Increases in Hydrophilicity and Charge on the Polar Face of Alyteserin 1c Helix Change its Selectivity towards Gram-Positive Bacteria


 ,
, 
Abstract
1. Introduction
2. Results
2.1. Peptide Design and Sequence Characteristics
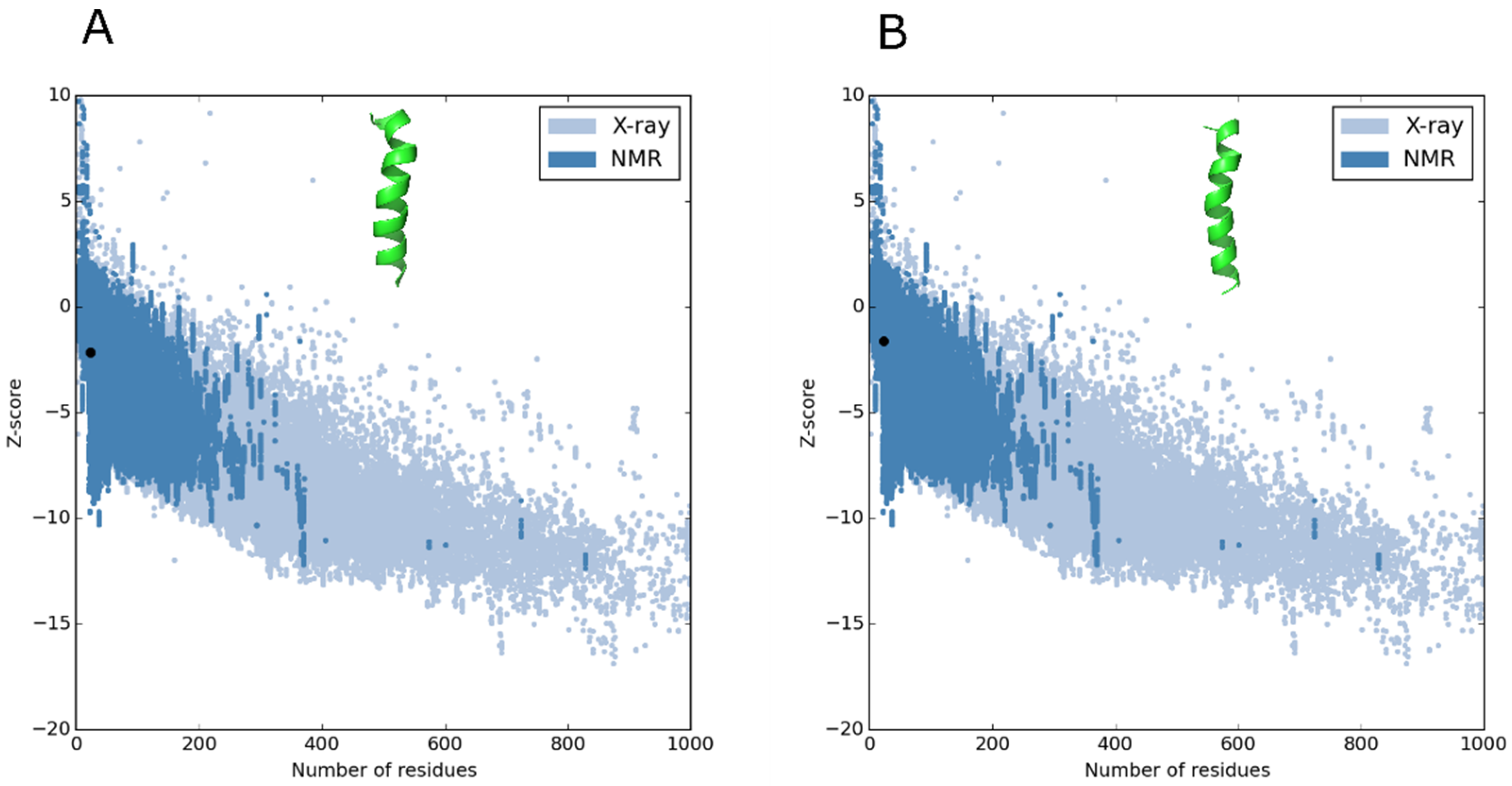
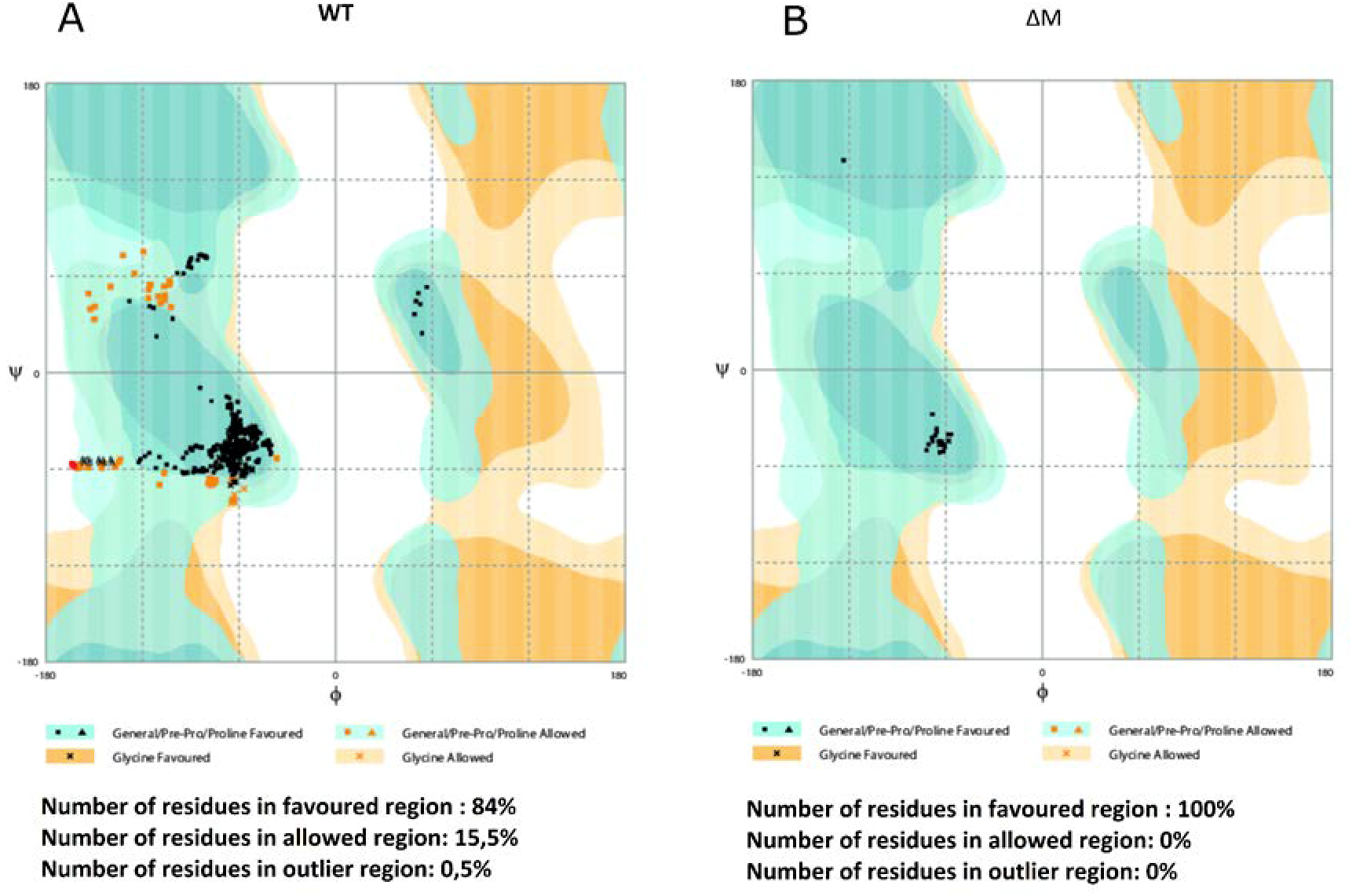
2.2. Molecular Modeling of the Peptides and Quality Testing
2.3. Antibacterial Activity
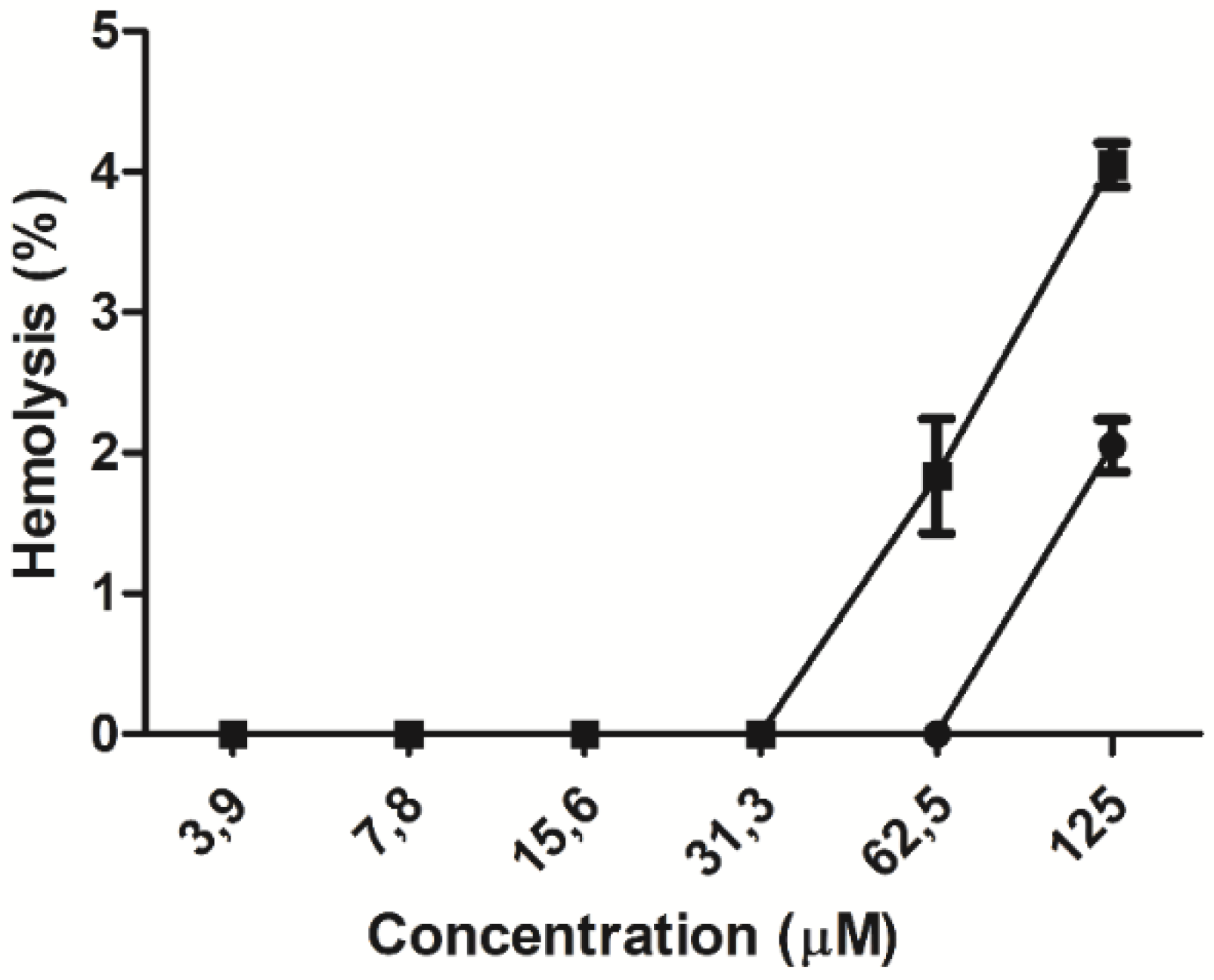
2.4. Hemolytic Effect
2.5. Prediction of Cleavage Sites of Staphylococcal Peptidase I
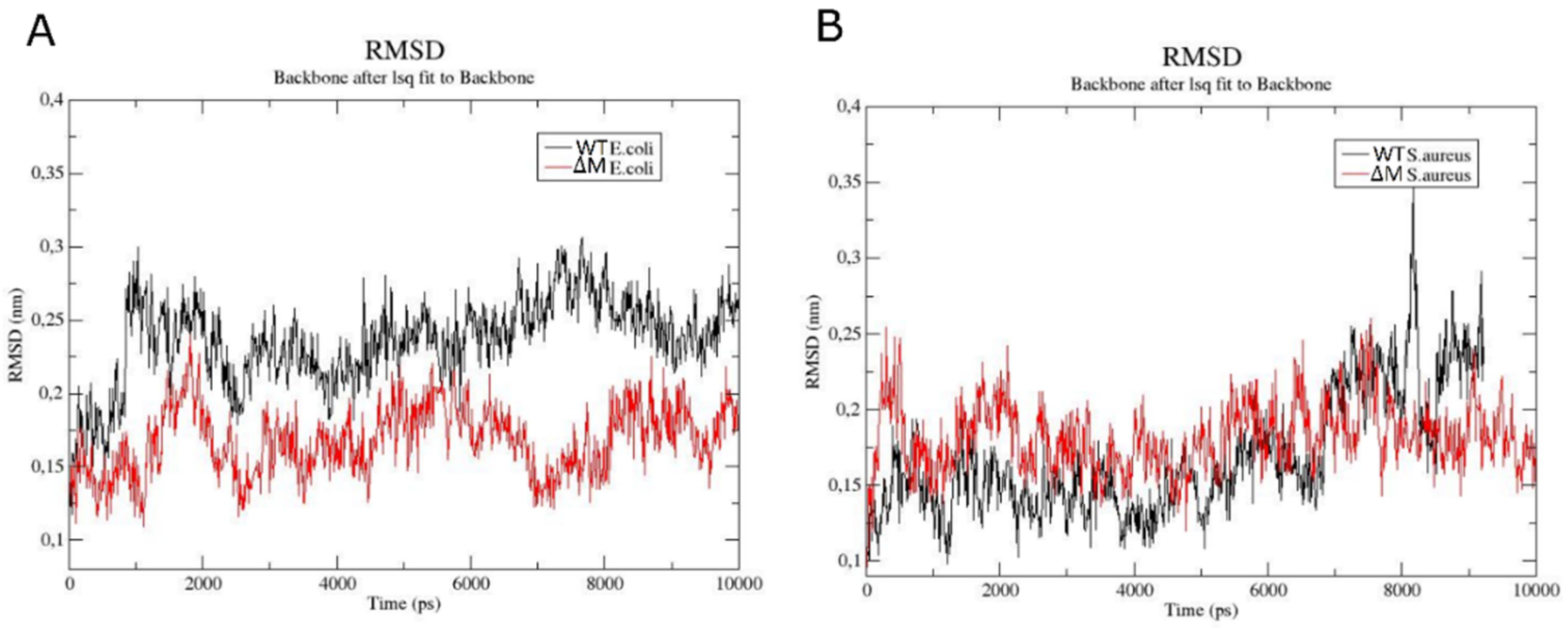
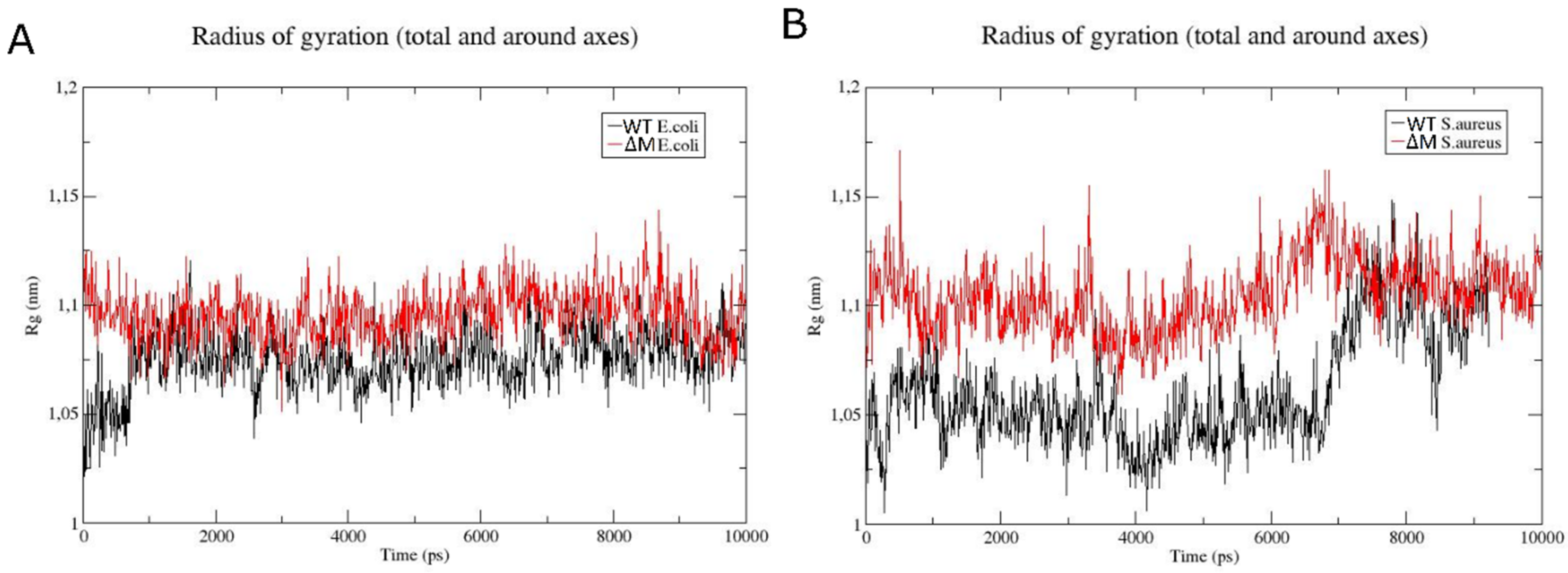
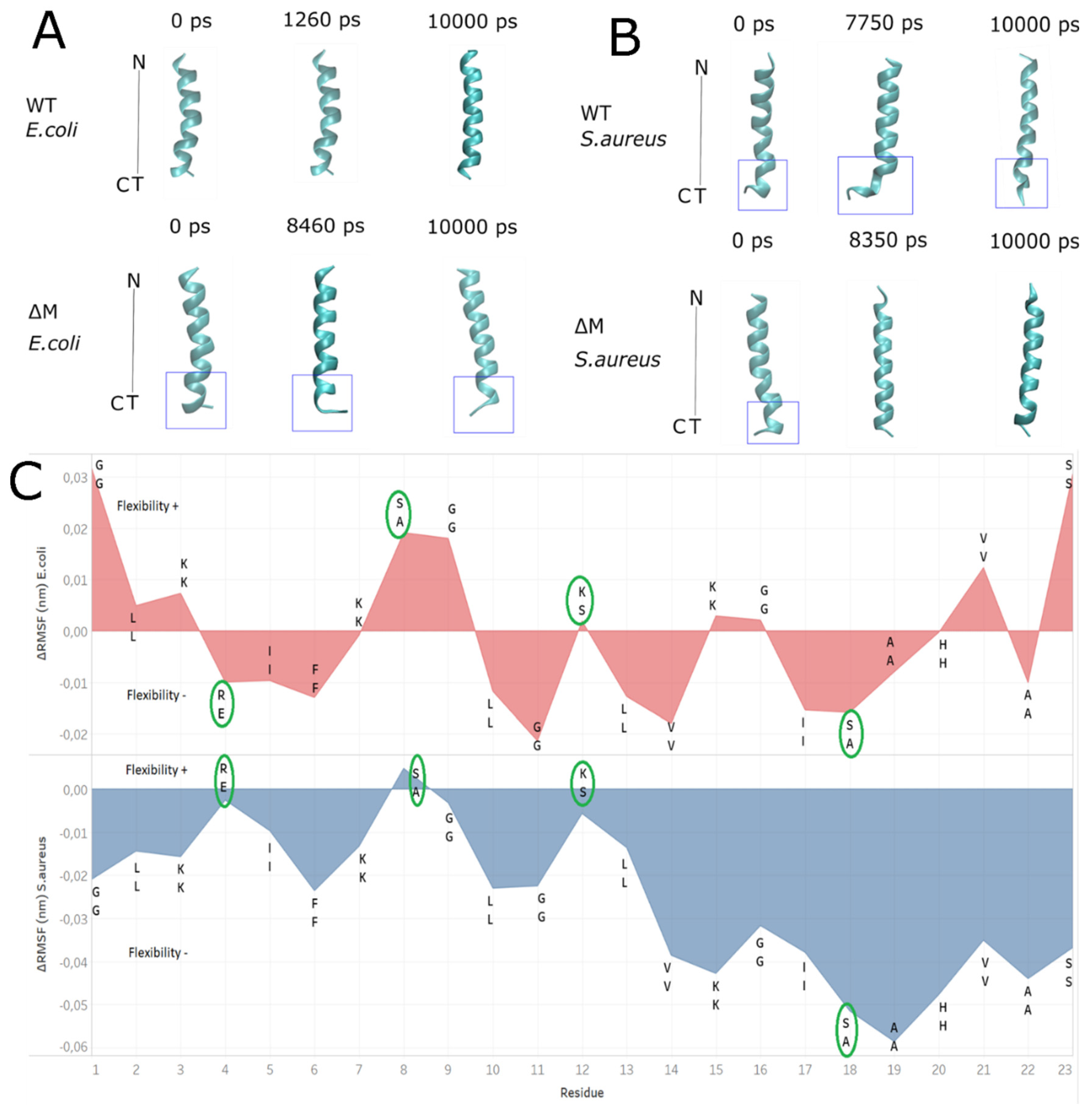
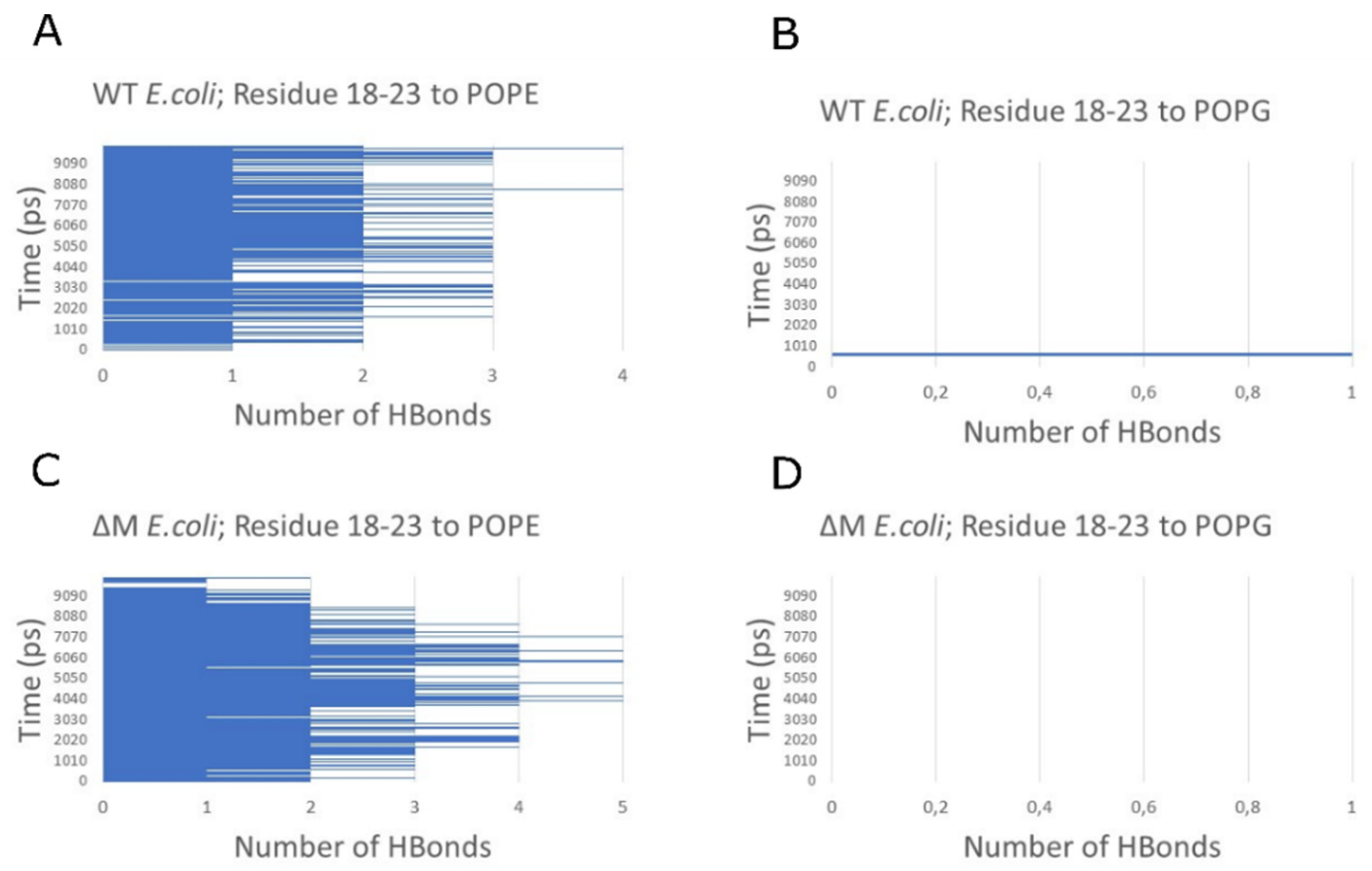
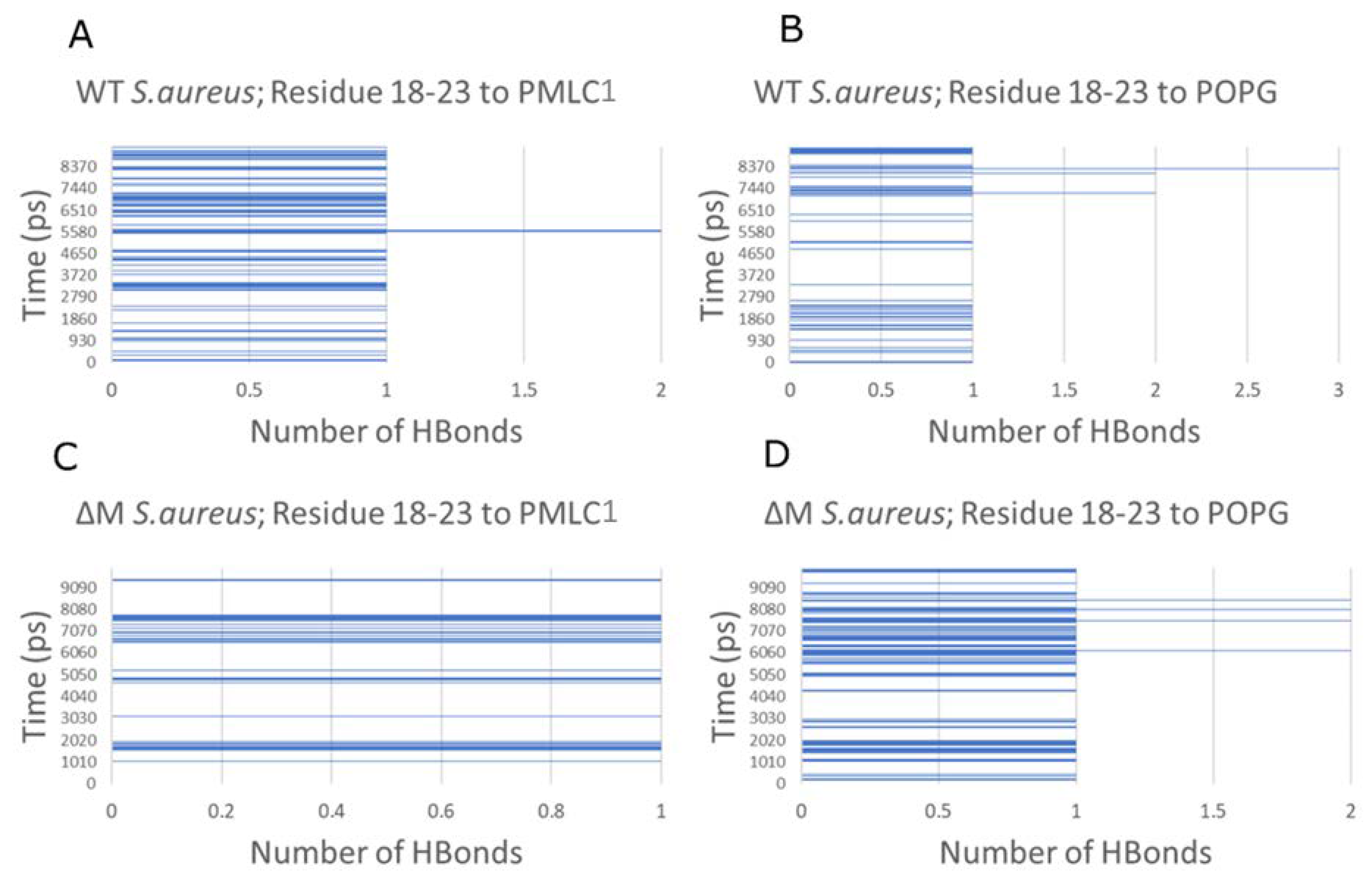
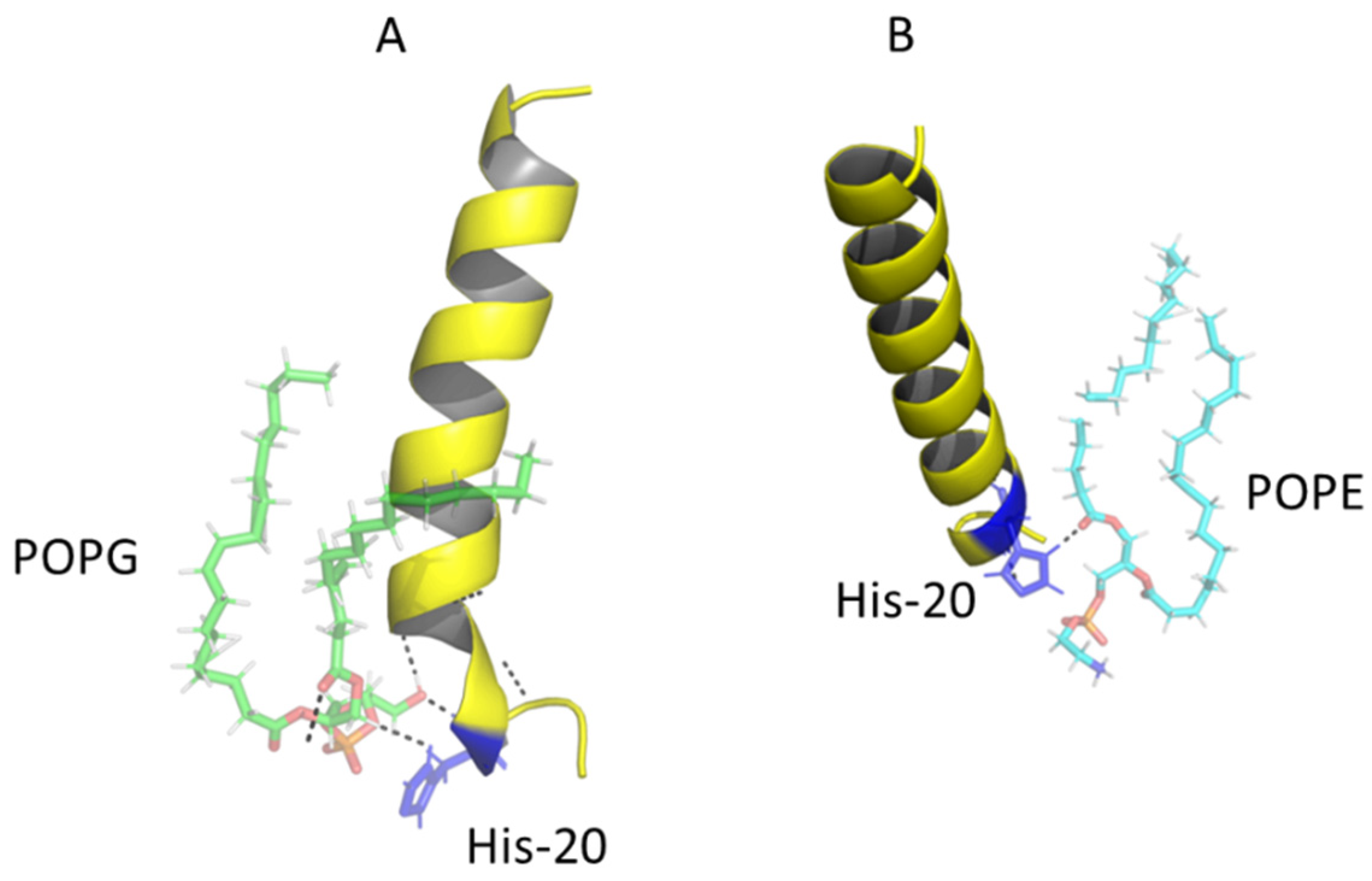
2.6. Modeling the Interaction of Peptides with Membranes of E. coli and S. aureus
3. Discussion
4. Materials and Methods
4.1. Strains and Reagents
4.2. Design and Synthesis of Peptides
4.3. Structural Modeling of Peptides
4.4. Antimicrobial Activity
4.5. Prediction of Peptide Digestion by Staphylococcal Enzyme
4.6. Hemolytic Effect
4.7. Construction of Gram-Negative and Gram-Positive Bacterial Membrane Models
4.8. Molecular Dynamic Simulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hiemstra, P.S.; Zaat, S.A.J. Antimicrobial Peptides and Innate Immunity; Springer: Basel, Switzerland, 2013; ISBN 9783034805414. [Google Scholar]
- Oñate-Garzón, J.; Manrique-Moreno, M.; Trier, S.; Leidy, C.; Torres, R.; Patiño, E. Antimicrobial activity and interactions of cationic peptides derived from Galleria mellonella cecropin D-like peptide with model membranes. J. Antibiot. 2017, 70, 238–245. [Google Scholar]
- Li, W.; Tailhades, J.; O’Brien-Simpson, N.M.; Separovic, F.; Otvos, L.; Hossain, M.A.; Wade, J.D. Proline-rich antimicrobial peptides: Potential therapeutics against antibiotic-resistant bacteria. Amino Acids 2014, 46, 2287–2294. [Google Scholar] [CrossRef] [PubMed]
- Fjell, C.D.; Hiss, J.A.; Hancock, R.E.; Schneider, G. Designing antimicrobial peptides: Form follows function. Nat. Rev. Drug Discov. 2012, 11, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Mangoni, M.L.; Mcdermott, A.M.; Zasloff, M. Antimicrobial peptides and wound healing: Biological and therapeutic considerations. Exp. Dermatol. 2016, 25, 167–173. [Google Scholar] [CrossRef]
- Strempel, N.; Strehmel, J.; Overhage, J. Potential Application of Antimicrobial Peptides in the Treatment of Bacterial Biofilm Infections. Curr. Pharm. Des. 2014, 21, 67–84. [Google Scholar] [CrossRef]
- Lee, T.H.; Hall, K.N.; Aguilar, M.I. Antimicrobial Peptide Structure and Mechanism of Action: A Focus on the Role of Membrane Structure. Curr. Top. Med. Chem. 2016, 16, 25–39. [Google Scholar] [CrossRef]
- Oñate-Garzón, J.; Ausili, A.; Manrique-Moreno, M.; Torrecillas, A.; Aranda, F.J.; Patiño, E.; Gomez-Fernández, J.C. The increase in positively charged residues in cecropin D-like Galleria mellonella favors its interaction with membrane models that imitate bacterial membranes. Arch. Biochem. Biophys. 2017, 629, 54–62. [Google Scholar]
- Subasinghage, A.P.; O’Flynn, D.; Conlon, J.M.; Hewage, C.M. Conformational and membrane interaction studies of the antimicrobial peptide alyteserin-1c and its analogue [E4K]alyteserin-1c. Biochim. Biophys. Acta 2011, 1808, 1975–1984. [Google Scholar] [CrossRef]
- Conlon, J.M.; Demandt, A.; Nielsen, P.F.; Leprince, J.; Vaudry, H.; Woodhams, D.C. The alyteserins: Two families of antimicrobial peptides from the skin secretions of the midwife toad Alytes obstetricans (Alytidae). Peptides 2009, 30, 1069–1073. [Google Scholar] [CrossRef]
- Aragón-Muriel, A.; Ausili, A.; Sánchez, K.; Rojas, A.; Oscar, E.; Londoño Mosquera, J.; Polo-Cerón, D.; Oñate-Garzón, J. Studies on the Interaction of Alyteserin 1c Peptide and Its Cationic Analogue with Model Membranes Imitating Mammalian and Bacterial Membranes. Biomolecules 2019, 9, 527–545. [Google Scholar]
- Berglund, N.A.; Piggot, T.J.; Jefferies, D.; Sessions, R.B.; Bond, P.J.; Khalid, S. Interaction of the antimicrobial peptide polymyxin B1 with both membranes of E. coli: A molecular dynamics study. PLoS Comput. Biol. 2015, 11, e1004180. [Google Scholar] [CrossRef] [PubMed]
- Ulmschneider, J.P.; Ulmschneider, M.B. Molecular Dynamics Simulations Are Redefining Our View of Peptides Interacting with Biological Membranes. Acc. Chem. Res. 2018, 51, 1106–1116. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Bolom, J.L.; Corzo, G.; Garduño-Juárez, R. Molecular dynamics simulation of the membrane binding and disruption mechanisms by antimicrobial scorpion venom-derived peptides. J. Biomol. Struct. Dyn. 2018, 36, 2070–2084. [Google Scholar] [CrossRef] [PubMed]
- Bordo, D.; Argos, P. Suggestions for “safe” residue substitutions in site-directed mutagenesis. J. Mol. Biol. 1991, 217, 721–729. [Google Scholar] [CrossRef]
- Cantor, S.; Vargas, L.; Rojas, O.E.A.; Yarce, C.J.; Salamanca, C.H.; Oñate-Garzón, J. Evaluation of the antimicrobial activity of cationic peptides loaded in surface-modified nanoliposomes against foodborne bacteria. Int. J. Mol. Sci. 2019, 20, 680. [Google Scholar] [CrossRef] [PubMed]
- Würz, J.M.; Güntert, P. Peak picking multidimensional NMR spectra with the contour geometry based algorithm CYPICK. J. Biomol. NMR 2017, 67, 63–76. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 17, 355–362. [Google Scholar] [CrossRef]
- Beg, M.A.; Shivangi; Thakur, S.C.; Meena, L.S. Structural Prediction and Mutational Analysis of Rv3906c Gene of Mycobacterium tuberculosis H37Rv to Determine Its Essentiality in Survival. Adv. Bioinform. 2018. [Google Scholar] [CrossRef]
- Rhodes, G. Other Diffraction Methods. In Crystallogr. Made Cryst. Clear; Elsevier: Amsterdam, The Netherlands, 2006. [Google Scholar]
- Lobanov, M.I.; Bogatyreva, N.S.; Galzitskaia, O.V. Radius of gyration is indicator of compactness of protein structure. Mol. Biol. (Mosk). 2008, 42, 701–706. [Google Scholar] [CrossRef]
- Waghu, F.H.; Joseph, S.; Ghawali, S.; Martis, E.A.; Madan, T.; Venkatesh, K.V.; Idicula-Thomas, S. Designing antibacterial peptides with enhanced killing kinetics. Front. Microbiol. 2018, 9, 325. [Google Scholar] [CrossRef]
- Vishnepolsky, B.; Zaalishvili, G.; Karapetian, M.; Nasrashvili, T.; Kuljanishvili, N.; Gabrielian, A.; Rosenthal, A.; Hurt, D.E.; Tartakovsky, M.; Grigolava, M.; et al. De Novo Design and In Vitro Testing of Antimicrobial Peptides against Gram-Negative Bacteria. Pharmaceuticals 2019, 12, 82. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, V.; Feio, M.J.; Bastos, M. Role of lipids in the interaction of antimicrobial peptides with membranes. Prog. Lipid Res. 2012, 51, 149–177. [Google Scholar] [CrossRef] [PubMed]
- Giangaspero, A.; Sandri, L.; Tossi, A. Amphipathic alpha helical antimicrobial peptides. Eur. J. Biochem. 2001, 268, 5589–5600. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Vasil, A.I.; Hale, J.D.; Hancock, R.E.; Vasil, M.L.; Hodges, R.S. Effects of net charge and the number of positively charged residues on the biological activity of amphipathic alpha-helical cationic antimicrobial peptides. Biopolymers 2008, 90, 369–383. [Google Scholar] [CrossRef]
- Oñate-Garzón, J.F.; Manrique-Moreno, M.; Patiño González, E. Actividad antimicrobiana de péptidos catiónicos diseñados a partir de un péptido neutro. Acta Biológica Colomb. 2017, 22, 157–164. [Google Scholar] [CrossRef]
- Abraham, T.; Lewis, R.N.; Hodges, R.S.; McElhaney, R.N. Isothermal titration calorimetry studies of the binding of a rationally designed analogue of the antimicrobial peptide gramicidin s to phospholipid bilayer membranes. Biochemistry 2005, 44, 2103–2112. [Google Scholar] [CrossRef]
- Sonnenfeld, E.M.; Beveridge, T.J.; Koch, A.L.; Doyle, R.J. Asymmetric distribution of charge on the cell wall of Bacillus subtilis. J. Bacteriol. 1985, 163, 1167–1171. [Google Scholar]
- Hancock, R.E. Peptide antibiotics. Lancet 1997, 349, 418–422. [Google Scholar] [CrossRef]
- Papo, N.; Shai, Y. Can we predict biological activity of antimicrobial peptides from their interactions with model phospholipid membranes? Peptides 2003, 24, 1693–1703. [Google Scholar] [CrossRef]
- Chen, Y.; Guarnieri, M.T.; Vasil, A.I.; Vasil, M.L.; Mant, C.T.; Hodges, R.S. Role of peptide hydrophobicity in the mechanism of action of alpha-helical antimicrobial peptides. Antimicrob. Agents Chemother. 2007, 51, 1398–1406. [Google Scholar] [CrossRef]
- Chou, H.T.; Kuo, T.Y.; Chiang, J.C.; Pei, M.J.; Yang, W.T.; Yu, H.C.; Lin, S.B.; Chen, W.J. Design and synthesis of cationic antimicrobial peptides with improved activity and selectivity against Vibrio spp. Int. J. Antimicrob. Agents 2008, 32, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Zelezetsky, I.; Tossi, A. Alpha-helical antimicrobial peptides—Using a sequence template to guide structure-activity relationship studies. Biochim. Biophys. Acta 2006, 1758, 1436–1449. [Google Scholar] [CrossRef] [PubMed]
- Yakimov, A.P.; Afanaseva, A.S.; Khodorkovskiy, M.A.; Petukhov, M.G. Design of Stable a-Helical Peptides and Thermostable Proteins in Biotechnology and Biomedicine. Acta Naturae 2016, 80, 70–81. [Google Scholar] [CrossRef]
- Bondar, A.N.; White, S.H. Hydrogen bond dynamics in membrane protein function. Biochim. Biophys. Acta Biomembr. 2012, 1818, 942–950. [Google Scholar] [CrossRef]
- Liu, L.; Fang, Y.; Wu, J. Flexibility is a mechanical determinant of antimicrobial activity for amphipathic cationic α-helical antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 2013, 1828, 2479–2486. [Google Scholar] [CrossRef]
- Mamonova, T.; Hespenheide, B.; Straub, R.; Thorpe, M.F.; Kurnikova, M. Protein flexibility using constraints from molecular dynamics simulations. Proc. Phys. Biol. 2005, 2, S137. [Google Scholar] [CrossRef]
- Pink, D.A.; Belaya, M.; Levadny, V.; Quinn, B. A model of polar group statics in lipid bilayers and monolayers. Langmuir 1997, 13, 1701–1711. [Google Scholar] [CrossRef]
- Krishna Deepak, R.N.V.; Sankararamakrishnan, R. N-H···N Hydrogen Bonds Involving Histidine Imidazole Nitrogen Atoms: A New Structural Role for Histidine Residues in Proteins. Biochemistry 2016, 55, 3774–3783. [Google Scholar] [CrossRef]
- Tossi, A.; Scocchi, M.; Skerlavaj, B.; Gennaro, R. Identification and characterization of a primary antibacterial domain in CAP18, a lipopolysaccharide binding protein from rabbit leukocytes. FEBS Lett. 1994, 14, 108–112. [Google Scholar] [CrossRef]
- Teilum, K.; Olsen, J.G.; Kragelund, B.B. Protein stability, flexibility and function. Biochim. Biophys. Acta Proteins Proteom. 2011, 1814, 969–976. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinforma. 2016, 47, 5.6.1–5.6.32. [Google Scholar]
- Clinical and Laboratory Standards Institute (CLSI). Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standards—Tenth Edition; CLSI document M07-A10 (ISBN 1-56238-987-4); CLSI: Wayne, PA, USA, 2015. [Google Scholar]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.F.; Savage, P.B.; Epand, R.M. Bacterial lipid composition and the antimicrobial efficacy of cationic steroid compounds (Ceragenins). Biochim. Biophys. Acta Biomembr. 2007, 1768, 2500–2509. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2016, 14, 71–73. [Google Scholar] [CrossRef]
- Legge, F.S.; Treutlein, H.; Howlett, G.J.; Yarovsky, I. Molecular dynamics simulations of a fibrillogenic peptide derived from apolipoprotein C-II. Biophys. Chem. 2007, 130, 102–113. [Google Scholar] [CrossRef]
- Ciccotti, G.; Ryckaert, J.P. Molecular dynamics simulation of rigid molecules. Comput. Phys. Reports 1986, 4, 346–392. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Frishman, D.; Argos, P. Knowledge-based protein secondary structure assignment. Proteins Struct. Funct. Bioinform. 1995, 23, 566–579. [Google Scholar] [CrossRef]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernández, C.X.; Schwantes, C.R.; Wang, L.P.; Lane, T.J.; Pande, V.S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. PyMOL: An open-source molecular graphics tool. Ccp4 Newslett. Protein Crystallogr. 2002, 40, 11. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | MHC | S. aureus ATCC25923 | S. aureus ATCC29213 | S. aureus ATCC43300 | L. monocytogenes ATCCbaa751 | B. cereus ATCC11778 | E. coli ATCC25922 | P. aeruginosa ATCC9027 | S. typhimurium ATCC14028 |
|---|---|---|---|---|---|---|---|---|---|
| MIC | MIC | MIC | MIC | MIC | MIC | MIC | MIC | ||
| WT | 62.5 | 250 | ND | ND | 125 | ND | 15.2 | 31.3 | 62.5 |
| ΔM | 31.3 | 62.5 | 250 | 250 | 62.5 | 125 | 62.5 | 250 | 125 |
| Peptide | Cut Numbers | Residue at the Cut Site |
|---|---|---|
| WT | 1 | 4 |
| ΔM | 0 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liscano, Y.; Salamanca, C.H.; Vargas, L.; Cantor, S.; Laverde-Rojas, V.; Oñate-Garzón, J. Increases in Hydrophilicity and Charge on the Polar Face of Alyteserin 1c Helix Change its Selectivity towards Gram-Positive Bacteria. Antibiotics 2019, 8, 238. https://doi.org/10.3390/antibiotics8040238
Liscano Y, Salamanca CH, Vargas L, Cantor S, Laverde-Rojas V, Oñate-Garzón J. Increases in Hydrophilicity and Charge on the Polar Face of Alyteserin 1c Helix Change its Selectivity towards Gram-Positive Bacteria. Antibiotics. 2019; 8(4):238. https://doi.org/10.3390/antibiotics8040238
Chicago/Turabian StyleLiscano, Yamil, Constain H. Salamanca, Lina Vargas, Stefania Cantor, Valentina Laverde-Rojas, and José Oñate-Garzón. 2019. "Increases in Hydrophilicity and Charge on the Polar Face of Alyteserin 1c Helix Change its Selectivity towards Gram-Positive Bacteria" Antibiotics 8, no. 4: 238. https://doi.org/10.3390/antibiotics8040238
APA StyleLiscano, Y., Salamanca, C. H., Vargas, L., Cantor, S., Laverde-Rojas, V., & Oñate-Garzón, J. (2019). Increases in Hydrophilicity and Charge on the Polar Face of Alyteserin 1c Helix Change its Selectivity towards Gram-Positive Bacteria. Antibiotics, 8(4), 238. https://doi.org/10.3390/antibiotics8040238