Mutually Isomeric 2- and 4-(3-Nitro-1,2,4-triazol-1-yl)pyrimidines Inspired by an Antimycobacterial Screening Hit: Synthesis and Biological Activity against the ESKAPE Panel of Pathogens

 ,
,  ,
,  ,
,
Abstract
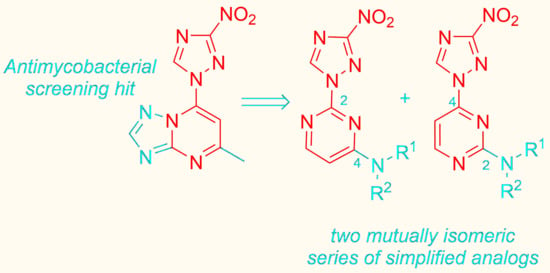
1. Introduction
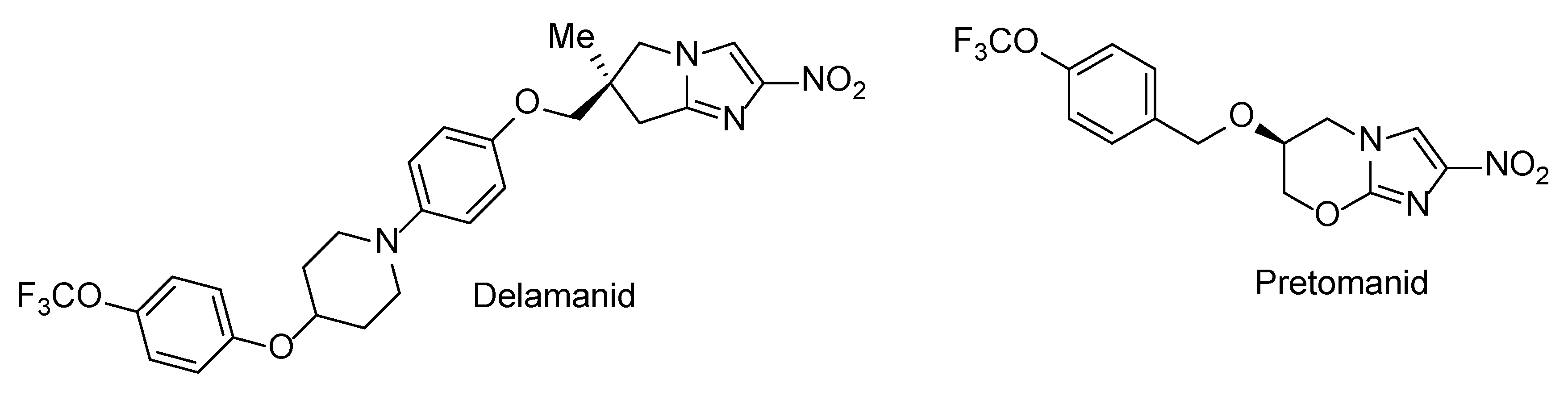
2. Results and Discussion
2.1. Synthesis
2.1.1. 2-(3-Nitro-1,2,4-triazol-1-yl)pyrimidines 1
2.1.2. 4-(3-Nitro-1,2,4-triazol-1-yl)pyrimidines 2
2.2. In Vitro Biological Evaluation
2.3. Electrochemical Behavior
3. Conclusions
4. Materials and Methods
4.1. General Experimental
4.2. Synthetic Organic Chemistry
4.2.1. Preparation of 2,4-bis(3-nitro-1H-1,2,4-triazol-1-yl)pyrimidine (4)
4.2.2. General Procedure 2 (GP2) for the Preparation of Compounds 1a-d, 1f-l and 1n-s
2-((2-(3-Nitro-1H-1,2,4-triazol-1-yl)pyrimidin-4-yl)amino)ethan-1-ol (1a)
N,N-Dimethyl-2-(3-nitro-1H-1,2,4-triazol-1-yl)pyrimidin-4-amine (1b)
N-Methyl-2-(3-nitro-1H-1,2,4-triazol-1-yl)pyrimidin-4-amine (1c)
N-Benzyl-N-methyl-2-(3-nitro-1H-1,2,4-triazol-1-yl)pyrimidin-4-amine (1d)
3-((2-(3-Nitro-1H-1,2,4-triazol-1-yl)pyrimidin-4-yl)amino)propan-1-ol (1f)
N-Ethyl-2-(3-nitro-1H-1,2,4-triazol-1-yl)pyrimidin-4-amine (1g)
4-(2-(3-Nitro-1H-1,2,4-triazol-1-yl)pyrimidin-4-yl)morpholine (1h)
4-(4-Methylpiperazin-1-yl)-2-(3-nitro-1H-1,2,4-triazol-1-yl)pyrimidine (1i)
N-Benzyl-2-(3-nitro-1H-1,2,4-triazol-1-yl)pyrimidin-4-amine (1j)
N-Isobutyl-2-(3-nitro-1H-1,2,4-triazol-1-yl)pyrimidin-4-amine (1k)
2,2′-((2-(3-Nitro-1H-1,2,4-triazol-1-yl)pyrimidin-4-yl)azanediyl)bis(ethan-1-ol) (1l)
N-Cyclopropyl-2-(3-nitro-1H-1,2,4-triazol-1-yl)pyrimidin-4-amine (1n)
N-Isopropyl-2-(3-nitro-1H-1,2,4-triazol-1-yl)pyrimidin-4-amine (1o)
N-(3-(1H-Imidazol-1-yl)propyl)-2-(3-nitro-1H-1,2,4-triazol-1-yl)pyrimidin-4-amine (1p)
2-(3-Nitro-1H-1,2,4-triazol-1-yl)-4-(pyrrolidin-1-yl)pyrimidine (1q)
2-(3-Nitro-1H-1,2,4-triazol-1-yl)-N-propylpyrimidin-4-amine (1r)
N-(4-Methoxybenzyl)-2-(3-nitro-1H-1,2,4-triazol-1-yl)pyrimidin-4-amine (1s)
N-(4-Methoxyphenyl)-2-(3-nitro-1H-1,2,4-triazol-1-yl)pyrimidin-4-amine (1e)
2-(3-Nitro-1H-1,2,4-triazol-1-yl)-N-(p-tolyl)pyrimidin-4-amine (1m)
4.2.3. Preparation of 2-chloro-4-(3-nitro-1H-1,2,4-triazol-1-yl)pyrimidine (5)
4.2.4. General Procedure 1 (GP1) for Preparation of Compounds 1a-d, 1f-l and 1n-s
2-((4-(3-Nitro-1H-1,2,4-triazol-1-yl)pyrimidin-2-yl)amino)ethan-1-ol (2a)
N,N-Dimethyl-4-(3-nitro-1H-1,2,4-triazol-1-yl)pyrimidin-2-amine (2b)
N-Methyl-4-(3-nitro-1H-1,2,4-triazol-1-yl)pyrimidin-2-amine (2c)
N-Benzyl-N-methyl-4-(3-nitro-1H-1,2,4-triazol-1-yl)pyrimidin-2-amine (2d)
3-((4-(3-Nitro-1H-1,2,4-triazol-1-yl)pyrimidin-2-yl)amino)propan-1-ol (2f)
N-Ethyl-4-(3-nitro-1H-1,2,4-triazol-1-yl)pyrimidin-2-amine (2g)
4-(4-(3-Nitro-1H-1,2,4-triazol-1-yl)pyrimidin-2-yl)morpholine (2h)
2-(4-Methylpiperazin-1-yl)-4-(3-nitro-1H-1,2,4-triazol-1-yl)pyrimidine (2i)
N-Benzyl-4-(3-nitro-1H-1,2,4-triazol-1-yl)pyrimidin-2-amine (2j)
N-Isobutyl-4-(3-nitro-1H-1,2,4-triazol-1-yl)pyrimidin-2-amine (2k)
2,2′-((4-(3-nitro-1H-1,2,4-triazol-1-yl)pyrimidin-2-yl)azanediyl)bis(ethan-1-ol) (2l)
N-cyclopropyl-4-(3-nitro-1H-1,2,4-triazol-1-yl)pyrimidin-2-amine (2n)
N-Isopropyl-4-(3-nitro-1H-1,2,4-triazol-1-yl)pyrimidin-2-amine (2o)
N-(3-(1H-Imidazol-1-yl)propyl)-4-(3-nitro-1H-1,2,4-triazol-1-yl)pyrimidin-2-amine (2p)
4-(3-Nitro-1H-1,2,4-triazol-1-yl)-2-(pyrrolidin-1-yl)pyrimidine (2q)
4-(3-Nitro-1H-1,2,4-triazol-1-yl)-N-propylpyrimidin-2-amine (2r)
N-(4-Methoxybenzyl)-4-(3-nitro-1H-1,2,4-triazol-1-yl)pyrimidin-2-amine (2s)
N-(4-Methoxyphenyl)-4-(3-nitro-1H-1,2,4-triazol-1-yl)pyrimidin-2-amine (2e)
4-(3-Nitro-1H-1,2,4-triazol-1-yl)-N-(p-tolyl)pyrimidin-2-amine (2m)
4.3. Evaluation of Antimycobacterial Activity
4.4. Screening against ESKAPE Pathogens
4.5. Cyclic Voltammetry
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Watanabe, Y.; Arimori, S. Preparation of tetrazolinone derivatives as agrochemical fungicides and pesticides. Chem. Abstr. 2015, 162, 418609. [Google Scholar]
- Jones, R.G.; Terando, N.H. Antibacterial compositions containing 3-nitropyrazoles. Chem. Abstr. 1978, 88, 152614. [Google Scholar]
- Srinivas, D.; Ghule, V.D.; Tewari, S.P.; Muralidharan, K. Synthesis of Amino, Azido, Nitro, and Nitrogen-Rich Azole-Substituted Derivatives of 1H-Benzotriazole for High-Energy Materials Applications. Chem. Eur. J. 2012, 18, 15031. [Google Scholar] [CrossRef] [PubMed]
- Kommu, N.; Ghule, V.D.; Kumar, A.S.; Sahoo, A.K. Triazole-Substituted Nitroarene Derivatives: Synthesis, Characterization, and Energetic Studies. Chem. Asian J. 2014, 9, 166. [Google Scholar] [CrossRef] [PubMed]
- Duddu, R.; Dave, P.R.; Damavarapu, R.; Surapaneni, R.; Parrish, D. Nucleophilic Substitution Reactions of 1-Methyl-2,4,5-trinitroimidazole (MTNI). Synth. Commun. 2009, 39, 4282. [Google Scholar] [CrossRef]
- Chuprun, S.S.; Kantin, G.; Krasavin, M. Synthesis and Medicinal Applications of N-Aryl-C-nitroazoles. Mini Rev. Med. Chem. 2018, 18, 1733. [Google Scholar] [CrossRef] [PubMed]
- Olive, P.L. Correlation between the half-wave reduction potentials of nitroheterocycles and their mutagenicity in Chinese hamster V79 spheroids. Mutat. Res. Fundam. Mol. Mech. Mutagen. 1981, 82, 137. [Google Scholar] [CrossRef]
- Krasavin, M.; Parchinsky, V.; Kantin, G.; Manicheva, O.; Dogonadze, M.; Vinogradova, T.; Karge, B.; Brönstrup, M. New nitrofurans amenable by isocyanide multicomponent chemistry are active against multidrug-resistant and poly-resistant Mycobacterium tuberculosis. Bioorg. Med. Chem. 2017, 25, 1867. [Google Scholar] [CrossRef]
- Krasavin, M.; Lukin, A.; Vedekhina, T.; Manicheva, O.; Dogonadze, M.; Vinogradova, T.; Zabolotnykh, N.; Rogacheva, E.; Kraeva, L.; Sharoyko, V.; et al. Attachment of a 5-nitrofuroyl moiety to spirocyclic piperidines produces non-toxic nitrofurans that are efficacious in vitro against multidrug-resistant Mycobacterium tuberculosis. Eur. J. Med. Chem. 2019, 166, 125. [Google Scholar] [CrossRef]
- Krasavin, M.; Lukin, A.; Vedekhina, T.; Manicheva, O.; Dogonadze, M.; Vinogradova, T.; Zabolotnykh, N.; Rogacheva, E.; Kraeva, L.; Yablonsky, P. Conjugation of a 5-nitrofuran-2-oyl moiety to aminoalkylimidazoles produces non-toxic nitrofurans that are efficacious in vitro and in vivo against multidrug-resistant Mycobacterium tuberculosis. Eur. J. Med. Chem. 2018, 157, 1115. [Google Scholar] [CrossRef]
- Matsumoto, M.; Hashizume, H.; Tomishige, T.; Kawasaki, M.; Tsubouchi, H.; Sasaki, H.; Shimokawa, Y.; Komatsu, M. OPC-67683, a Nitro-Dihydro-Imidazooxazole Derivative with Promising Action against Tuberculosis In Vitro and In Mice. PLoS Med. 2006, 3, 2131. [Google Scholar] [CrossRef] [PubMed]
- Stover, C.K.; Warrener, P.; VanDevanter, D.R.; Sherman, D.R.; Arain, T.M.; Langhorne, M.H.; Anderson, S.W.; Towell, J.A.; Yuan, Y.; McMurray, D.N.; et al. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 2000, 405, 962. [Google Scholar] [CrossRef] [PubMed]
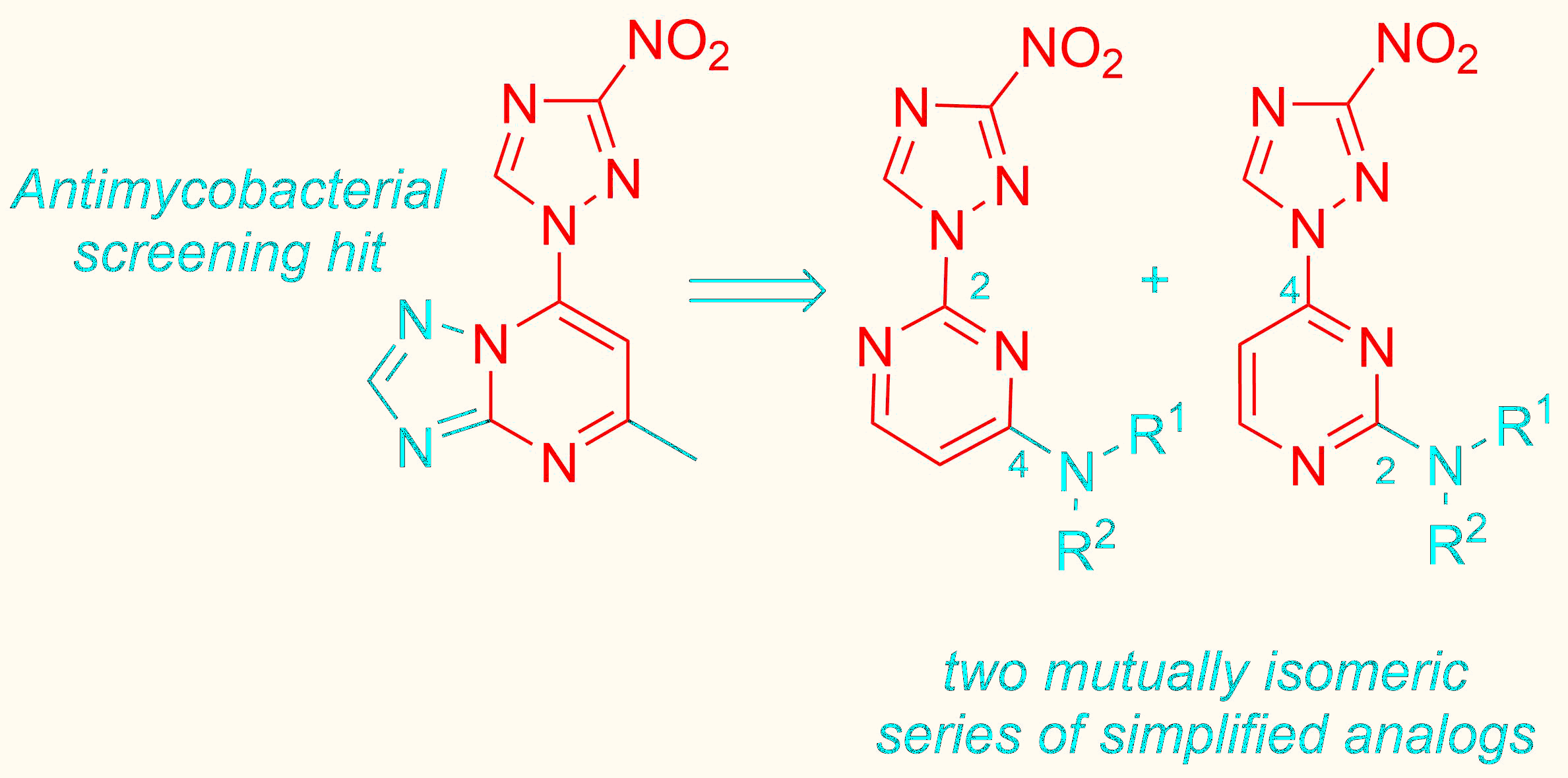
- Nepali, K.; Lee, H.-Y.; Liou, J.-P. Nitro-Group-Containing Drugs. J. Med. Chem. 2019, 62, 2851. [Google Scholar] [CrossRef] [PubMed]
- Krasavin, M.; Trifonov, R.E.; Tolstyakov, V.V.; Dar’in, D.V.; Vinogradova, T.I.; Manicheva, O.A.; Dogonadze, M.Z.; Zabolotnykh, N.V.; Vitovskaya, M.L.; Yablonskii, P.K. 5-Methyl-7-(3-nitro-[1,2,4]triazol-1-yl)-[1,2,4]triazolo[1,5-a]pyrimidine, having anti-tuberculosis activity against the agent with multiple drug resistance, and a method for production thereof. Chem. Abstr. 2019, 171, 563769. [Google Scholar]
- Pendleton, J.N.; Gorman, S.P.; Gilmore, B.F. Clinical relevance of the ESKAPE pathogens. Expert Rev. Anti Infect. Ther. 2013, 11, 297. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, M.V.; Bloomer, W.D.; Rosenzweig, H.S.; Wilkinson, S.R.; Szular, J.; Kaiser, M. Nitrotriazole-based acetamides and propanamides with broad spectrum antitrypanosomal activity. Eur. J. Med. Chem. 2016, 123, 895. [Google Scholar] [CrossRef]
- Lagoja, I.M.; Pochet, S.; Boudou, V.; Little, R.; Lescrinier, E.; Rozenski, J.; Herdewijn, P. A Short Path Synthesis of [13C/15N] Multilabeled Pyrimidine Nucleosides Starting from Glucopyranose Nucleosides. J. Org. Chem. 2003, 68, 1867. [Google Scholar] [CrossRef]
- Goldstein, B.P.; Vidal-Plana, R.R.; Cavalleri, B.; Zerilli, L.; Carniti, G.; Silvestri, L.G. The Mechanism of Action of Nitro-heterocyclic Antimicrobial Drugs. Metabolic Activation by Micro-organisms. J. Gen. Microbiol. 1977, 100, 283. [Google Scholar] [CrossRef]
- Edwards, D.I.J. Nitroimidazole drugs-action and resistance mechanisms I. Mechanism of action. Antimicrob. Chemother. 1993, 31, 9. [Google Scholar] [CrossRef]
- Olender, D.; Zwawiak, J.; Zaprutko, L. Multidirectional Efficacy of Biologically Active Nitro Compounds Included in Medicines. Pharmaceuticals 2018, 11, 54. [Google Scholar] [CrossRef]
- Paula, F.R.; Trossini, G.H.; Ferreira, E.I.; Serrano, S.H.P.; Menezes, C.M.S.; Tavares, L.C. Theoretical and voltammetric studies of 5-nitro-heterocyclic derivatives with potential trypanocidal activities. J. Braz. Chem. Soc. 2010, 21, 740. [Google Scholar] [CrossRef]
- Haeili, M.; Moore, C.; Davis, C.; Cochran, J.; Shah, S.; Shrestha, T.; Zhang, Y.; Bossmann, S.; Benjamin, W.; Kutsch, O.; et al. Copper Complexation Screen Reveals Compounds with Potent Antibiotic Properties against Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2014, 58, 3727. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Camacho, M.; Portaels, F.; Palomino, C.C. Resazurin microtiter assay plate testing of Mycobacterium tuberculosis susceptibilities to second-line drugs: Rapid, simple, and inexpensive method. Antimicrob. Agents Chemother. 2003, 47, 3616. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.W.; Kirby, W.M.; Sherris, J.C.; Turck, M. Antibiotic Susceptibility Testing by a Standardized Single Disk Method. Am. J. Clin. Pathol. 1966, 45, 493. [Google Scholar] [CrossRef]
- Standard Operating Procedure: Procedure for Establishing Zone Diameter Breakpoints and Quality Control Criteria for New Antimicrobial Agents. EUCAST SOP 9.1. 2018. Available online: https://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/EUCAST_SOPs/2018/EUCAST_SOP_9.1_Disk_diffusion_breakpoints_and_QC_ranges_20180123.pdf (accessed on 23 January 2018).
- Wiegand, I.; Hilpert, K.; Hancock, R.E. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
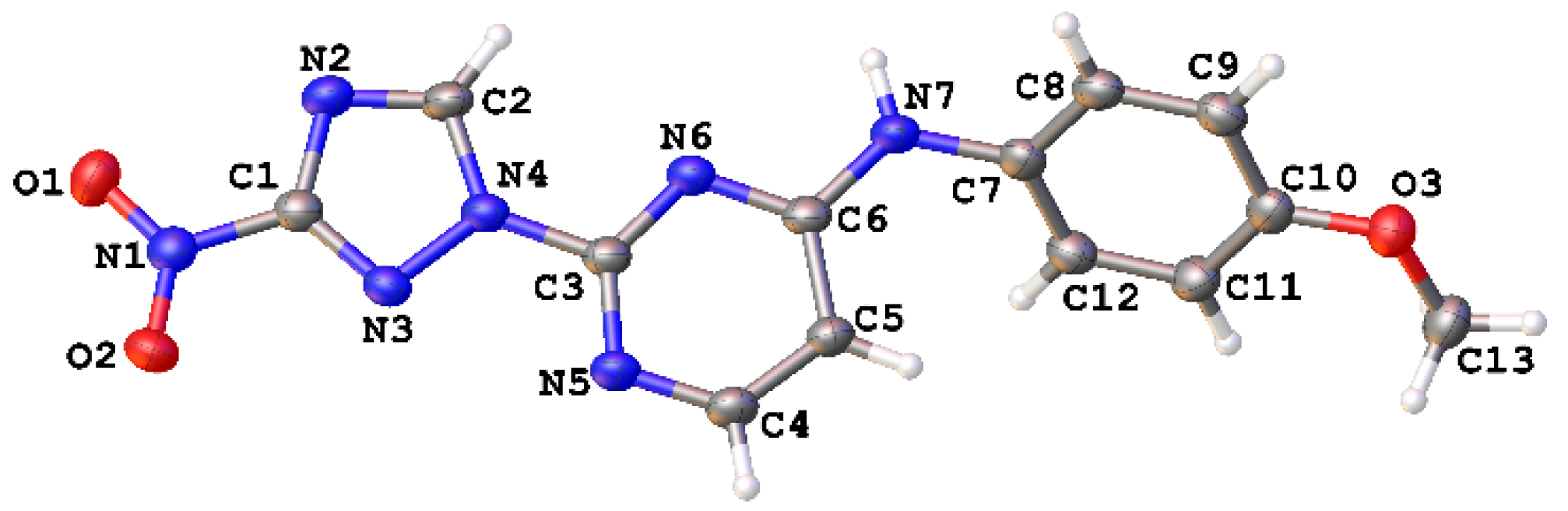{kind=link}
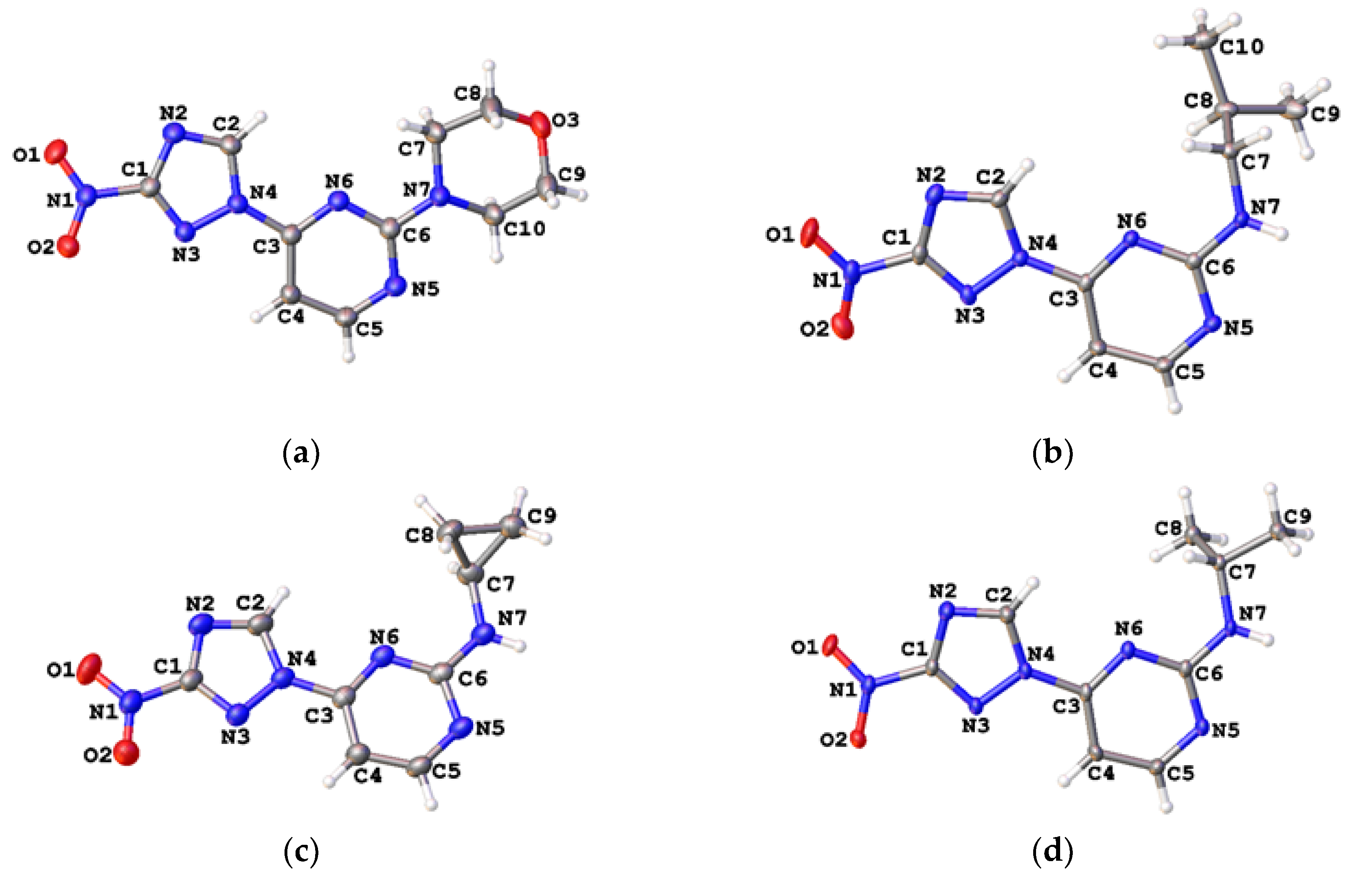{kind=link}
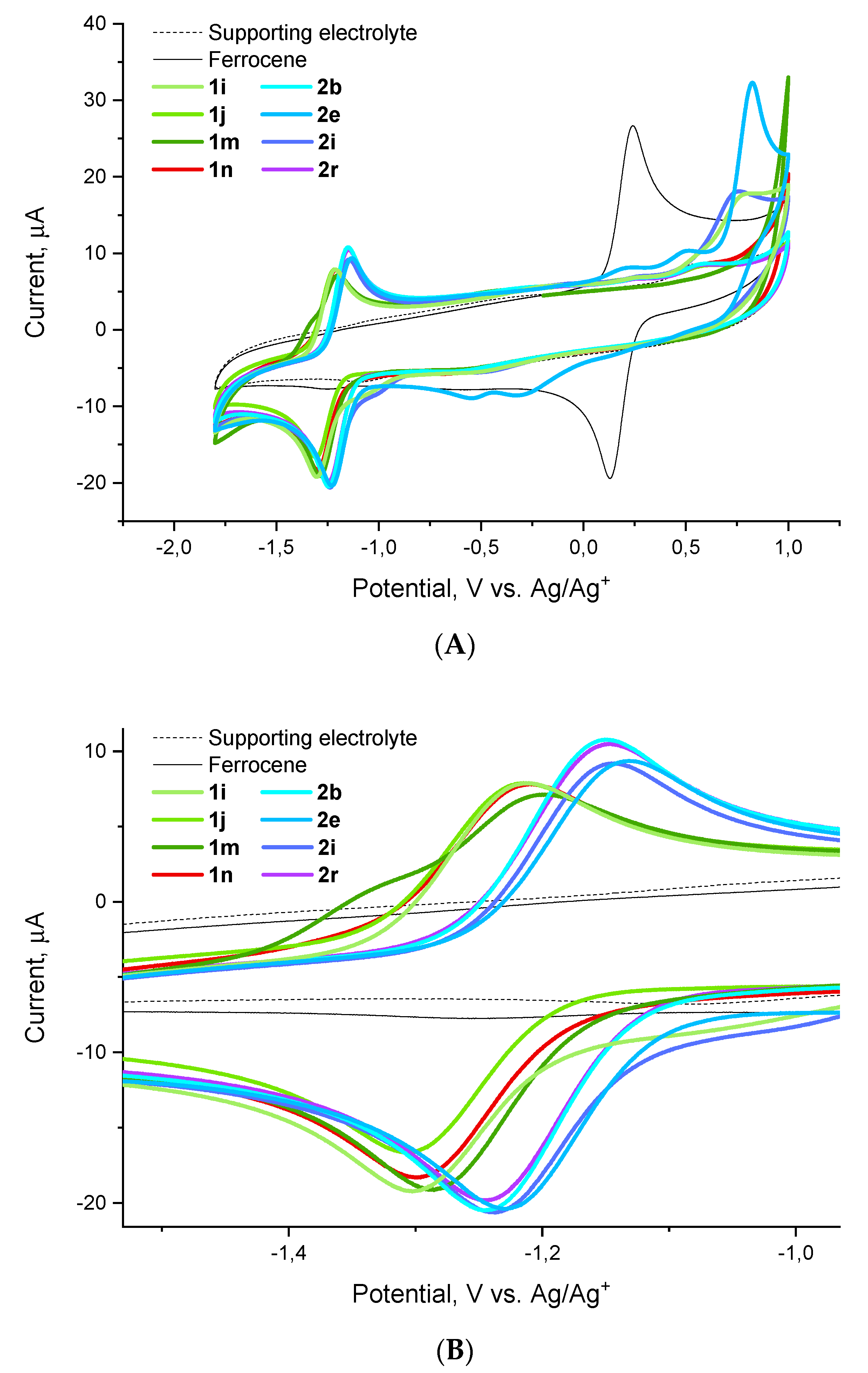{kind=link}
{kind=link}
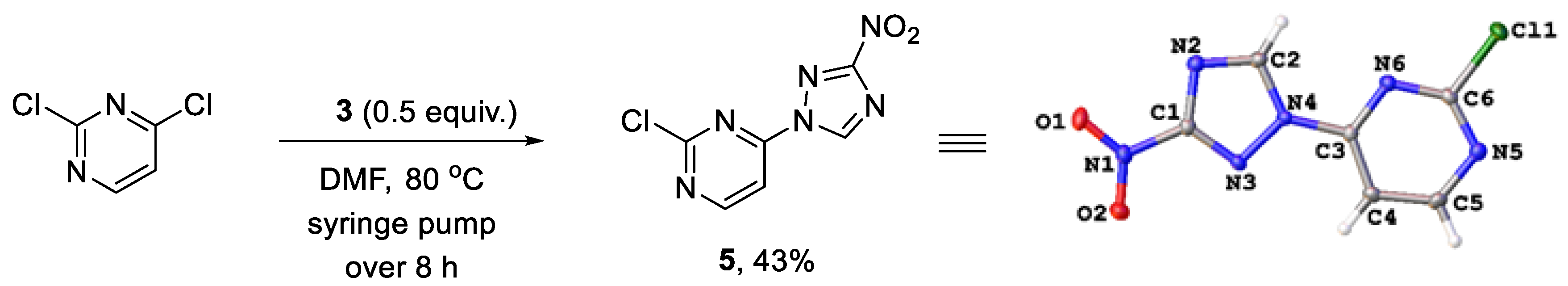{kind=link}

| Entry | Compound | Product | Ratio 1:2 | Isolated Yield of 1 (%) |
|---|---|---|---|---|
| 1 | 1a |  | 4.6:1 | 65 |
| 2 | 1b |  | 2.5:1 | 48 |
| 3 | 1c |  | 3.2:1 | 61 |
| 4 | 1d |  | 4:1 | 68 |
| 5 | 1e |  | 11.2:1 a | 45 |
| 6 | 1f |  | 4.6:1 | 35 |
| 7 | 1g |  | 2.2:1 | 46 |
| 8 | 1h |  | 3.5:1 | 38 |
| 9 | 1i |  | 22:1 | 66 |
| 10 | 1j |  | 3.5:1 | 39 |
| 11 | 1k |  | 1.4:1 | 32 |
| 12 | 1l |  | 1.2:1 | 28 |
| 13 | 1m |  | 1.6:1 a | 35 |
| 14 | 1n |  | 3.5:1 | 47 |
| 15 | 1o |  | 2.4:1 | 43 |
| 16 | 1p |  | 7.2:1 | 66 |
| 17 | 1q |  | 4:1 | 44 |
| 18 | 1r |  | - b | 61 |
| 19 | 1s |  | - b | 19 |

| Entry | Compound | Product | Yield (%) |
|---|---|---|---|
| 1 | 2a |  | 39 |
| 2 | 2b |  | 54 |
| 3 | 2c |  | 62 |
| 4 | 2d |  | 23 |
| 5 | 2e |  | 41 a |
| 6 | 2f |  | 58 |
| 7 | 2g |  | 75 |
| 8 | 2h |  | 83 |
| 9 | 2i |  | 76 |
| 10 | 2j |  | 81 |
| 11 | 2k |  | 44 |
| 12 | 2l |  | 71 |
| 13 | 2m |  | 62 a |
| 14 | 2n |  | 49 |
| 15 | 2o |  | 59 |
| 16 | 2p |  | 26 |
| 17 | 2q |  | 40 |
| 18 | 2r |  | 49 |
| 19 | 2s |  | 46 |

| Entry | Compound | R1NR2 Side Chain | MIC, μg/mL | |||||
|---|---|---|---|---|---|---|---|---|
| E1 | S | K | A | P | E2 | |||
| 1 | 1a |  | 16 | >100 | >100 | 16 | >100 | >100 |
| 2 | 2a | 8 | 63 | 8 | >100 | >100 | 63 | |
| 3 | 1b |  | 8 | >100 | >100 | 4 | >100 | >100 |
| 4 | 2b | 8 | >100 | 2 | 2 | >100 | 8 | |
| 5 | 1c |  | 16 | >100 | >100 | 4 | >100 | >100 |
| 6 | 2c | 63 | >100 | >100 | 8 | >100 | >100 | |
| 7 | 1d |  | 8 | 16 | 32 | >100 | >100 | 63 |
| 8 | 2d | 8 | 63 | 16 | >100 | >100 | 63 | |
| 9 | 1e |  | 4 | >100 | 63 | >100 | >100 | >100 |
| 10 | 2e | 8 | 8 | >100 | 4 | >100 | 63 | |
| 11 | 1f |  | 4 | >100 | >100 | 8 | >100 | >100 |
| 12 | 2f | 8 | 63 | 8 | 32 | >100 | 16 | |
| 13 | 1g |  | 4 | >100 | >100 | 32 | >100 | >100 |
| 14 | 2g | 32 | >100 | >100 | 8 | >100 | 63 | |
| 15 | 1h |  | 16 | >100 | >100 | 32 | >100 | >100 |
| 16 | 2h | 4 | 16 | >100 | 63 | >100 | >100 | |
| 17 | 1i |  | 2 | 4 | 32 | 32 | >100 | 32 |
| 18 | 2i | 2 | >100 | >100 | >100 | >100 | >100 | |
| 19 | 1j |  | 4 | >100 | >100 | 4 | >100 | >100 |
| 20 | 2j | 8 | >100 | >100 | 63 | >100 | >100 | |
| 21 | 1k |  | 2 | 63 | >100 | >100 | >100 | >100 |
| 22 | 2k | >100 | >100 | >100 | 4 | >100 | >100 | |
| 23 | 1l |  | 63 | >100 | >100 | 4 | >100 | >100 |
| 24 | 2l | 16 | 16 | >100 | 8 | >100 | >100 | |
| 25 | 1m |  | 8 | 8 | 32 | 2 | >100 | 16 |
| 26 | 2m | 16 | >100 | >100 | 8 | >100 | >100 | |
| 27 | 1n |  | >100 | >100 | >100 | >100 | >100 | >100 |
| 28 | 2n | 4 | 32 | >100 | 63 | >100 | >100 | |
| 29 | 1o |  | 2 | 4 | 16 | 32 | >100 | 32 |
| 30 | 2o | >100 | >100 | >100 | 8 | >100 | >100 | |
| 31 | 1p |  | 32 | >100 | >100 | 8 | >100 | >100 |
| 32 | 2p | 4 | >100 | >100 | 8 | >100 | >100 | |
| 33 | 1q |  | 16 | >100 | >100 | 4 | >100 | >100 |
| 34 | 2q | 32 | 63 | >100 | 16 | >100 | >100 | |
| 35 | 1r |  | 16 | >100 | >100 | 8 | >100 | 63 |
| 36 | 2r | 16 | 63 | >100 | >100 | >100 | >100 | |
| 37 | 1s |  | 16 | 63 | 63 | >100 | >100 | >100 |
| 38 | 2s | 2 | >100 | >100 | 4 | >100 | >100 | |
| 39 | 4 |  | 32 | >100 | >100 | >100 | >100 | >100 |
| 40 | ciprofloxacin | 0.3 | 1.25 | 0.6 | 2.5 | 0.6 | 1.25 | |
| Entry | Compound | E1/2 (V vs. Ag/Ag+) | E1/2 (V vs. Fc/Fc+) | E. faecium MIC (μg/mL) |
|---|---|---|---|---|
| 1 | 1i | −1.253 | −1.439 | 2 |
| 2 | 1j | −1.262 | −1.447 | 4 |
| 3 | 1m | −1.246 | −1.432 | 8 |
| 4 | 1n | −1.255 | −1.441 | >100 |
| 5 | 2b | −1.195 | 1.381 | 8 |
| 6 | 2e | −1.178 | −1.364 | 8 |
| 7 | 2i | −1.192 | −1.378 | 2 |
| 8 | 2r | −1.196 | −1.382 | 16 |
| 9 | ferrocene | 0.186 | 0.000 | – |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chuprun, S.; Dar’in, D.; Rogacheva, E.; Kraeva, L.; Levin, O.; Manicheva, O.; Dogonadze, M.; Vinogradova, T.; Bakulina, O.; Krasavin, M. Mutually Isomeric 2- and 4-(3-Nitro-1,2,4-triazol-1-yl)pyrimidines Inspired by an Antimycobacterial Screening Hit: Synthesis and Biological Activity against the ESKAPE Panel of Pathogens. Antibiotics 2020, 9, 666. https://doi.org/10.3390/antibiotics9100666
Chuprun S, Dar’in D, Rogacheva E, Kraeva L, Levin O, Manicheva O, Dogonadze M, Vinogradova T, Bakulina O, Krasavin M. Mutually Isomeric 2- and 4-(3-Nitro-1,2,4-triazol-1-yl)pyrimidines Inspired by an Antimycobacterial Screening Hit: Synthesis and Biological Activity against the ESKAPE Panel of Pathogens. Antibiotics. 2020; 9(10):666. https://doi.org/10.3390/antibiotics9100666
Chicago/Turabian StyleChuprun, Sergey, Dmitry Dar’in, Elizaveta Rogacheva, Liudmila Kraeva, Oleg Levin, Olga Manicheva, Marine Dogonadze, Tatiana Vinogradova, Olga Bakulina, and Mikhail Krasavin. 2020. "Mutually Isomeric 2- and 4-(3-Nitro-1,2,4-triazol-1-yl)pyrimidines Inspired by an Antimycobacterial Screening Hit: Synthesis and Biological Activity against the ESKAPE Panel of Pathogens" Antibiotics 9, no. 10: 666. https://doi.org/10.3390/antibiotics9100666
APA StyleChuprun, S., Dar’in, D., Rogacheva, E., Kraeva, L., Levin, O., Manicheva, O., Dogonadze, M., Vinogradova, T., Bakulina, O., & Krasavin, M. (2020). Mutually Isomeric 2- and 4-(3-Nitro-1,2,4-triazol-1-yl)pyrimidines Inspired by an Antimycobacterial Screening Hit: Synthesis and Biological Activity against the ESKAPE Panel of Pathogens. Antibiotics, 9(10), 666. https://doi.org/10.3390/antibiotics9100666