Mitochondrial Toxicity of Azithromycin Results in Aerobic Glycolysis and DNA Damage of Human Mammary Epithelia and Fibroblasts

Abstract
1. Introduction
2. Results
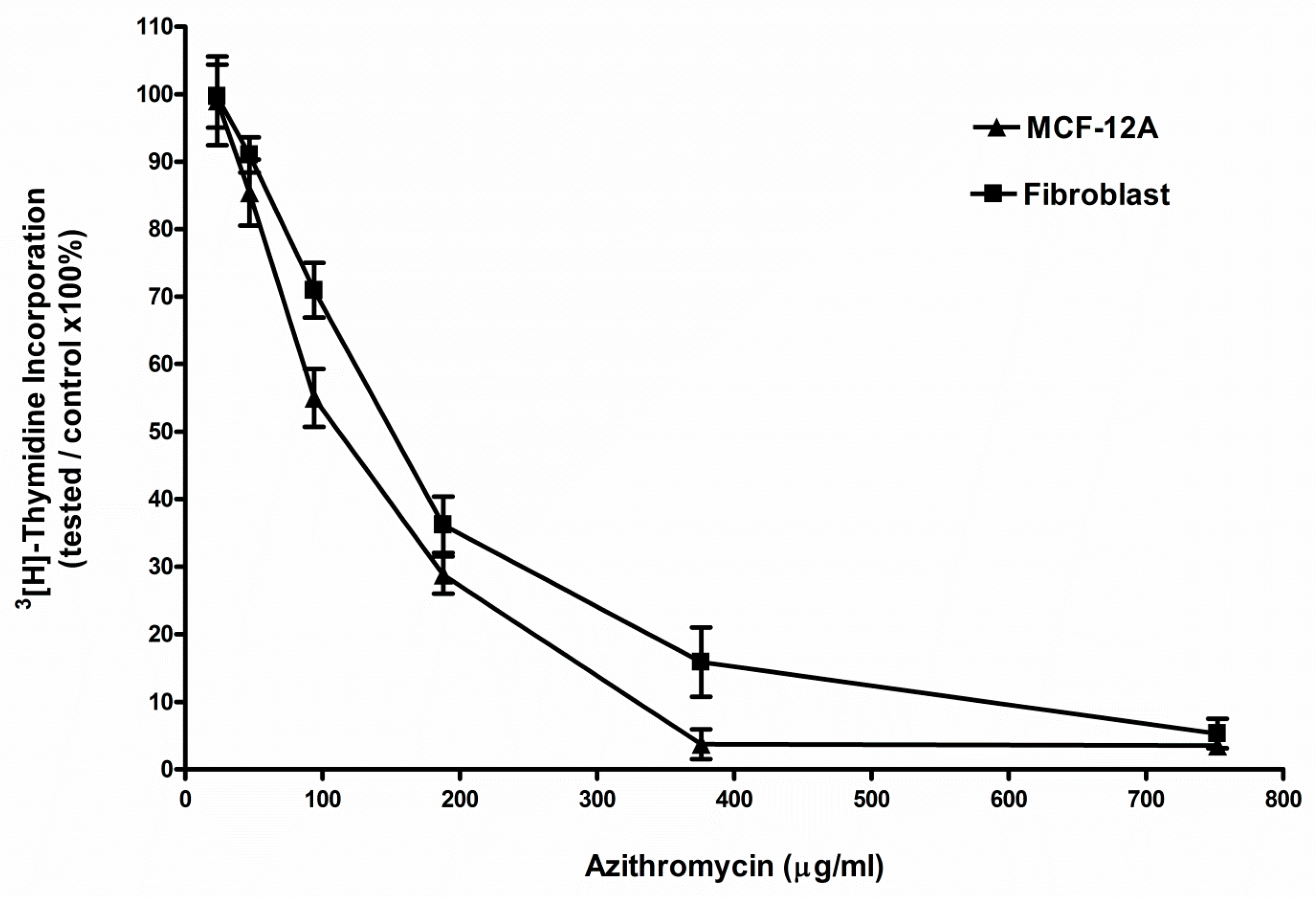
2.1. Azithromycin Inhibited the Proliferation of MCF-12A and Fibroblast Cells
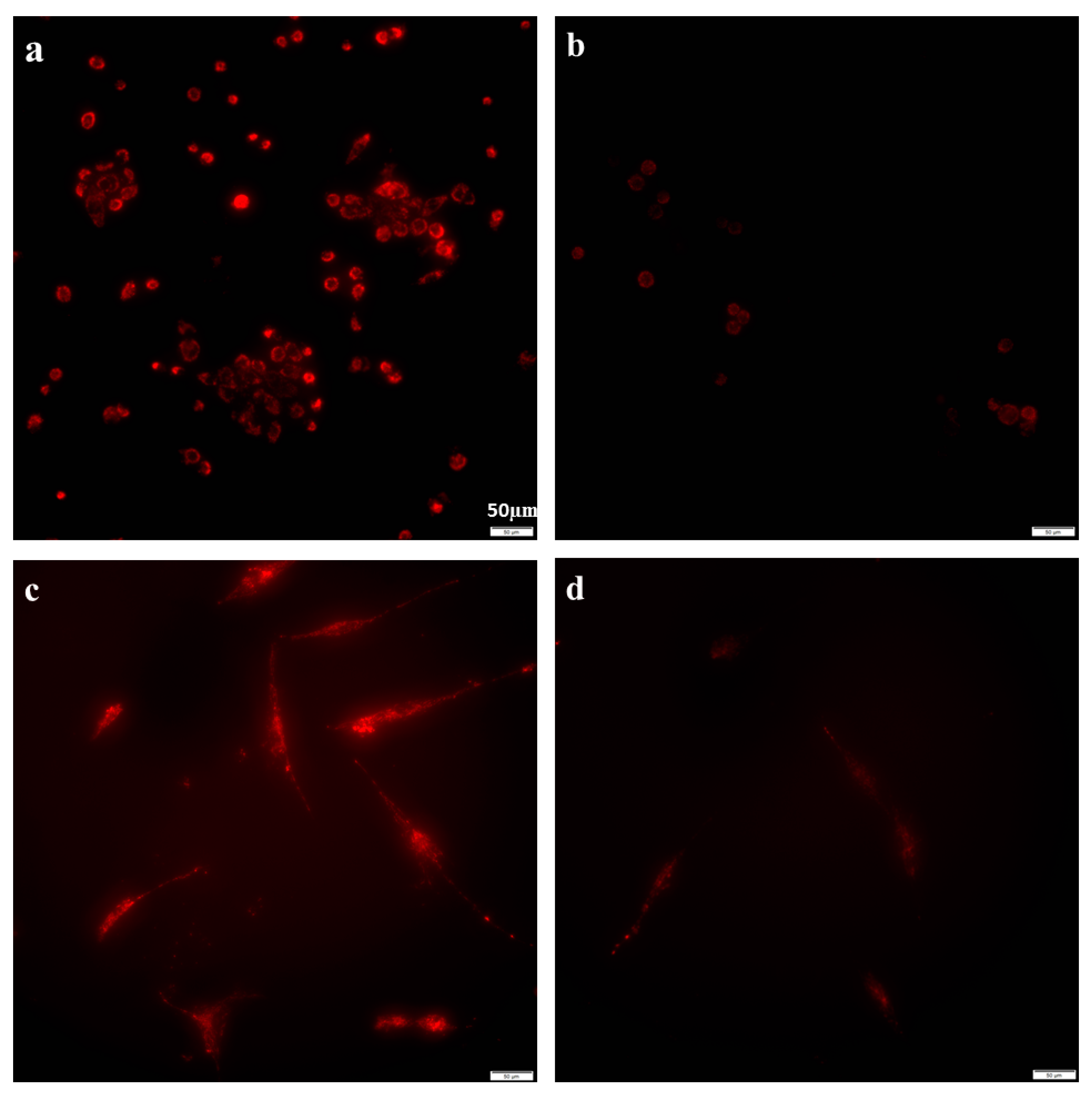
2.2. Azithromycin Suppressed Mitochondrial Membrane Potential Gradient
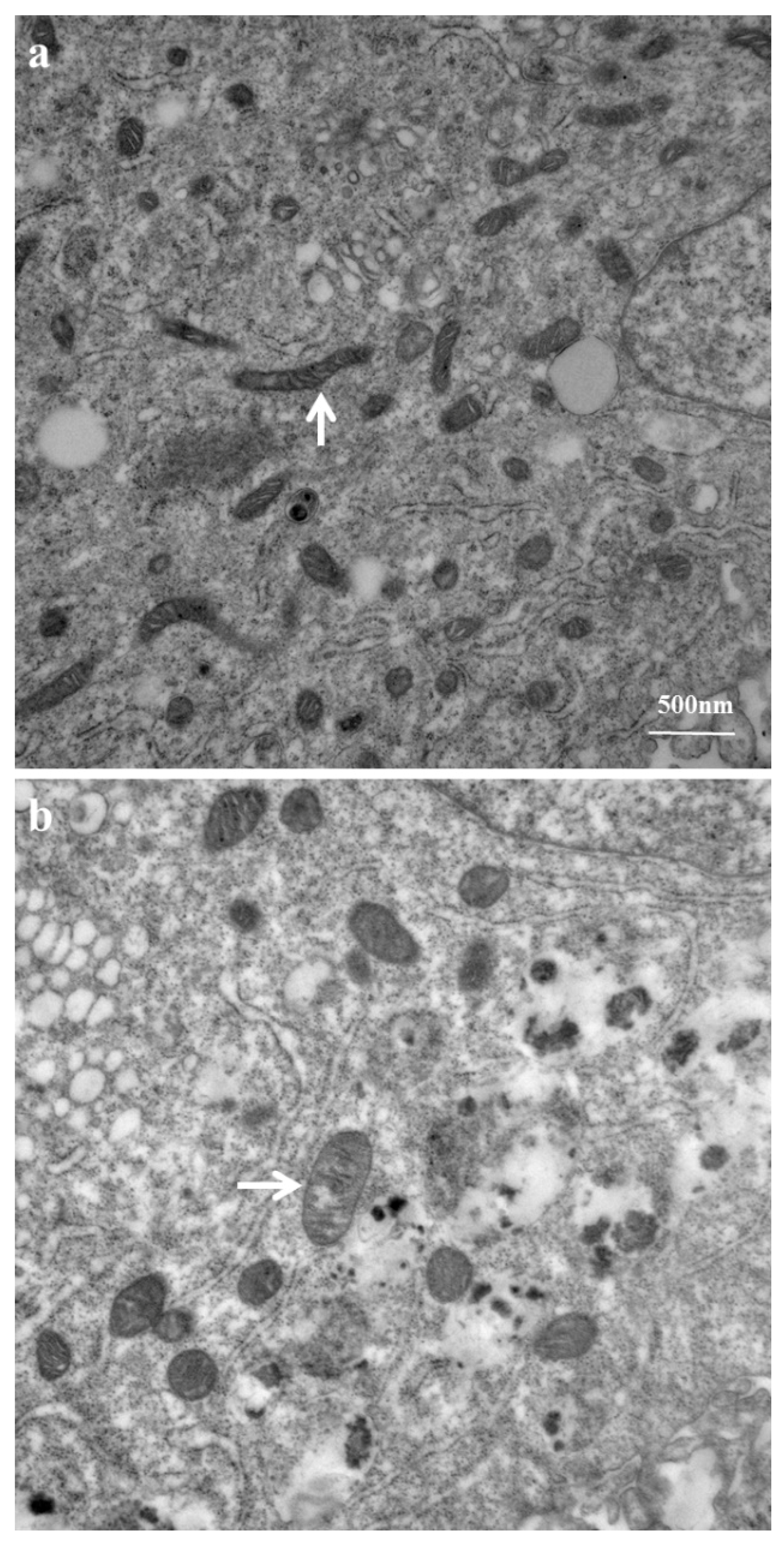
2.3. Mitochondrial Ultrastructure Damage by Azithromycin
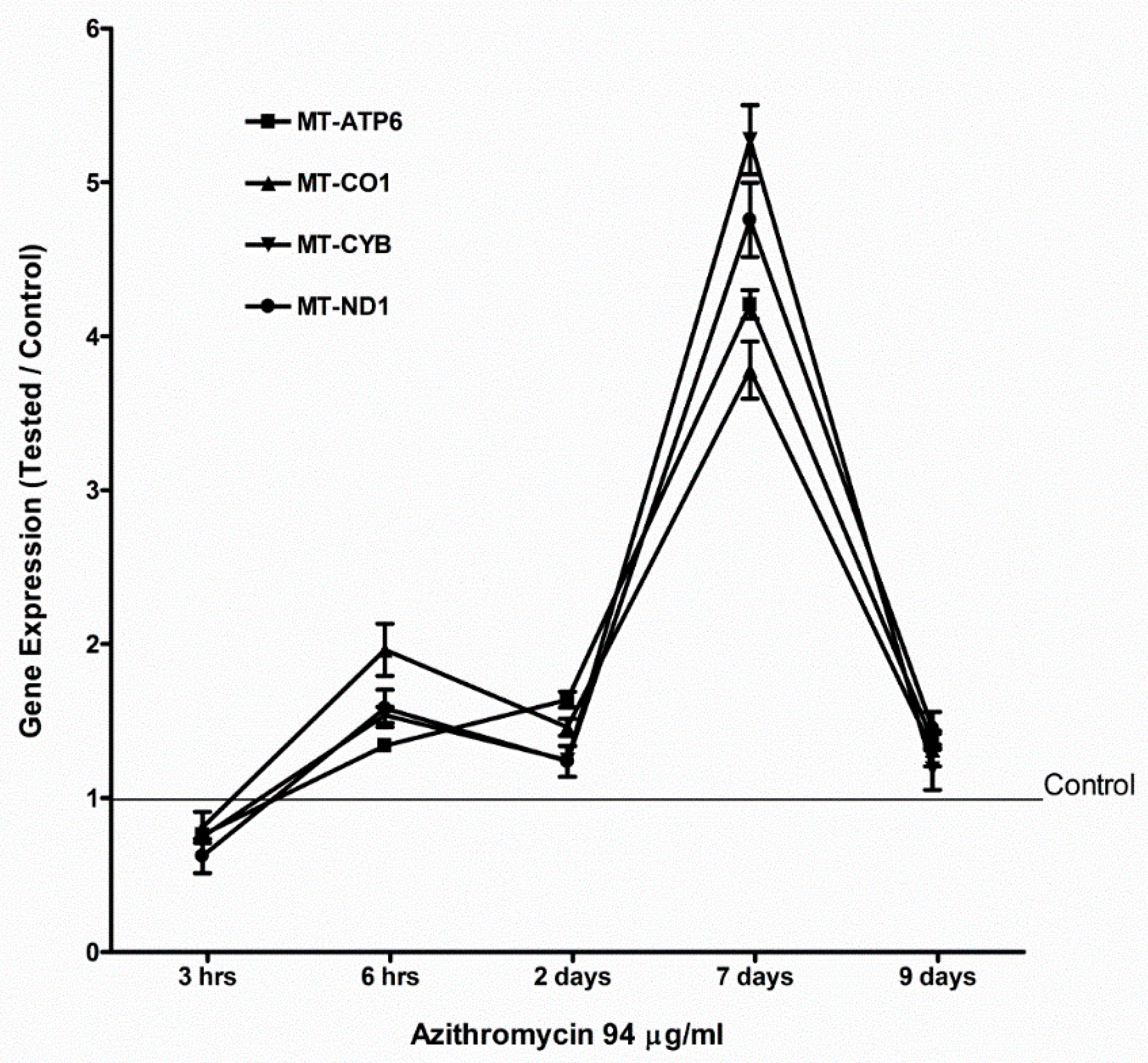
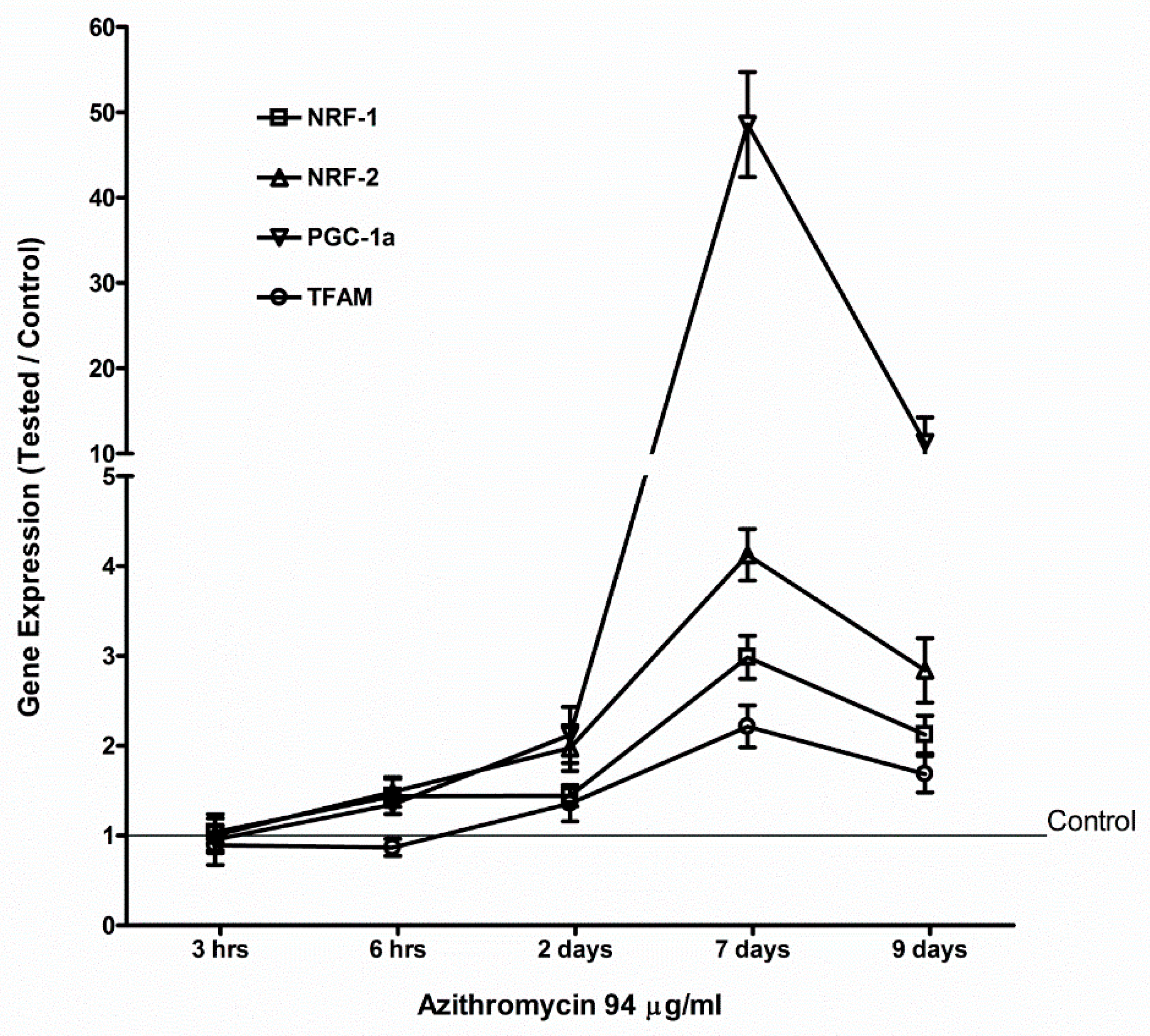
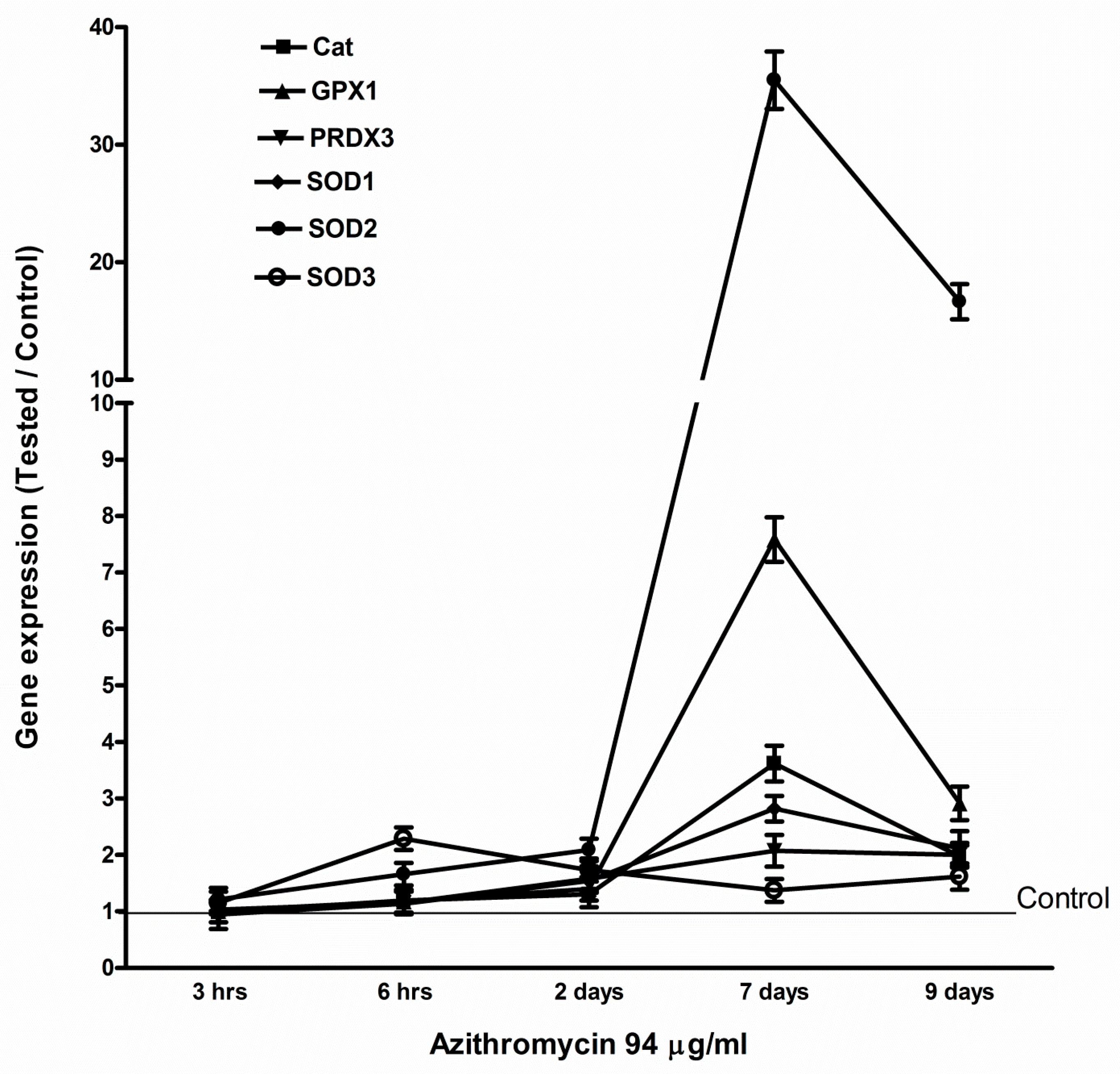
2.4. Azithromycin Upregulated Expression of Mitochondrial OXPHOS, Mitochondrial Biogenesis, and Antioxidant Genes
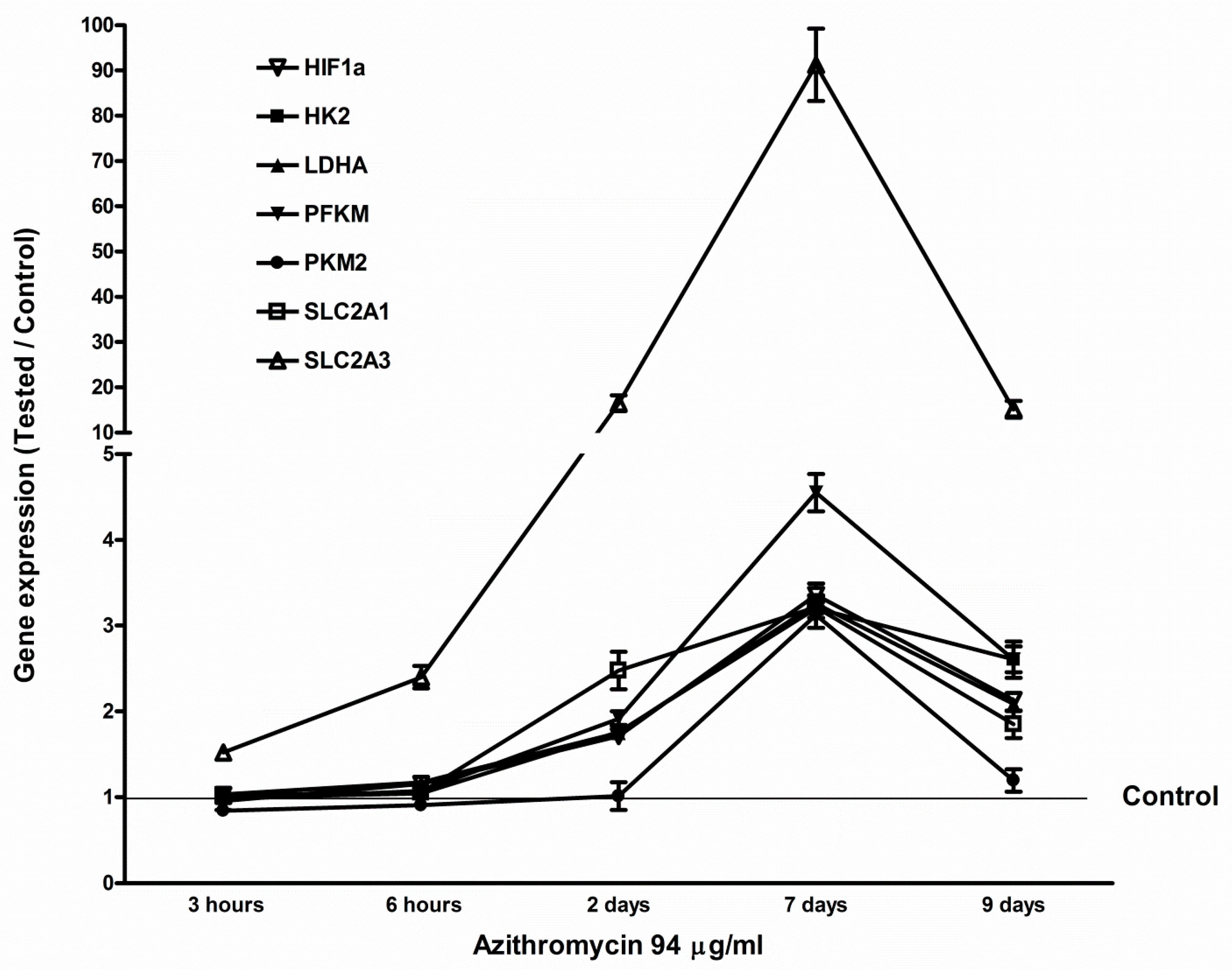
2.5. Azithromycin-Induced Gene Expression of HIF1a, Glycolytic Enzymes, and Glucose Transporters
2.6. Azithromycin Increased Lactate Production
2.7. Azithromycin Induces Mitochondrial Superoxide
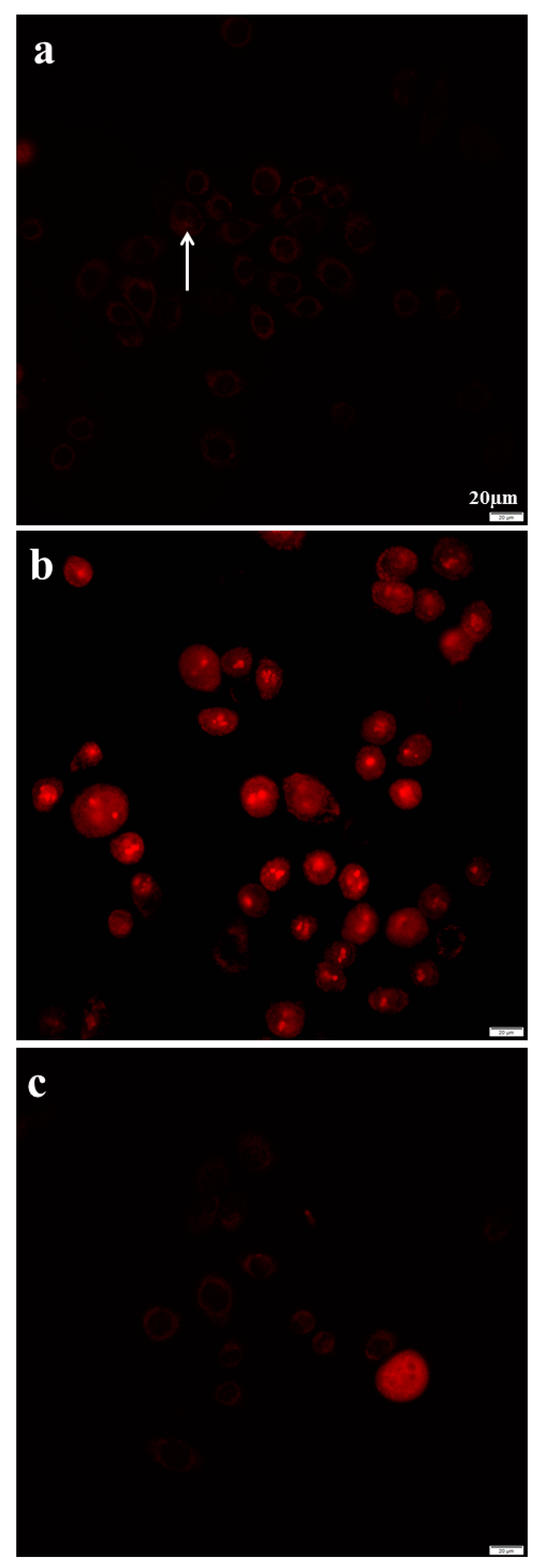
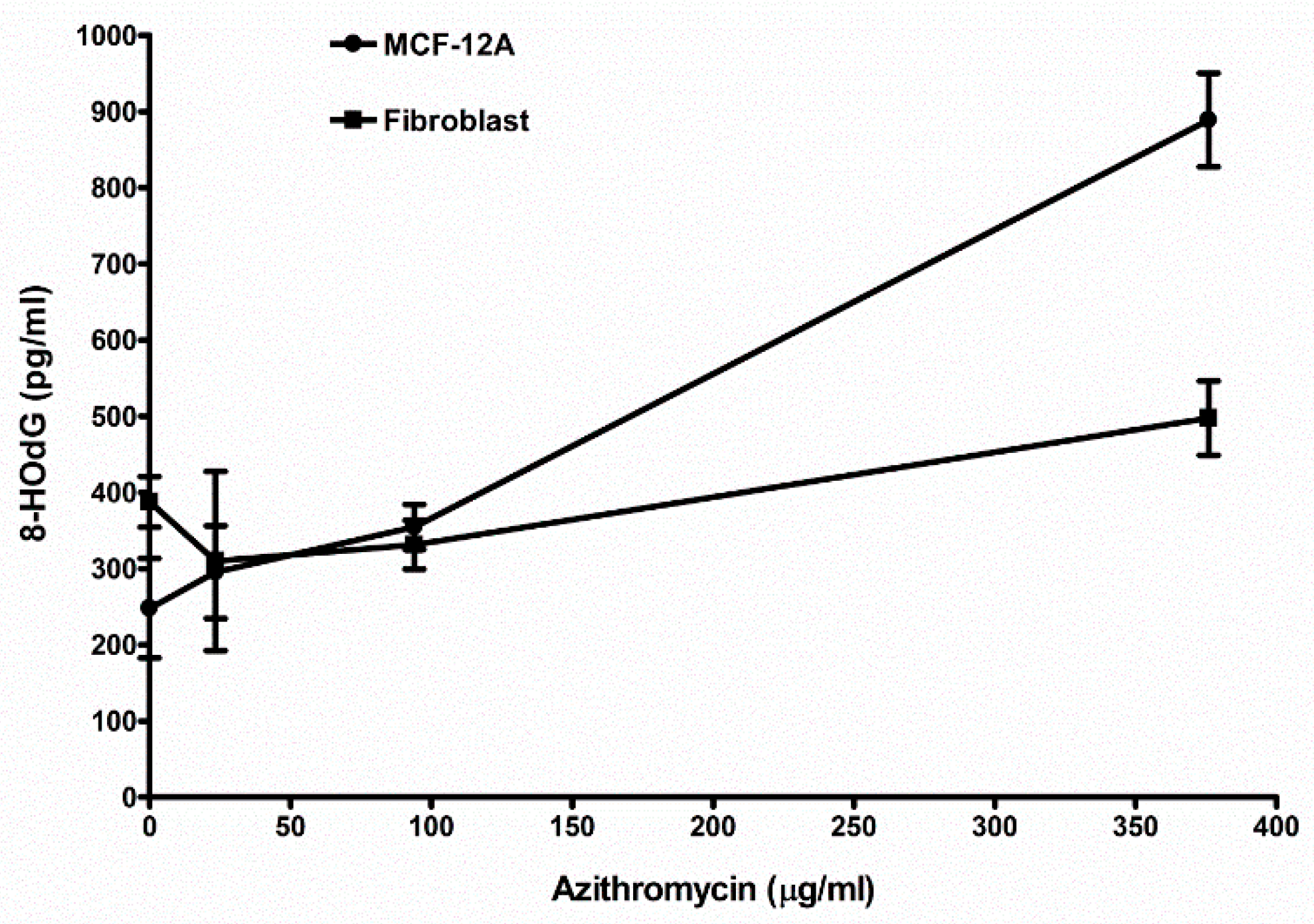
2.8. Azithromycin Causes Cell DNA Oxidative Damage
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. 3(H)-Thymidine Incorporation Assay
4.3. Mitochondria Staining for Mitochondrial Membrane Potential
4.4. Mitochondrial Ultrastructure Examination
4.5. Real-Time Polymerase Chain Reaction (qPCR)
4.6. Measurement of Lactate
4.7. Mitochondrial Superoxide Detection
4.8. DNA Oxidative Damage Examination
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Peters, D.H.; Friedel, H.A.; McTavish, D. Azithromycin. A review of its antimicrobial activity, pharmacokinetic properties and clinical efficacy. Drugs 1992, 44, 750–799. [Google Scholar] [CrossRef] [PubMed]
- Hand, W.L.; Hand, D.L. Characteristics and mechanisms of azithromycin accumulation and efflux in human polymorphonuclear leukocytes. Int. J. Antimicrob. Agents 2001, 18, 419–425. [Google Scholar] [CrossRef]
- Kagkelaris, K.A.; Makri, O.E.; Georgakopoulos, C.D.; Panayiotakopoulos, G.D. An eye for azithromycin: Review of the literature. Ther. Adv. Ophthalmol. 2018, 10, 2515841418783622. [Google Scholar] [CrossRef] [PubMed]
- McDonald, P.J.; Pruul, H. Phagocyte uptake and transport of azithromycin. Eur. J. Clin. Microbiol. Infect. Dis. 1991, 10, 828–833. [Google Scholar] [CrossRef]
- Matzneller, P.; Krasniqi, S.; Kinzig, M.; Sörgel, F.; Hüttner, S.; Lackner, E.; Müller, M.; Zeitlinger, M. Blood, tissue, and intracellular concentrations of azithromycin during and after end of therapy. Antimicrob. Agents Chemother. 2013, 57, 1736–1742. [Google Scholar] [CrossRef] [PubMed]
- Margulis, L. Origin of Eukaryotic Cells; Yale University Press: New Haven, CT, USA, 1970. [Google Scholar]
- Gray, M.W. Mitochondrial evolution. Cold Spring Harb. Perspect. Biol. 2012, 4, a011403. [Google Scholar] [CrossRef]
- Hopkin, S. Clinical toleration and safety of azithromycin. Am. J. Med. 1991, 91, 40S–45S. [Google Scholar] [CrossRef]
- Li, H.; Liu, D.H.; Chen, L.L.; Zhao, Q.; Yu, Y.Z.; Ding, J.J.; Miao, L.Y.; Xiao, Y.L.; Cai, H.R.; Zhang, D.P.; et al. Meta-analysis of the adverse effects of long-term azithromycin use in patients with chronic lung diseases. Antimicrob. Agents Chemother. 2014, 58, 511–517. [Google Scholar] [CrossRef]
- Salimi, A.; Eybagi, S.; Seydi, E.; Naserzadeh, P.; Kazerouni, N.P.; Pourahmad, J. Toxicity of macrolide antibiotics on isolated heart mitochondria: A justification for their cardiotoxic adverse effect. Xenobiotica 2016, 46, 82–93. [Google Scholar] [CrossRef]
- Romanowska-Sarlej, J.; Matysiak, W.; Kifer-Wysocka, E.; Czerny, K.; Masłyk, T.; Piskórz, J. Ultrastructure of the hepatocytes after application of azithromycin (Sumamed). Ann. Univ. Mariae Curie Sklodowska Med. 2004, 59, 57–60. [Google Scholar]
- Gray, M.W.; Burger, G.; Lang, B.F. Mitochondrial evolution. Science 1999, 283, 1476. [Google Scholar] [CrossRef] [PubMed]
- Carter, A.P.; Clemons, W.M.; Brodersen, D.E.; Morgan-Warren, R.J.; Wimberly, B.T.; Ramakrishnan, V. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature 2000, 407, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Barnhill, A.E.; Brewer, M.T.; Carlson, S.A. Adverse effects of antimicrobials via predictable or idiosyncratic inhibition of host mitochondrial components. Antimicrob. Agents Chemother. 2012, 56, 4046–4051. [Google Scholar] [CrossRef] [PubMed]
- Kalghatgi, S.; Spina, C.S.; Costello, J.C.; Liesa, M.; Morones-Ramirez, J.R.; Slomovic, S.; Molina, A.; Shirihai, O.S.; Collins, J.J. Bactericidal antibiotics induce mitochondrial dysfunction and oxidative damage in mammalian cells. Sci. Transl. Med. 2013, 5, 192ra85. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Kohanski, M.A.; Dwyer, D.J.; Wierzbowski, J.; Cottarel, G.; Collins, J.J. Mistranslation of membrane proteins and two-component system activation trigger antibiotic-mediated cell death. Cell 2008, 135, 679. [Google Scholar] [CrossRef]
- Dröse, S.; Brandt, U. The Mechanism of Mitochondrial Superoxide Production by the Cytochrome bc1 Complex. J. Biol. Chem. 2008, 283, 21649. [Google Scholar] [CrossRef]
- Waris, G.; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinog. 2006, 5, 14. [Google Scholar] [CrossRef]
- Federico, A.; Cardaioli, E.; Da Pozzo, P.; Formichi, P.; Gallus, G.N.; Radi, E. Mitochondria, oxidative stress and neurodegeneration. J. Neurol. Sci. 2012, 322, 254–262. [Google Scholar] [CrossRef]
- Shimura, T.; Sasatani, M.; Kawai, H.; Kamiya, K.; Kobayashi, J.; Komatsu, K.; Kunugita, N. Radiation-Induced Myofibroblasts Promote Tumor Growth via Mitochondrial ROS-Activated TGFβ Signaling. Mol. Cancer Res. 2018, 16, 1676–1686. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Karin, M. Inflammation and oncogenesis: A vicious connection. Curr. Opin. Genet. Dev. 2010, 20, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Velicer, C.M.; Heckbert, S.R.; Lampe, J.W.; Potter, J.D.; Robertson, C.A.; Taplin, S.H. Antibiotic use in relation to the risk of breast cancer. JAMA 2004, 291, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer. 2008, 8, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Makanji, Y.; Tagler, D.; Pahnke, J.; Shea, L.D.; Woodruff, T.K. Hypoxia-mediated carbohydrate metabolism and transport promote early-stage murine follicle growth and survival. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E893–E903. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Dai, J.; Park, Y.H.; Fai, L.Y.; Wang, L.; Pratheeshkumar, P.; Son, Y.O.; Kondo, K.; Xu, M.; Luo, J.; et al. Activation of Epidermal Growth Factor Receptor/p38/Hypoxia-inducible Factor-1α Is Pivotal for Angiogenesis and Tumorigenesis of Malignantly Transformed Cells Induced by Hexavalent Chromium. J. Biol. Chem. 2016, 291, 16271–16281. [Google Scholar] [CrossRef]
- Kraus, R.J.; Yu, X.; Cordes, B.A.; Sathiamoorthi, S.; Iempridee, T.; Nawandar, D.M.; Ma, S.; Romero-Masters, J.C.; McChesney, K.G.; Lin, Z.; et al. Hypoxia-inducible factor-1α plays roles in Epstein-Barr virus’s natural life cycle and tumorigenesis by inducing lytic infection through direct binding to the immediate-early BZLF1 gene promoter. PLoS Pathog. 2017, 13, e1006404. [Google Scholar] [CrossRef] [PubMed]
- Tarrado-Castellarnau, M.; de Atauri, P.; Cascante, M. Oncogenic regulation of tumor metabolic reprogramming. Oncotarget 2016, 7, 62726–62753. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar] [CrossRef]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef]
- Austin, S.; St-Pierre, J. PGC1α and mitochondrial metabolism--emerging concepts and relevance in ageing and neurodegenerative disorders. J. Cell Sci. 2012, 125 Pt 21, 4963–4971. [Google Scholar] [CrossRef]
- Ceriello, A.; Morocutti, A.; Mercuri, F.; Quagliaro, L.; Moro, M.; Damante, G.; Viberti, G.C. Defective intracellular antioxidant enzyme production in type 1 diabetic patients with nephropathy. Diabetes 2000, 49, 2170–2177. [Google Scholar] [CrossRef] [PubMed]
- van Zwieten, R.; Verhoeven, A.J.; Roos, D. Inborn defects in the antioxidant systems of human red blood cells. Free Radic Biol. Med. 2014, 67, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Huang, Z. Variations in Antioxidant Genes and Male Infertility. Biomed Res. Int. 2015, 2015, 513196. [Google Scholar] [CrossRef] [PubMed]
- Elliott, R.L.; Jiang, X.P. The adverse effect of gentamicin on cell metabolism in three cultured mammary cell lines: “Are cell culture data skewed?”. PLoS ONE 2019, 14, e0214586. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.P.; Elliott, R.L.; Head, J.F. Exogenous normal mammary epithelial mitochondria suppress glycolytic metabolism and glucose uptake of human breast cancer cells. Breast Cancer Res. Treat. 2015, 153, 519–529. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MCF-12A | Fibroblast | |
|---|---|---|
| Cell number | 10,000 | 10,000 |
| Control (media without any antibiotic) | 2.29 ± 0.41 (3) 1 | 3.01 ± 0.65 (3) |
| 47 µg/mL azithromycin | 3.03 ± 0.64 (3) NS 2 | 3.51 ± 0.59 (3) NS |
| 94 µg/mL azithromycin | 3.45 ± 0.56 (3) p < 0.05 | 4.38 ± 0.54 (3) p < 0.05 |
| 188 µg/mL azithromycin | 3.91 ± 0.49 (3) p < 0.05 | 4.68 ± 0.75 (3) p < 0.05 |
| MCF-12A | Fibroblast | |
|---|---|---|
| Control | 25.5 ± 4.8 (3) 1 | 19.6 ± 5.6 (3) |
| Azithromycin 188 µg/mL | 57.4 ± 13.6 p < 0.05 2 | 45.3 ± 10.5 (3) p < 0.05 |
| Azithromycin 188 µg/mL +SOD 100 u/mL | 35.3 ± 5.4 NS | 27.8 ± 8.9 (3) NS |
| MCF-12A | Fibroblast | |
|---|---|---|
| Cell amount | 10,000 | 10,000 |
| Control (media without any antibiotic) | 248.2 ± 65.6 (6) 1 | 388.0 ± 33.2 (3) |
| 23.5 µg/mL azithromycin | 295.7 ± 60.9 (3) NS 2 | 330.3 ± 117.6 (3) NS |
| 94 µg/mL azithromycin | 355.0 ± 29.6 (3) p < 0.05 | 331.3 ± 32.0 (3) NS |
| 376 µg/mL azithromycin | 889.0 ± 61.5 (3) p < 0.01 | 497.7 ± 48.5 (3) p < 0.05 |
| Gene | Pair of Primers (FWD and REV) | Probe |
|---|---|---|
| ACTB | GGATCAGCAAGCAGGAGTATG; | AGAAAGGGTGTAACGCAACTAA |
| TCGTCCACCGCAAATGCTTCTAGG | ||
| HIF1a | GTCTGCAACATGGAAGGTATTG; | GCAGGTCATAGGTGGTTTCT |
| ACTGCACAGGCCACATTCACGTAT | ||
| HK2 | GCAGAAGGTTGACCAGTATCTC; | CCAAGCCCTTTCTCCATCTC |
| CACATGCGCCTCTCTGATGAGACC | ||
| PFKM | GCATCCCATTTGTGGTCATTC; | GTCACAGGTTGTGCAGATAGT |
| AATGTCCCTGGCTCAGACTTCAGC | ||
| PKM2 | CTGTGGCTGGACTACAAGAA; | CTGCTTCACCTGGAGAGAAATA |
| AAGTGGGCAGCAAGATCTACGTGG | ||
| SLC2A1 | CTGGGCAAGTCCTTTGAGAT; | GTGACACTTCACCCACATACA |
| AGTACACACCGATGATGAAGCGGC | ||
| SLC2A3 | AGGATGCAGGTGTTCAAGAG; | GCCCTTTCCACCAGAAATAGA |
| CGGCGCGGGTGTGGTTAATACTAT | ||
| LDHA | AGATTCCAGTGTGCCTGTATG; | ACCTCTTTCCACTGTTCCTTATC |
| AGTGGAATGAATGTTGCTGGTGTCTCT | ||
| PGC-1a | AGAGCGCCGTGTGATTTAT; | CTCCATCATCCCGCAGATTTA |
| ACCTGACACAACACGGACAGAACT | ||
| NRF1 | GTATCTCACCCTCCAAACCTAAC; | CCAGGATCATGCTCTTGTACTT |
| TGCAGCACCTTTGGAGAATGTGGT | ||
| NRF2 | GCAATCTGCTACACCTACTACC; | TCCCAGGTGAGCTTCTATCT |
| AAGCAGCCAAAGTACAAAGAGCGC | ||
| TFAM | GTTGGAGGGAACTTCCTGATT; | CTGACTTGGAGTTAGCTGTTCT |
| AAGATGCTTATAGGGCGGAGTGGC | ||
| SOD1 | GTGCAGGGCATCATCAATTTC; | GGCCTTCAGTCAGTCCTTTAAT |
| AAGTAATGGACCAGTGAAGGTGTGGG | ||
| SOD2 | GCCTACGTGAACAACCTGAA; | GAAGAGCTATCTGGGCTGTAAC |
| TCACCGAGGAGAAGTACCAGGAGG | ||
| SOD3 | GCTGGCGCTACTGTGTT; | CGTGACCTTGGCGTACAT |
| AACTCTGACTCGGCGGAGTGGATC | ||
| CAT | CTGGAGCACAGCATCCAATA; | TCATTCAGCACGTTCACATAGA |
| ATTGGCAGTGTTGAATCTCCGCAC | ||
| GPX1 | CAGTCGGTGTATGCCTTCTC; | GCTCGTTCATCTGGGTGTAG |
| TATCGAGAATGTGGCGTCCCTCTGA | ||
| PRDX3 | CCTTTGTGTGTCCTACAGAAATTG; | TCCAGGCAAGATGGCTAAAG |
| TGTGAAGTTGTCGCAGTCTCAGTGG |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, X.; Baucom, C.; Elliott, R.L. Mitochondrial Toxicity of Azithromycin Results in Aerobic Glycolysis and DNA Damage of Human Mammary Epithelia and Fibroblasts. Antibiotics 2019, 8, 110. https://doi.org/10.3390/antibiotics8030110
Jiang X, Baucom C, Elliott RL. Mitochondrial Toxicity of Azithromycin Results in Aerobic Glycolysis and DNA Damage of Human Mammary Epithelia and Fibroblasts. Antibiotics. 2019; 8(3):110. https://doi.org/10.3390/antibiotics8030110
Chicago/Turabian StyleJiang, Xianpeng, Catherine Baucom, and Robert L. Elliott. 2019. "Mitochondrial Toxicity of Azithromycin Results in Aerobic Glycolysis and DNA Damage of Human Mammary Epithelia and Fibroblasts" Antibiotics 8, no. 3: 110. https://doi.org/10.3390/antibiotics8030110
APA StyleJiang, X., Baucom, C., & Elliott, R. L. (2019). Mitochondrial Toxicity of Azithromycin Results in Aerobic Glycolysis and DNA Damage of Human Mammary Epithelia and Fibroblasts. Antibiotics, 8(3), 110. https://doi.org/10.3390/antibiotics8030110