1, 2-benzisothiazol-3 (2H)-one derivatives. Bioorg. Med. Chem. 2011, 19, 5782–5787. [Google Scholar] [CrossRef] [PubMed]
- Mathew, V.; Keshavayya, J.; Vaidya, V.P.; Giles, D. Studies on synthesis and pharmacological activities of 3, 6-disubstituted-1, 2, 4-triazolo [3, 4-b]-1, 3, 4-thiadiazoles and their dihydro analogues. Eur. J. Med. Chem. 2007, 42, 823–840. [Google Scholar] [CrossRef] [PubMed]
- Elguero, J.; Jaequier, R.; Tarrago, J.G. Structure des produits de copulation du chlorurre de phényldiazonium avec les β-dicétones e les β-cétoesters. Bull. Soc. Chim. Fr. 1966, 2981–2989. [Google Scholar]
- Bose, A.K.; Kugajevsky, I. NMR spectral studies—IV: Some 15NH coupling constants. Tetrahedron 1967, 23, 1489–1497. [Google Scholar] [CrossRef]
- Bandyopadhyay, P.; Guha, L.; Seenivasagan, T.; Sathe, M.; Sharma, P.; Parashar, B.D.; Kaushik, M.P. Synthesis and bio-evaluation of aryl hydrazono esters for oviposition responses in Aedes albopictus. Bioorg. Med. Chem. Lett. 2011, 21, 794–797. [Google Scholar] [CrossRef]
- Ferguson, G.N.; Valant, C.; Horne, J.; Figler, H.; Flynn, B.L.; Linden, J.; Chalmers, D.K.; Sexton, P.M.; Christopoulos, A.; Scammells, P.J. 2-Aminothienopyridazines as novel adenosine A1 receptor allosteric modulators and antagonists. J. Med. Chem. 2008, 51, 6165–6172. [Google Scholar] [CrossRef]
- Pareek, A.K.; Joseph, P.E.; Seth, D.S. A convenient route for the synthesis and spectral characterization of substituted pyrazolones. Orient. J. Chem. 2009, 25, 735–738. [Google Scholar]
- Ballatore, C.; Brunden, K.; Crowe, A.; Huryn, D.; Lee, V.; Trojanowski, J.; Smith, A.; Huang, R.; Huang, W.; Johnson, R.; et al. Aminothienopyridazine Inhibitors of Tau Assembly. Patent WO2011037985 A8, 31 March 2011. [Google Scholar]
- Gupta, S.C.; Mandal, D.K.; Rani, A.; Sahay, A.; Prasad, S.M. Ethyl 3-oxo-2-(2-phenyl-hydrazinylidene) butanoate: A re-determination. Acta Crystallogr. Sect. E Struct. Rep. 2011, 67, o470. [Google Scholar] [CrossRef] [PubMed]
- Khudina, O.G.; Burgart, Y.V.; Shchegol’kov, E.V.; Saloutin, V.I.; Kazheva, O.N.; Chekhlov, A.N.; D’yachenko, O.A. Steric structure of alkyl 2-aryl(hetaryl)hydrazono-3-fluoroalkyl-3-oxopropionates. Russ. J. Org. Chem. 2009, 45, 801–809. [Google Scholar] [CrossRef]
- Mitchell, A.; Nonhebel, D.C. Spectroscopic studies of tautomeric systems—III: 2-Arylhydrazones of 1,2,3-triketones. Tetrahedron 1979, 35, 2013–2019. [Google Scholar] [CrossRef]
- Saez, R.; Otero, M.D.; Batanero, B.; Barba, F. Microwave reaction of diazonium salts with nitriles. J. Chem. Res. 2008, 2008, 492–494. [Google Scholar] [CrossRef]
- Jirman, J.; Lyˇcka, A. 13C-and 15N-NMR spectra of phenylazoacetoacetamides and similar compounds. Dyes Pigment. 1987, 8, 55–62. [Google Scholar] [CrossRef]
- Yan, Y.; Qin, B.; Ren, C.L.; Chen, X.Y.; Yip, Y.K.; Ye, R.J.; Zhang, D.W.; Su, H.B.; Zeng, H.Q. Synthesis, structural investigations, hydrogen–deuterium exchange studies, and molecular modeling of conformationally stablilized aromatic oligoamides. J. Am. Chem. Soc. 2010, 132, 5869–5879. [Google Scholar] [CrossRef]
- Janetzko, J.; Batey, R.A. Organoboron-based allylation approach to the total synthesis of the medium-ring dilactone (+)-antimycin A1b. J. Org. Chem. 2014, 79, 7415–7424. [Google Scholar] [CrossRef]
- Reid, E.E. Esterification a review of the recent past and a look towards the future. Ind. Eng. Chem. 1937, 29, 1344–1350. [Google Scholar] [CrossRef]
- Okamiya, J. The preparation and the rates of the reaction of heterocyclic (thiophen and thiazole) bromoketones with thioamides. Nippon Kagaku Zasshi 1966, 87, 594–600. [Google Scholar] [CrossRef]
- Talath, S.; Gadad, A.K. Synthesis, antibacterial and antitubercular activities of some 7-[4-(5-amino-[1, 3, 4] thiadiazole-2-sulfonyl)-piperazin-1-yl] fluoroquinolonic derivatives. Eur. J. Med. Chem. 2006, 41, 918–924. [Google Scholar] [CrossRef]
- Guan, P.; Hou, X.; Wang, F.; Yi, F.; Xu, W.; Fang, H. Design, synthesis and preliminary bioactivity studies of 1,3,4-thiadiazole hydroxamic acid derivatives as novel histone deacetylase inhibitors. Bioorgan. Med. Chem. 2012, 20, 3865–3872. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.T.; Jung, C.H.; Kim, K.W.; Kim, H.S. Synthesis of 2-pyridinylbenzoxazole: Mechanism for the intramolecular photosubstitution of the haloarene with the carbonyl oxygen of the amide bond in basic medium. J. Org. Chem. 1999, 64, 8546–8556. [Google Scholar] [CrossRef]
- Study of tideglusib in adolescent and adult patients with myotonic dystrophy (NCT02858908). ClinicalTrials. 2016. Available online: https://clinicaltrials.gov/ct2/show/NCT02858908 (accessed on 3 September 2020).
- Turner, E.M.; Blazer, L.L.; Neubig, R.R.; Husbands, S.M. Small molecule inhibitors of regulator of G protein signalling (RGS) proteins. ACS Med. Chem. Lett. 2012, 3, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Maresso, A.W.; Wu, R.; Kern, J.W.; Zhang, R.; Janik, D.; Missiakas, D.M.; Duban, M.-E.; Joachimiak, A.; Schneewind, O. Activation of inhibitors by sortase triggers irreversible modification of the active site. J. Biol. Chem. 2007, 282, 23129–23139. [Google Scholar] [CrossRef]
- Naidu, B.N.; Sorenson, M.E.; Connolly, T.P.; Ueda, Y. Michael addition of amines and thiols to dehydroalanine amides: A remarkable rate acceleration in water. J. Org. Chem. 2003, 68, 10098–10102. [Google Scholar] [CrossRef]
- Davioud-Charvet, E.; McLeish, M.J.; Veine, D.M.; Giegel, D.; Arscott, L.D.; Andricopulo, A.D.; Becker, K.; Muller, S.; Schirmer, R.H.; Williams, C.H., Jr.; et al. Mechanism-based inactivation of thioredoxin reductase from Plasmodium falciparum by Mannich bases. Implication for cytotoxicity. Biochemistry 2003, 42, 13319–13330. [Google Scholar] [CrossRef]
- Mollica, J.A.; Smith, J.B.; Nunes, I.M.; Govan, H.K. Kinetics of the decomposition of a Mannich base. J. Pharm. Sci. 1970, 59, 1770–1774. [Google Scholar] [CrossRef]
- Andrisano, R.; Angeloni, A.S.; De Maria, P.; Tramontini, M. Reactivity of mannich bases. Part X. The mechanism of the reaction between β-amino-ketones and thiophenols. J. Chem. Soc. C 1967, 43, 2307–2311. [Google Scholar] [CrossRef]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ha, M.W.; Yi, S.W.; Paek, S.-M. Design and Synthesis of Small Molecules as Potent Staphylococcus aureus Sortase A Inhibitors. Antibiotics 2020, 9, 706. https://doi.org/10.3390/antibiotics9100706
Ha MW, Yi SW, Paek S-M. Design and Synthesis of Small Molecules as Potent Staphylococcus aureus Sortase A Inhibitors. Antibiotics. 2020; 9(10):706. https://doi.org/10.3390/antibiotics9100706
Chicago/Turabian StyleHa, Min Woo, Sung Wook Yi, and Seung-Mann Paek. 2020. "Design and Synthesis of Small Molecules as Potent Staphylococcus aureus Sortase A Inhibitors" Antibiotics 9, no. 10: 706. https://doi.org/10.3390/antibiotics9100706
APA StyleHa, M. W., Yi, S. W., & Paek, S.-M. (2020). Design and Synthesis of Small Molecules as Potent Staphylococcus aureus Sortase A Inhibitors. Antibiotics, 9(10), 706. https://doi.org/10.3390/antibiotics9100706