Optimization and Validation of a Chromatographic Method for the Quantification of Isoniazid in Urine of Tuberculosis Patients According to the European Medicines Agency Guideline



Abstract

1. Introduction
2. Experimental
2.1. Chemical and Reagents
2.2. Apparatus and Instrumentation
2.3. Preparation of Solutions
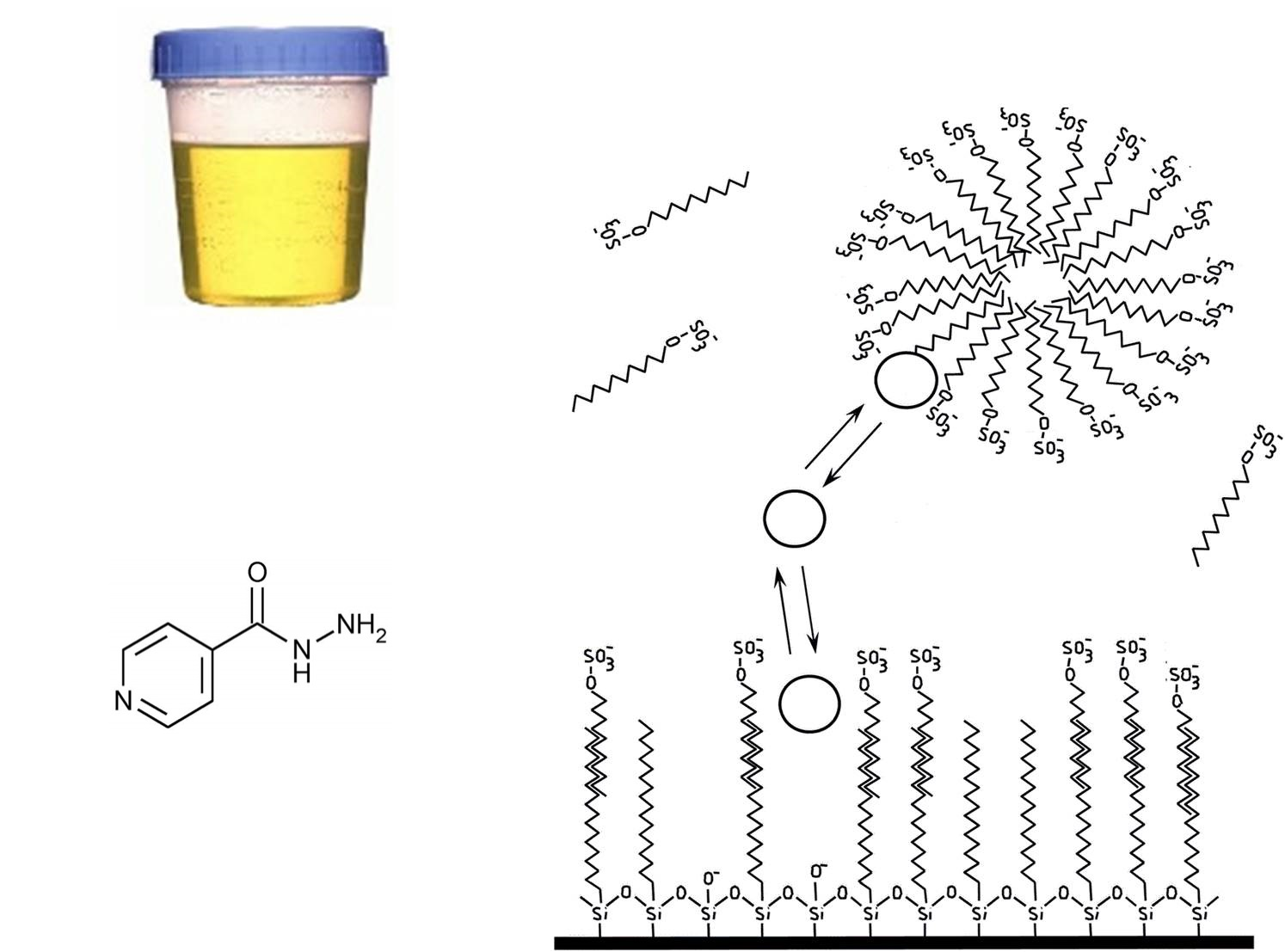
2.4. Sample Collection and Processing
3. Results and Discussion
3.1. Optimization of Micellar Chromatographic Conditions
3.1.1. Optimization of the pH
3.1.2. Optimization of SDS and Organic Modifier
3.1.3. Selection of the Optimal Detection Wavelength
3.2. Method Validation
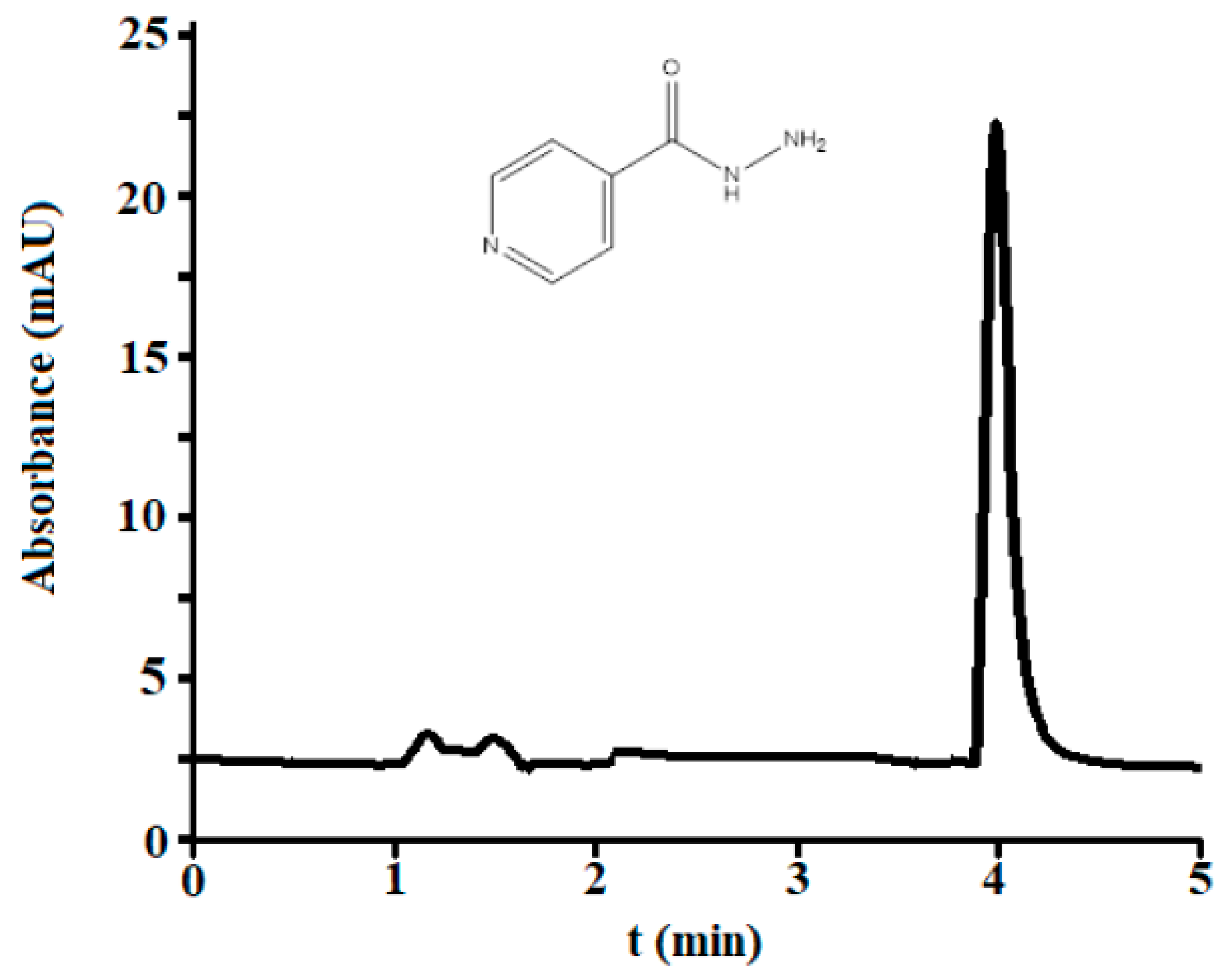
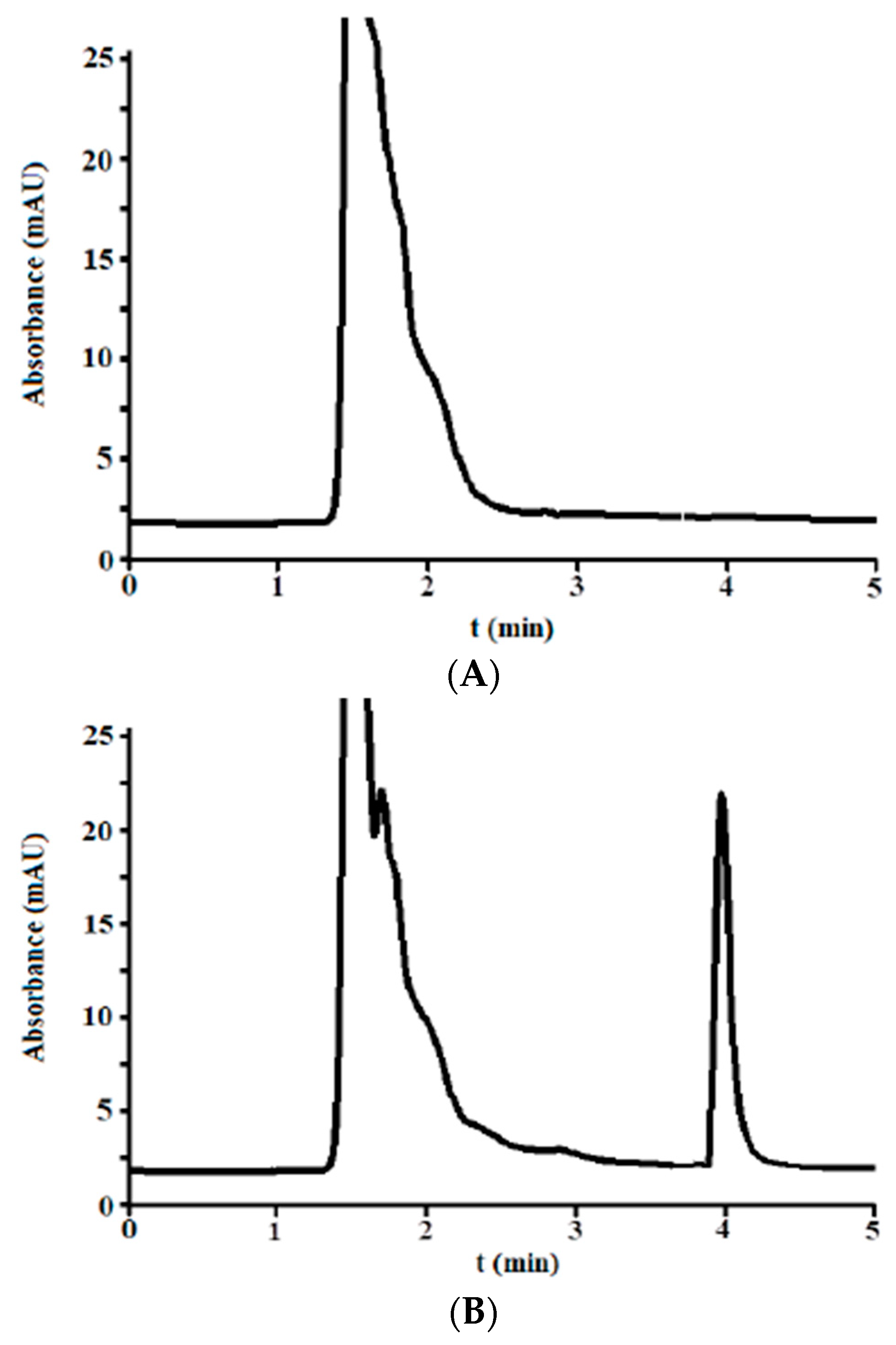
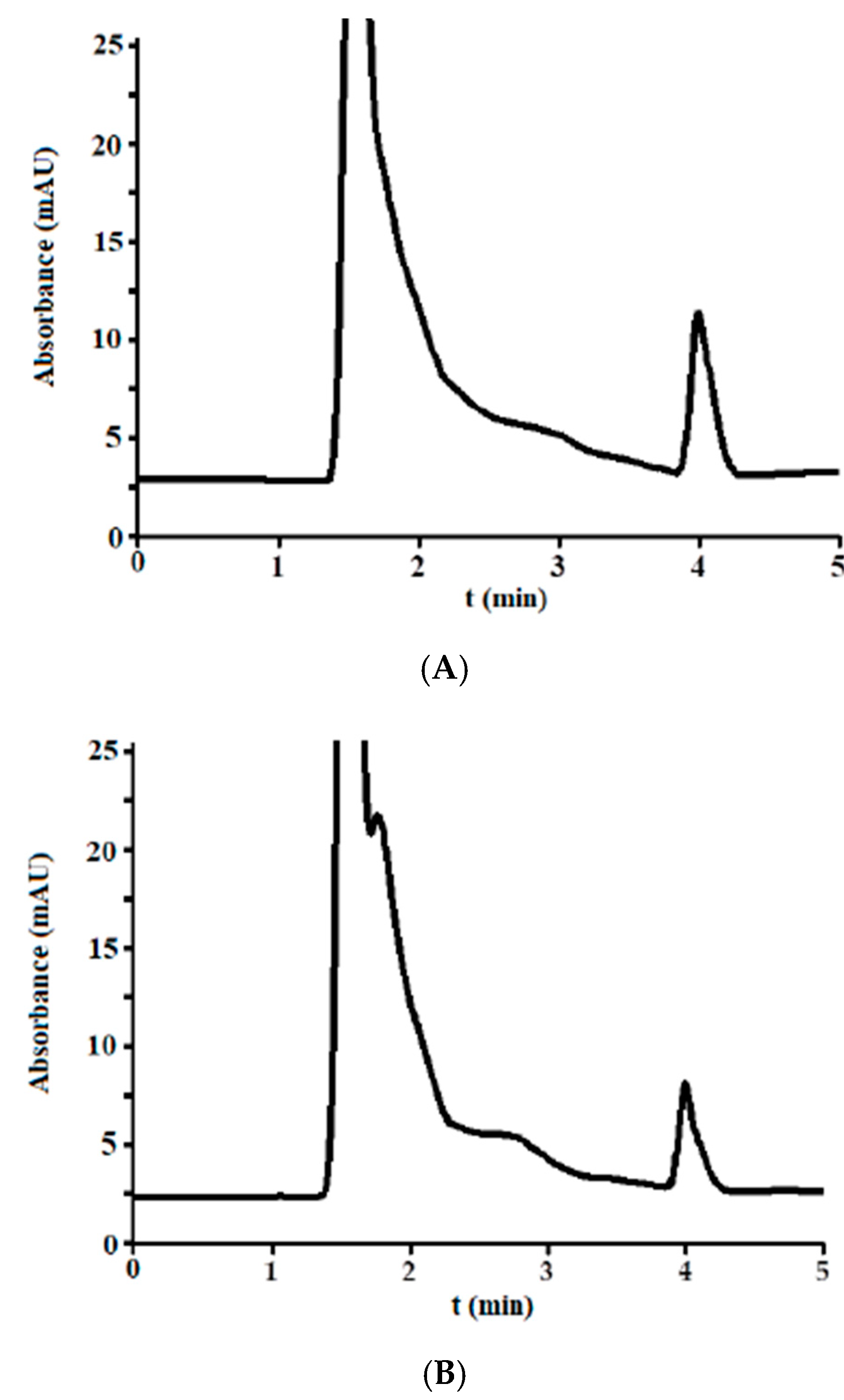
3.2.1. Selectivity
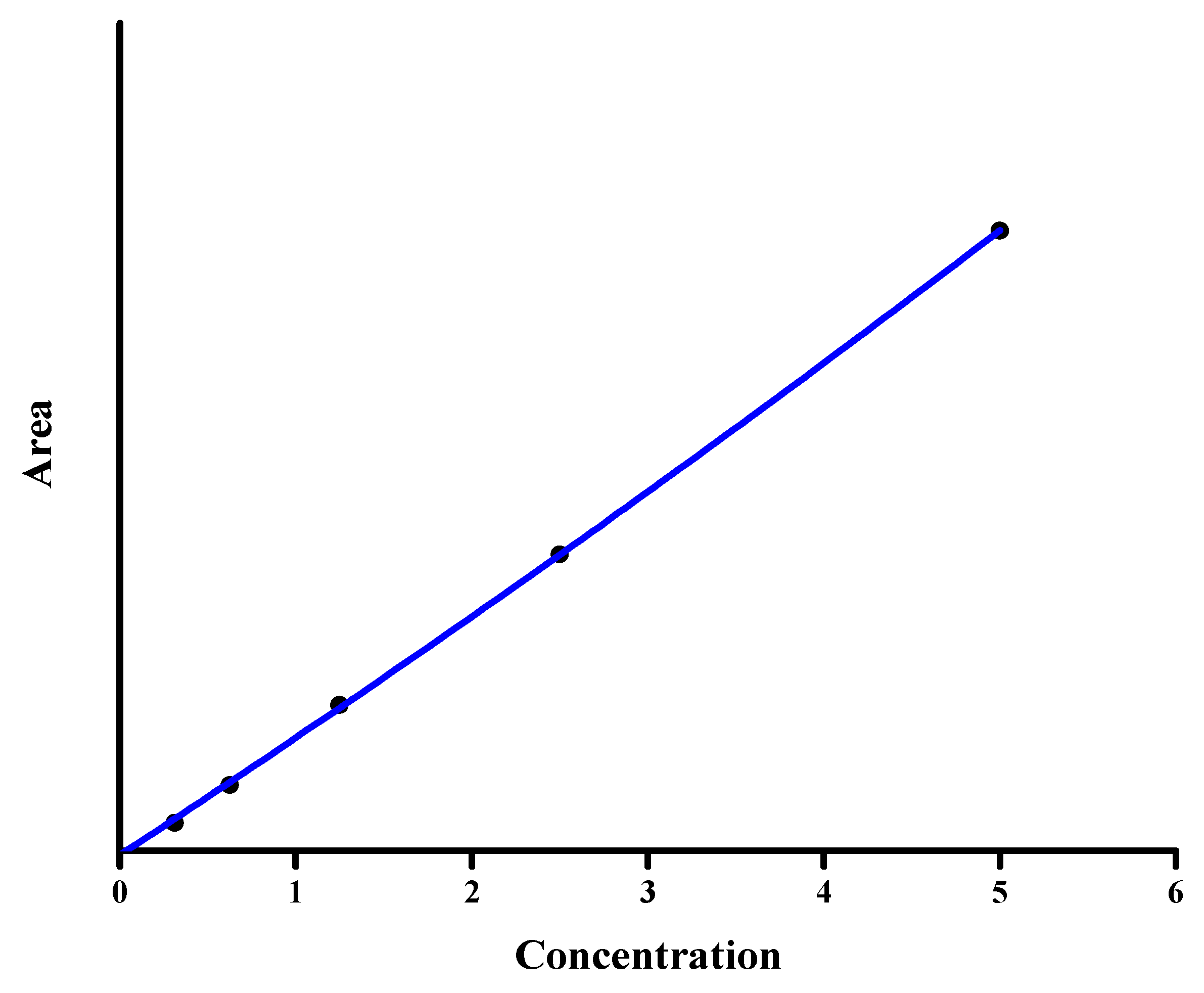
3.2.2. Linearity and Sensitivity
3.2.3. Detection and Quantification Limit
3.2.4. Precision and Accuracy
3.2.5. Carry-Over Effect
3.2.6. Matrix Effects
3.2.7. Dilution Integrity
3.2.8. Robustness
3.3. Analysis of Incurred Urine Samples
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Badou, F.; Raynaud, C.; Ramos, C.; Laneelle, M.A.; Laneelle, G. Mechanism of isoniazid uptake in Mycobacterium tuberculosis. Microbiology 1998, 144, 2539–2544. [Google Scholar]
- Catherine, V.F.W.; Masayoshi, A.; Hernando Hazbón, M.; Colangeli, R.; Kremer, L.; Weisbrod, R.; Alland, D.; Sacchettini, J.C.; Jacobs, W.R. Transfer of a point mutation in Mycobacterium tuberculosis inhA resolves the target of isoniazid. Nat. Med. 2006, 12, 1027–1029. [Google Scholar]
- Ruth Amos, I.J.; Gourlay, S.B.; Schiesser, H.C.; Smith, A.J.; Yatesa, F.B. A mechanistic study on the oxidation of hydrazides: Application to the tuberculosis drug isoniazid. Chem. Commun. 2008, 14, 1695–1697. [Google Scholar] [CrossRef]
- Gordan, A.; Ellardand, T.; Gammon, P. Pharmacokinetics of isoniazid metabolism in man. J. Pharmacokinet. Biopharm. 1976, 42, 83–113. [Google Scholar]
- Addington, W.W. The Side Effects and Interactions of Antituberculosis Drugs. CHEST J. 1979, 76, 782–784. [Google Scholar] [CrossRef]
- Shewiyo, D.H.; Kaale, E.; Risha, P.G.; Dejaegher, B.; Smeyers-Verbeke, J.; Heyden, Y.V. Optimization of a reversed-phase-high-performance thin-layer chromatography method for the separation of isoniazid, ethambutol, rifampicin and pyrazinamide in fixed-dose combination antituberculosis tablets. J. Chromatogr. A 2012, 60, 232–238. [Google Scholar] [CrossRef]
- Ali, J.; Ali, N.; Sultana, Y.; Baboota, S.; Faiyaz, S. Development and validation of stability indicating HPTLC method for analysis of antitubercular drugs. Acta Chromatogr. 2007, 18, 168–179. [Google Scholar]
- Dhal, S.K.; Sharma, R. Development and Validation of RP HPLC Method for Simultaneous Determination of Pyridoxine Hydrochrloride, Isoniazid, Pyrazinamide and Rifampicin in pharmaceutical Formulation. Chem. Anal. 2009, 54, 1487–1490. [Google Scholar]
- Prasanthi, B.; Vijaya Ratna, J.; Phani, R.S.C. Development and validation of RP-HPLC method for simultaneous estimation of rifampicin, isoniazid and pyrazinamide in human plasma. J. Anal. Chem. 2015, 70, 1015–1022. [Google Scholar] [CrossRef]
- Calleri, E.; De Lorenzi, E.; Furlanetto, S.; Massolini, G.; Caccialanza, G. Validation of a RP-LC method for the simultaneous determination of isoniazid, pyrazinamide and rifampicin in a pharmaceutical formulation. J. Pharm. Biomed. Anal. 2002, 29, 1089–1096. [Google Scholar] [CrossRef]
- Hemnath, A.K.; Sudha, V.; Ramchandran, G. Simple and rapid liquid chromatography method for simultaneous determination of isoniazid and pyrazinamide in plasma. SAARC J. Tuberc. 2012, 9, 13–18. [Google Scholar] [CrossRef]
- Hee, K.H.; Seo, J.J.; Lee, L.S. Development and validation of liquid chromatography tandem mass spectrometry method for simultaneous quantification of first line tuberculosis drugs and metabolites in human plasma and its application in clinical study. J. Pharm. Biomed. Anal. 2015, 102, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Song, B.; Jiang, H.; Yu, K.; Zhong, D. A liquid chromatography/tandem mass spectrometry method for the simultaneous quantification of isoniazid and ethambutol in human plasma. Rapid Commun. Mass Spectrom. 2005, 19, 2591–2596. [Google Scholar] [CrossRef] [PubMed]
- Esteve-Romero, J.; Albiol-Chiva, J.; Peris-Vicente, J. A review on development of analytical methods to determine monitorable drugs in serum and urine by micellar liquid chromatography using direct injection. Anal. Chim. Acta 2016, 926, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Juan Peris-Vicente, J.; Albiol-Chiva, J.; Roca-Genovés, P.; Esteve-Romero, J. Advances on melamine determination by micellar liquid chromatography: A review. J. Liq. Chromatogr. Relat. Technol. 2016, 39, 325–338. [Google Scholar] [CrossRef]
- Ochoa-Aranda, E.; Esteve-Romero, J.; Rambla-Alegre, M.; Peris-Vicente, J.; Bose, D. Development of a methodology to quantify tamoxifen and endoxifen in breast cancer patients by micellar liquid chromatography and validation according to the ICH guidelines. Talanta 2011, 84, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Dubey, S.; Shukla, S.K.; Durgbanshi, A.; Esteve-Romero, J.; Bose, D. Simultaneous determination of three traditional and two novel Antiepilectic Drugs using Micellar Liquid Chromatography. Int. J. Anal. Bioanal. Chem. 2013, 3, 1–8. [Google Scholar]
- Agrawal, N.; Esteve-Romero, J.; Bose, D.; Dubey, N.P.; Peris-Vicente, J.; Carda-Broch, S. Determination of selective serotonin reuptake inhibitors in plasma and urine by micellar liquid chromatography coupled to fluorescence detection. J. Chromatogr. B 2014, 965, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Marco-Peiró, S.; Esteve-Romero, J.; Durgbanshi, A.; Bose, D.; Peris-Vicente, J.; Carda-Broch, S. Determination of Paroxetine in Blood and Urine Using Micellar Liquid Chromatography with Electrochemical Detection. J. Chromatogr. Sci. 2014, 52, 1217–1223. [Google Scholar] [CrossRef]
- Peris-Vicente, J.; Ochoa-Aranda, E.; Bose, D.; Esteve-Romero, J. Determination of tamoxifen and its main metabolites in plasma samples from breast cancer patients by micellar liquid chromatography. Talanta 2015, 131, 535–540. [Google Scholar] [CrossRef]
- Committee for Medicinal Products for Human Use. Guideline on Bioanalytical Method Validation; European Medicines Agency: London, UK, 2011; Available online: https://www.ema.europa.eu/en/bioanalytical-method-validation (accessed on 27 November 2018).
- Peris-Vicente, J.; Esteve-Romero, J.; Carda-Broch, S. Validation of analytical methods based on chromatographic techniques: An overview. Anal. Sep. Sci. 2015, 5, 1757–1808. [Google Scholar]
- Garrido-Cano, I.; García-García, A.; Peris-Vicente, J.; Ochoa-Aranda, E.; Esteve-Romero, J. A method to quantify several tyrosine kinase inhibitors in plasma by micellar liquid chromatography and validation according to the European Medicines Agency guidelines. Talanta 2015, 144, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Snyder, L.R.; Kirkland, J.J. Introduction to Modern Liquid Chromatography, 2nd ed.; Wiley: New York, NY, USA, 1979. [Google Scholar]
- Stets, S.; Tavares, T.M.; Peralta-Zamora, P.G.; Pessoa, C.A.; Nagata, N. Simultaneous Determination of Rifampicin and Isoniazid in Urine and Pharmaceutical Formulations by Multivariate Visible Spectrophotometry. J. Braz. Chem. Soc. 2013, 24, 1198–1205. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Trung Dung, N. Determination of isoniazid in human urine by spectrophotometric method. Vietnam J. Sci. Technol. 2015, 53, 780–788. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Matrix | Concentration (μg/mL) | Intraday a | Interday b | ||
|---|---|---|---|---|---|
| Accuracy (ε, %) | Precision (RSD, %) | Accuracy (ε, %) | Precision (RSD, %) | ||
| Urine | 0.03 | +8.5 | 9.3 | +6.9 | 16.0 |
| 0.25 | +3.2 | 6.6 | +2.5 | 7.5 | |
| 2.5 | −0.9 | 2.4 | −0.4 | 2.9 | |
| Urine (dilution integrity) | 15 | +1.9 | 3.2 | - | - |
| Working solution | 0.006 | +7.3 | 7.5 | - | - |
| 0.05 | +3.0 | 5.2 | - | - | |
| 0.5 | −0.2 | 2.0 | - | - | |
| Chromatographic Condition | Interval | tR (min) | N | B/A |
|---|---|---|---|---|
| Flow rate (mL/min) | 0.9–1.1 | 12.1 | 0.9 | 1.2 |
| pH | 6.8–7.2 | 0.1 | 2.6 | 1.6 |
| 1-propanol proportion (%) | 12–13 | 0.1 | 1.5 | 2.3 |
| SDS concentration (M) | 0.14–0.16 | 0.7 | 0.7 | 2.0 |
| Patient Number | Amount of Isoniazid µg/mL |
|---|---|
| Patient 1 | 2.9 |
| Patient 2 | 1.34 |
| Patient 3 | 4.31 |
| Patient 4 | 8.45 |
| Patient 5 | 2.98 |
| Patient 6 | 3.45 |
| Patient 7 | 5.34 |
| Patient 8 | 2.84 |
| Patient 9 | 7.23 |
| Patient 10 | 6.89 |
| Patient 11 | 3.14 |
| Patient 12 | 5.48 |
| Patient 13 | 5.39 |
| Patient 14 | 6.78 |
| Patient 15 | 3.87 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mishra, P.; Albiol-Chiva, J.; Bose, D.; Durgbanshi, A.; Peris-Vicente, J.; Carda-Broch, S.; Esteve-Romero, J. Optimization and Validation of a Chromatographic Method for the Quantification of Isoniazid in Urine of Tuberculosis Patients According to the European Medicines Agency Guideline. Antibiotics 2018, 7, 107. https://doi.org/10.3390/antibiotics7040107
Mishra P, Albiol-Chiva J, Bose D, Durgbanshi A, Peris-Vicente J, Carda-Broch S, Esteve-Romero J. Optimization and Validation of a Chromatographic Method for the Quantification of Isoniazid in Urine of Tuberculosis Patients According to the European Medicines Agency Guideline. Antibiotics. 2018; 7(4):107. https://doi.org/10.3390/antibiotics7040107
Chicago/Turabian StyleMishra, Pooja, Jaume Albiol-Chiva, Devasish Bose, Abhilasha Durgbanshi, Juan Peris-Vicente, Samuel Carda-Broch, and Josep Esteve-Romero. 2018. "Optimization and Validation of a Chromatographic Method for the Quantification of Isoniazid in Urine of Tuberculosis Patients According to the European Medicines Agency Guideline" Antibiotics 7, no. 4: 107. https://doi.org/10.3390/antibiotics7040107
APA StyleMishra, P., Albiol-Chiva, J., Bose, D., Durgbanshi, A., Peris-Vicente, J., Carda-Broch, S., & Esteve-Romero, J. (2018). Optimization and Validation of a Chromatographic Method for the Quantification of Isoniazid in Urine of Tuberculosis Patients According to the European Medicines Agency Guideline. Antibiotics, 7(4), 107. https://doi.org/10.3390/antibiotics7040107