Establishing Genotype-to-Phenotype Relationships in Bacteria Causing Hospital-Acquired Pneumonia: A Prelude to the Application of Clinical Metagenomics

 ,
,
Abstract
1. Introduction
2. From Genotype to Phenotype
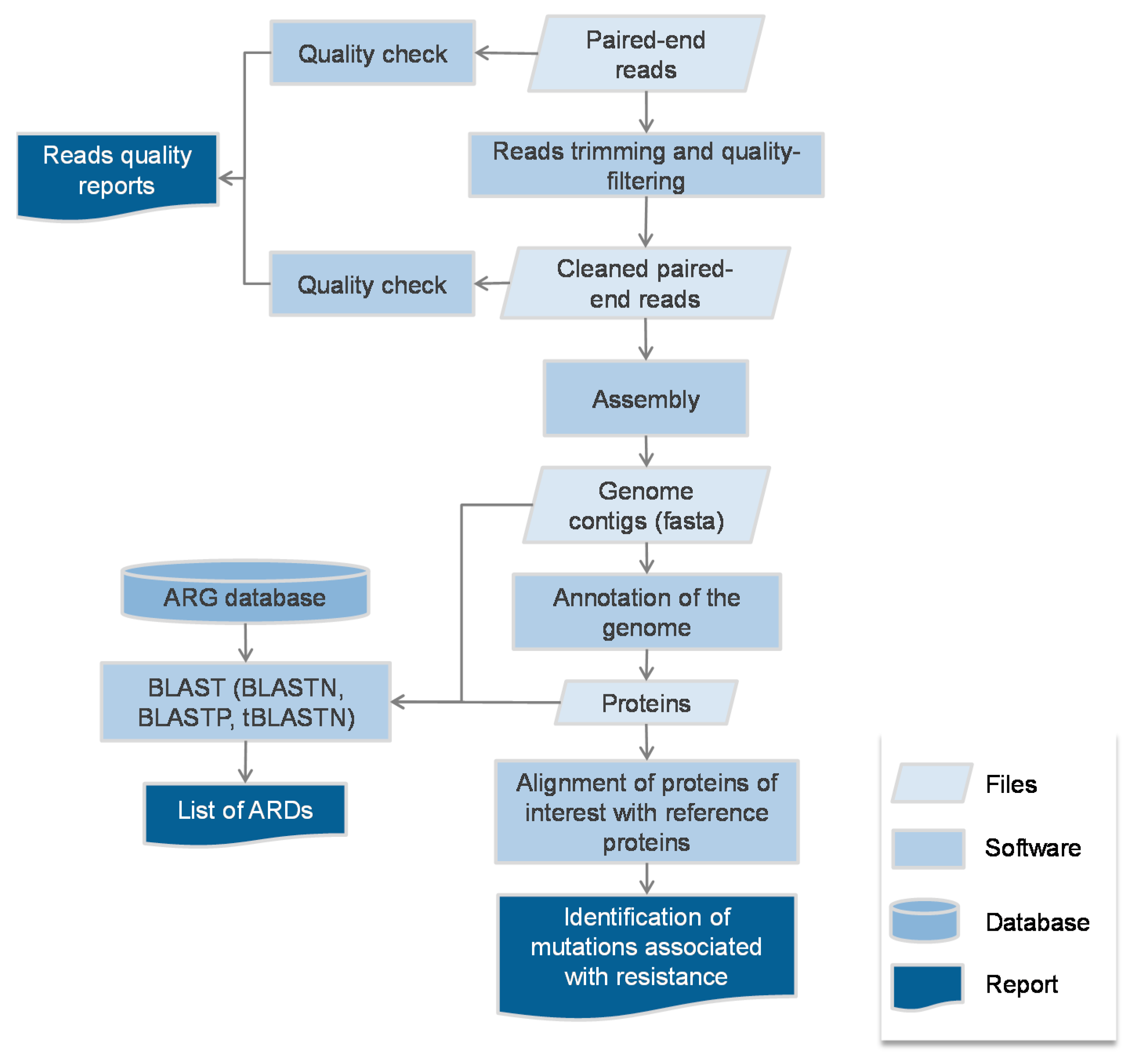
2.1. Protocol
2.2. Escherichia coli
2.3. Klebsiella pneumoniae
2.4. Other Enterobacteriaceae Involved in HAP
2.5. Pseudomonas aeruginosas
2.6. Acinetobacter baumannii
2.7. Stenotrophomonas maltophilia
2.8. Staphylococcus aureus
3. Discussion
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Ruppé, E.; Baud, D.; Schicklin, S.; Guigon, G.; Schrenzel, J. Clinical metagenomics for the management of hospital- and healthcare-acquired pneumonia. Future Microbiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Pendleton, K.M.; Erb-Downward, J.R.; Bao, Y.; Branton, W.R.; Falkowski, N.R.; Newton, D.W.; Huffnagle, G.B.; Dickson, R.P. Rapid Pathogen Identification in Bacterial Pneumonia Using Real-time Metagenomics. Am. J. Respir. Crit. Care Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Langelier, C.; Zinter, M.S.; Kalantar, K.; Yanik, G.A.; Christenson, S.; O’Donovan, B.; White, C.; Wilson, M.; Sapru, A.; Dvorak, C.C.; et al. Metagenomic Sequencing Detects Respiratory Pathogens in Hematopoietic Cellular Transplant Patients. Am. J. Respir. Crit. Care Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Leo, S.; Gaïa, N.; Ruppé, E.; Emonet, S.; Girard, M.; Lazarevic, V.; Schrenzel, J. Detection of Bacterial Pathogens from Broncho-Alveolar Lavage by Next-Generation Sequencing. Int. J. Mol. Sci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hasman, H.; Saputra, D.; Sicheritz-Ponten, T.; Lund, O.; Svendsen, C.A.; Frimodt-Møller, N.; Aarestrup, F.M. Rapid whole-genome sequencing for detection and characterization of microorganisms directly from clinical samples. J. Clin. Microbiol. 2014, 52, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.; Mwaigwisya, S.; Crossman, L.C.; Doumith, M.; Munroe, D.; Pires, C.; Khan, A.M.; Woodford, N.; Saunders, N.J.; Wain, J.; et al. Identification of bacterial pathogens and antimicrobial resistance directly from clinical urines by nanopore-based metagenomic sequencing. J. Antimicrob. Chemother. 2016. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.R.; Naccache, S.N.; Samayoa, E.; Biagtan, M.; Bashir, H.; Yu, G.; Salamat, S.M.; Somasekar, S.; Federman, S.; Miller, S.; et al. Actionable diagnosis of neuroleptospirosis by next-generation sequencing. N. Engl. J. Med. 2014, 370, 2408–2417. [Google Scholar] [CrossRef] [PubMed]
- Frémond, M.-L.; Pérot, P.; Muth, E.; Cros, G.; Dumarest, M.; Mahlaoui, N.; Seilhean, D.; Desguerre, I.; Hébert, C.; Corre-Catelin, N.; et al. Next-Generation Sequencing for Diagnosis and Tailored Therapy: A Case Report of Astrovirus-Associated Progressive Encephalitis. J. Pediatr. Infect. Dis. Soc. 2015, 4, e53–e57. [Google Scholar] [CrossRef] [PubMed]
- Gyarmati, P.; Kjellander, C.; Aust, C.; Song, Y.; Öhrmalm, L.; Giske, C.G. Metagenomic analysis of bloodstream infections in patients with acute leukemia and therapy-induced neutropenia. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Grumaz, S.; Stevens, P.; Grumaz, C.; Decker, S.O.; Weigand, M.A.; Hofer, S.; Brenner, T.; von Haeseler, A.; Sohn, K. Next-generation sequencing diagnostics of bacteremia in septic patients. Genome Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Parize, P.; Muth, E.; Richaud, C.; Gratigny, M.; Pilmis, B.; Lamamy, A.; Mainardi, J.L.; Cheval, J.; de Visser, L.; Jagorel, F.; et al. Untargeted next-generation sequencing-based first-line diagnosis of infection in immunocompromised adults: A multicentre, blinded, prospective study. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ruppe, E.; Lazarevic, V.; Girard, M.; Mouton, W.; Ferry, T.; Laurent, F.; Schrenzel, J. Clinical metagenomics of bone and joint infections: A proof of concept study. Sci. Rep. 2017. [Google Scholar] [CrossRef] [PubMed]
- Street, T.L.; Sanderson, N.D.; Atkins, B.L.; Brent, A.J.; Cole, K.; Foster, D.; McNally, M.A.; Oakley, S.; Peto, L.; Taylor, A.; et al. Molecular Diagnosis of Orthopedic-Device-Related Infection Directly from Sonication Fluid by Metagenomic Sequencing. J. Clin. Microbiol. 2017, 55, 2334–2347. [Google Scholar] [CrossRef] [PubMed]
- Thoendel, M.; Jeraldo, P.; Greenwood-Quaintance, K.E.; Chia, N.; Abdel, M.P.; Steckelberg, J.M.; Osmon, D.R.; Patel, R. A Possible Novel Prosthetic Joint Infection Pathogen, Mycoplasma salivarium, Identified by Metagenomic Shotgun Sequencing. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bodemer, C.; Sauvage, V.; Mahlaoui, N.; Cheval, J.; Couderc, T.; Leclerc-Mercier, S.; Debré, M.; Pellier, I.; Gagnieur, L.; Fraitag, S.; et al. Live rubella virus vaccine long-term persistence as an antigenic trigger of cutaneous granulomas in patients with primary immunodeficiency. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2014, 20, O656–O663. [Google Scholar] [CrossRef] [PubMed]
- Thoendel, M.; Jeraldo, P.R.; Greenwood-Quaintance, K.E.; Yao, J.Z.; Chia, N.; Hanssen, A.D.; Abdel, M.P.; Patel, R. Comparison of microbial DNA enrichment tools for metagenomic whole genome sequencing. J. Microbiol. Methods 2016. [Google Scholar] [CrossRef] [PubMed]
- Xavier, B.B.; Das, A.J.; Cochrane, G.; Ganck, S.D.; Kumar-Singh, S.; Aarestrup, F.M.; Goossens, H.; Malhotra-Kumar, S. Consolidating and Exploring Antibiotic Resistance Gene Data Resources. J. Clin. Microbiol. 2016, 54, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Thoendel, M.; Jeraldo, P.; Greenwood-Quaintance, K.E.; Yao, J.; Chia, N.; Hanssen, A.D.; Abdel, M.P.; Patel, R. Impact of Contaminating DNA in Whole-Genome Amplification Kits Used for Metagenomic Shotgun Sequencing for Infection Diagnosis. J. Clin. Microbiol. 2017, 55, 1789–1801. [Google Scholar] [CrossRef] [PubMed]
- Personne, Y.; Ozongwu, C.; Platt, G.; Basurto-Lozada, P.; Shamin, M.; Gant, V.A.; Zumla, A.; Enne, V.I. “Sample-in, answer-out”? Evaluation and comprehensive analysis of the Unyvero P50 pneumonia assay. Diagn. Microbiol. Infect. Dis. 2016, 86, 5–10. [Google Scholar]
- Altun, O.; Almuhayawi, M.; Ullberg, M.; Ozenci, V. Clinical evaluation of the FilmArray blood culture identification panel in identification of bacteria and yeasts from positive blood culture bottles. J. Clin. Microbiol. 2013, 51, 4130–4136. [Google Scholar] [CrossRef] [PubMed]
- American Thoracic Society, and Infectious Diseases Society of America. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am. J. Respir. Crit. Care Med. 2005, 171, 388–416. [Google Scholar]
- Barbier, F.; Andremont, A.; Wolff, M.; Bouadma, L. Hospital-acquired pneumonia and ventilator-associated pneumonia: Recent advances in epidemiology and management. Curr. Opin. Pulm. Med. 2013, 19, 216–228. [Google Scholar] [CrossRef] [PubMed]
- Emonet, S.; Lazarevic, V.; Pugin, J.; Schrenzel, J.; Ruppé, E. Clinical Metagenomics for the Diagnostic of Hospital-acquired Infections: Promises and Hurdles. Am. J. Respir. Crit. Care Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef] [PubMed]
- McArthur, A.G.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Bhullar, K.; Canova, M.J.; De Pascale, G.; Ejim, L.; et al. The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 2013, 57, 3348–3357. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Padmanabhan, B.R.; Diene, S.M.; Lopez-Rojas, R.; Kempf, M.; Landraud, L.; Rolain, J.-M. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother. 2014, 58, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Gordon, N.C.; Price, J.R.; Cole, K.; Everitt, R.; Morgan, M.; Finney, J.; Kearns, A.M.; Pichon, B.; Young, B.; Wilson, D.J.; et al. Prediction of Staphylococcus aureus antimicrobial resistance by whole-genome sequencing. J. Clin. Microbiol. 2014, 52, 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- Gibson, M.K.; Forsberg, K.J.; Dantas, G. Improved annotation of antibiotic resistance determinants reveals microbial resistomes cluster by ecology. ISME J. 2015, 9, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Pesesky, M.W.; Hussain, T.; Wallace, M.; Patel, S.; Andleeb, S.; Burnham, C.-A.D.; Dantas, G. Evaluation of Machine Learning and Rules-Based Approaches for Predicting Antimicrobial Resistance Profiles in Gram-negative Bacilli from Whole Genome Sequence Data. Front. Microbiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Hunt, M.; Mather, A.E.; Sánchez-Busó, L.; Page, A.J.; Parkhill, J.; Keane, J.A.; Harris, S.R. ARIBA: Rapid antimicrobial resistance genotyping directly from sequencing reads. Microb. Genom. 2017. [Google Scholar] [CrossRef] [PubMed]
- Lakin, S.M.; Dean, C.; Noyes, N.R.; Dettenwanger, A.; Ross, A.S.; Doster, E.; Rovira, P.; Abdo, Z.; Jones, K.L.; Ruiz, J.; et al. MEGARes: An antimicrobial resistance database for high throughput sequencing. Nucleic Acids Res. 2017, 45, D574–D580. [Google Scholar] [CrossRef] [PubMed]
- Zankari, E.; Hasman, H.; Kaas, R.S.; Seyfarth, A.M.; Agersø, Y.; Lund, O.; Larsen, M.V.; Aarestrup, F.M. Genotyping using whole-genome sequencing is a realistic alternative to surveillance based on phenotypic antimicrobial susceptibility testing. J. Antimicrob. Chemother. 2013, 68, 771–777. [Google Scholar] [CrossRef] [PubMed]
- Stoesser, N.; Batty, E.M.; Eyre, D.W.; Morgan, M.; Wyllie, D.H.; Del Ojo Elias, C.; Johnson, J.R.; Walker, A.S.; Peto, T.E.A.; Crook, D.W. Predicting antimicrobial susceptibilities for Escherichia coli and Klebsiella pneumoniae isolates using whole genomic sequence data. J. Antimicrob. Chemother. 2013, 68, 2234–2244. [Google Scholar] [CrossRef] [PubMed]
- Tyson, G.H.; McDermott, P.F.; Li, C.; Chen, Y.; Tadesse, D.A.; Mukherjee, S.; Bodeis-Jones, S.; Kabera, C.; Gaines, S.A.; Loneragan, G.H.; et al. WGS accurately predicts antimicrobial resistance in Escherichia coli. J. Antimicrob. Chemother. 2015. [Google Scholar] [CrossRef] [PubMed]
- Jaurin, B.; Grundström, T. ampC cephalosporinase of Escherichia coli K-12 has a different evolutionary origin from that of beta-lactamases of the penicillinase type. Proc. Natl. Acad. Sci. USA 1981, 78, 4897–4901. [Google Scholar] [CrossRef] [PubMed]
- Jaurin, B.; Grundström, T.; Normark, S. Sequence elements determining AmpC promoter strength in E. coli. EMBO J. 1982, 1, 875–881. [Google Scholar] [PubMed]
- Caroff, N.; Espaze, E.; Bérard, I.; Richet, H.; Reynaud, A. Mutations in the AmpC promoter of Escherichia coli isolates resistant to oxyiminocephalosporins without extended spectrum beta-lactamase production. FEMS Microbiol. Lett. 1999, 173, 459–465. [Google Scholar] [PubMed]
- Caroff, N.; Espaze, E.; Gautreau, D.; Richet, H.; Reynaud, A. Analysis of the effects of -42 and -32 AmpC promoter mutations in clinical isolates of Escherichia coli hyperproducing ampC. J. Antimicrob. Chemother. 2000, 45, 783–788. [Google Scholar] [CrossRef] [PubMed]
- Ruppé, E.; Olearo, F.; Pires, D.; Baud, D.; Renzi, G.; Cherkaoui, A.; Goldenberger, D.; Huttner, A.; François, P.; Harbarth, S.; et al. Clonal or not clonal? Investigating hospital outbreaks of KPC-producing Klebsiella pneumoniae with whole-genome sequencing. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kos, V.N.; Déraspe, M.; McLaughlin, R.E.; Whiteaker, J.D.; Roy, P.H.; Alm, R.A.; Corbeil, J.; Gardner, H. The resistome of Pseudomonas aeruginosa in relationship to phenotypic susceptibility. Antimicrob. Agents Chemother. 2015, 59, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Köser, C.U.; Holden, M.T.G.; Ellington, M.J.; Cartwright, E.J.P.; Brown, N.M.; Ogilvy-Stuart, A.L.; Hsu, L.Y.; Chewapreecha, C.; Croucher, N.J.; Harris, S.R.; et al. Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak. N. Engl. J. Med. 2012, 366, 2267–2275. [Google Scholar] [CrossRef] [PubMed]
- Bradley, P.; Gordon, N.C.; Walker, T.M.; Dunn, L.; Heys, S.; Huang, B.; Earle, S.; Pankhurst, L.J.; Anson, L.; de Cesare, M.; et al. Rapid antibiotic-resistance predictions from genome sequence data for Staphylococcus aureus and Mycobacterium tuberculosis. Nat. Commun. 2015. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.C.; Long, S.W.; Musser, J.M.; Beres, S.B.; Olsen, R.J.; Dallas, S.D.; Nunez, Y.O.; Frei, C.R. Comparative whole genome sequencing of community-associated methicillin-resistant Staphylococcus aureus sequence type 8 from primary care clinics in a texas community. Pharmacotherapy 2015, 35, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Haeggman, S.; Löfdahl, S.; Paauw, A.; Verhoef, J.; Brisse, S. Diversity and evolution of the class a chromosomal beta-lactamase gene in Klebsiella pneumoniae. Antimicrob. Agents Chemother. 2004, 48, 2400–2408. [Google Scholar] [CrossRef] [PubMed]
- Knott-Hunziker, V.; Petursson, S.; Waley, S.G.; Jaurin, B.; Grundström, T. The acyl-enzyme mechanism of beta-lactamase action. The evidence for class C Beta-lactamases. Biochem. J. 1982, 207, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Girlich, D.; Naas, T.; Nordmann, P. Biochemical characterization of the naturally occurring oxacillinase OXA-50 of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2004, 48, 2043–2048. [Google Scholar] [CrossRef] [PubMed]
- Hächler, H.; Santanam, P.; Kayser, F.H. Sequence and characterization of a novel chromosomal aminoglycoside phosphotransferase gene, aph (3’)-IIb, in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 1996, 40, 1254–1256. [Google Scholar] [PubMed]
- El Zowalaty, M.E.; Al Thani, A.A.; Webster, T.J.; El Zowalaty, A.E.; Schweizer, H.P.; Nasrallah, G.K.; Marei, H.E.; Ashour, H.M. Pseudomonas aeruginosa: Arsenal of resistance mechanisms, decades of changing resistance profiles, and future antimicrobial therapies. Future Microbiol. 2015, 10, 1683–1706. [Google Scholar] [CrossRef] [PubMed]
- Trias, J.; Nikaido, H. Outer membrane protein D2 catalyzes facilitated diffusion of carbapenems and penems through the outer membrane of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 1990, 34, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Jaillard, M.; van Belkum, A.; Cady, K.C.; Creely, D.; Shortridge, D.; Blanc, B.; Barbu, E.M.; Dunne, W.M.; Zambardi, G.; Enright, M.; et al. Correlation between phenotypic antibiotic susceptibility and the resistome in Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 2017, 50, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Bou, G.; Martínez-Beltrán, J. Cloning, Nucleotide Sequencing, and Analysis of the Gene Encoding an AmpC β-Lactamase in Acinetobacter baumannii. Antimicrob. Agents Chemother. 2000, 44, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.; Young, H.K.; Amyes, S.G.B. Characterisation of OXA-51, a novel class D carbapenemase found in genetically unrelated clinical strains of Acinetobacter baumannii from Argentina. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2005, 11, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Magnet, S.; Courvalin, P.; Lambert, T. Resistance-nodulation-cell division-type efflux pump involved in aminoglycoside resistance in Acinetobacter baumannii strain BM4454. Antimicrob. Agents Chemother. 2001, 45, 3375–3380. [Google Scholar] [CrossRef] [PubMed]
- Segal, H.; Jacobson, R.K.; Garny, S.; Bamford, C.M.; Elisha, B.G. Extended -10 promoter in ISAba-1 upstream of blaOXA-23 from Acinetobacter baumannii. Antimicrob. Agents Chemother. 2007, 51, 3040–3041. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, A.; Avison, M.B. Induction of L1 and L2 beta-lactamase production in Stenotrophomonas maltophilia is dependent on an AmpR-type regulator. Antimicrob. Agents Chemother. 2008, 52, 1525–1528. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.L.; Chen, H.F.; Chang, C.Y.; Lee, T.M.; Wu, W.J. Contribution of integrons, and SmeABC and SmeDEF efflux pumps to multidrug resistance in clinical isolates of Stenotrophomonas maltophilia. J. Antimicrob. Chemother. 2004, 53, 518–521. [Google Scholar] [CrossRef] [PubMed]
- Rahmati-Bahram, A.; Magee, J.T.; Jackson, S.K. Temperature-dependent aminoglycoside resistance in Stenotrophomonas (Xanthomonas) maltophilia; alterations in protein and lipopolysaccharide with growth temperature. J. Antimicrob. Chemother. 1996, 37, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Rahmati-Bahram, A.; Magee, J.T.; Jackson, S.K. Effect of temperature on aminoglycoside binding sites in Stenotrophomonas maltophilia. J. Antimicrob. Chemother. 1997, 39, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Ribera, A.; Doménech-Sanchez, A.; Ruiz, J.; Benedi, V.J.; Jimenez de Anta, M.T.; Vila, J. Mutations in gyrA and parC QRDRs are not relevant for quinolone resistance in epidemiological unrelated Stenotrophomonas maltophilia clinical isolates. Microb. Drug Resist. Larchmt. N. Y. 2002, 8, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Valdezate, S.; Vindel, A.; Saéz-Nieto, J.A.; Baquero, F.; Cantón, R. Preservation of topoisomerase genetic sequences during in vivo and in vitro development of high-level resistance to ciprofloxacin in isogenic Stenotrophomonas maltophilia strains. J. Antimicrob. Chemother. 2005, 56, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.F.; Chang, X.; Ye, Y.; Wang, Z.X.; Shao, Y.B.; Shi, W.; Li, X.; Li, J.B. Stenotrophomonas maltophilia resistance to trimethoprim/sulfamethoxazole mediated by acquisition of sul and dfrA genes in a plasmid-mediated class 1 integron. Int. J. Antimicrob. Agents 2011, 37, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, C.C.; Sevitt, S.; Topley, E. Acute osteomyelitis due to a penicillinase-producing Staphylococcus aureus. Lancet Lond. Engl. 1949, 1, 259–261. [Google Scholar] [CrossRef]
- Katayama, Y.; Ito, T.; Hiramatsu, K. A new class of genetic element, staphylococcus cassette chromosome mec, encodes methicillin resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 2000, 44, 1549–1555. [Google Scholar] [CrossRef] [PubMed]
- White, D.G.; Alekshun, M.N.; McDermott, P.F. Frontiers in Antimicrobial Resistance: A Tribute to Stuart B. Levy; ASM Press: Washington, DC, USA, 2005. [Google Scholar]
- Cui, L.; Ma, X.; Sato, K.; Okuma, K.; Tenover, F.C.; Mamizuka, E.M.; Gemmell, C.G.; Kim, M.-N.; Ploy, M.-C.; El-Solh, N.; et al. Cell wall thickening is a common feature of vancomycin resistance in Staphylococcus aureus. J. Clin. Microbiol. 2003, 41, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, M.; Kuroda, H.; Oshima, T.; Takeuchi, F.; Mori, H.; Hiramatsu, K. Two-component system VraSR positively modulates the regulation of cell-wall biosynthesis pathway in Staphylococcus aureus. Mol. Microbiol. 2003, 49, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Maki, H.; McCallum, N.; Bischoff, M.; Wada, A.; Berger-Bächi, B. tcaA inactivation increases glycopeptide resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 2004, 48, 1953–1959. [Google Scholar] [CrossRef] [PubMed]
- Vidaillac, C.; Gardete, S.; Tewhey, R.; Sakoulas, G.; Kaatz, G.W.; Rose, W.E.; Tomasz, A.; Rybak, M.J. Alternative Mutational Pathways to Intermediate Resistance to Vancomycin in Methicillin-Resistant Staphylococcus aureus. J. Infect. Dis. 2013, 208, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Toh, S.-M.; Xiong, L.; Arias, C.A.; Villegas, M.V.; Lolans, K.; Quinn, J.; Mankin, A.S. Acquisition of a natural resistance gene renders a clinical strain of methicillin-resistant Staphylococcus aureus resistant to the synthetic antibiotic linezolid. Mol. Microbiol. 2007, 64, 1506–1514. [Google Scholar] [CrossRef] [PubMed]
- Marshall, S.H.; Donskey, C.J.; Hutton-Thomas, R.; Salata, R.A.; Rice, L.B. Gene dosage and linezolid resistance in Enterococcus faecium and Enterococcus faecalis. Antimicrob. Agents Chemother. 2002, 46, 3334–3336. [Google Scholar] [CrossRef] [PubMed]
- EUCAST WGS Subcommitte Consultation on Report from the EUCAST Subcommittee on the Role of Whole Genome Sequencing (WGS) in Antimicrobial Susceptibility Testing of Bacteria. Available online: http://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Consultation/2016/EUCAST_WGS_report_consultation_20160511.pdf (accessed on 24 May 2016).
- Reuter, S.; Ellington, M.J.; Cartwright, E.J.P.; Köser, C.U.; Török, M.E.; Gouliouris, T.; Harris, S.R.; Brown, N.M.; Holden, M.T.G.; Quail, M.; et al. Rapid bacterial whole-genome sequencing to enhance diagnostic and public health microbiology. JAMA Intern. Med. 2013, 173, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Khaledi, A.; Schniederjans, M.; Pohl, S.; Rainer, R.; Bodenhofer, U.; Xia, B.; Klawonn, F.; Bruchmann, S.; Preusse, M.; Eckweiler, D.; et al. Transcriptome Profiling of Antimicrobial Resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2016, 60, 4722–4733. [Google Scholar] [CrossRef] [PubMed]
- Cabot, G.; López-Causapé, C.; Ocampo-Sosa, A.A.; Sommer, L.M.; Domínguez, M.Á.; Zamorano, L.; Juan, C.; Tubau, F.; Rodríguez, C.; Moyà, B.; et al. Deciphering the Resistome of the Widespread Pseudomonas aeruginosa Sequence Type 175 International High-Risk Clone through Whole-Genome Sequencing. Antimicrob. Agents Chemother. 2016, 60, 7415–7423. [Google Scholar] [PubMed]
- Pirnay, J.P.; Bilocq, F.; Pot, B.; Cornelis, P.; Zizi, M.; Van Eldere, J.; Deschaght, P.; Vaneechoutte, M.; Jennes, S.; Pitt, T.; et al. Pseudomonas aeruginosa population structure revisited. PLoS ONE 2009. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Species | Escherichia Coli | Klebsiella Pneumoniae | PseudomonaS Aeruginosa | Staphylococcus Aureus | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference | Zankari [33] | Stoesser [34] a | Tyson [35] | Stoesser [34] a | Ruppé [40] b | Kos [41] | Köser [42] | Gordon [28] Derivation set | Gordon [28] Validation set | Bradley [43] | Lee [44] | |
| N | 48 | 74 | 76 | 69 | 18 | 388 | 14 | 501 | 491 | 501 | 13 | |
| Origin | Danish pigs | Bloodstream infections | Cattle | Bloodstream infections | Infection and intestinal carriage | Infections | Infections and nasal carriage | Infections and nasal carriage | Infections and nasal carriage | Infections and nasal carriage | Infections | |
| ARG Database | Resfinder | In house | In house | In house | In house | In house | In house | In house | In house | In house | ARG-ANNOT | |
| Mutation Analysis | None | ampC, topoisomerases | ampC, topoisomerases | Topoisomerases | Topoisomerases, porins | Topoisomerases, oprD | None | Topoisomerases | Topoisomerases | Topoisomerases | Topoisomerases | |
| Ampicillin/Amoxi-Cillin/Penicillin | C | 100 | 98.6 | 100 | NR | NR | NR | 100 | 99.2 | 94.3 | 88 | NT |
| ME | 0 | 1.4 | 0 | NR | NR | NR | 0 | 0.8 | 5.1 | 11.7 | NT | |
| VME | 0 | 0 | 0 | NR | NR | NR | 0 | 0 | 0.6 | 0.3 | NT | |
| Methicillin | C | NT | NT | NT | NT | NT | NT | 100 | 99.6 | 99.2 | 100 | 100 |
| ME | NT | NT | NT | NT | NT | NT | 0 | 0.2 | 0.4 | 0 | 0 | |
| VME | NT | NT | NT | NT | NT | NT | 0 | 0.2 | 0.4 | 0 | 0 | |
| Co-Amoxiclav | C | NT | 100 | 100 | 98.6 | 100 | NR | NT | NT | NT | NT | NT |
| ME | NT | 0 | 0 | 1.4 | 0 | NR | NT | NT | NT | NT | NT | |
| VME | NT | 0 | 0 | 0 | 0 | NR | NT | NT | NT | NT | NT | |
| 3GC | C | 100 c | 97.2 d | 98.7 d | 97.2 d | 100 | NT | NT | NT | NT | NT | NT |
| ME | 0 c | 1.4 d | 0 d | 1.4 d | 0 | NT | NT | NT | NT | NT | NT | |
| VME | 0 c | 1.4 d | 1.3 d | 2.9 d | 0 | NT | NT | NT | NT | NT | NT | |
| Carbapenems | C | NT | 100 e | NT | 98.6 e | 100 | 92.4 e | NT | NT | NT | NT | NT |
| ME | NT | 0 e | NT | 0 e | 0 | 3.7 e | NT | NT | NT | NT | NT | |
| VME | NT | 0 e | NT | 1.4 e | 0 | 3.9 e | NT | NT | NT | NT | NT | |
| Fluoroquinolones | C | 97.9 | 100 f | 100 f | 91.3 f | 100 | 92.8 g | NT | 98.0 f | 98.0 f | 95.2 f | 100 |
| ME | 0 | 0 f | 0 f | 2.9 f | 0 | 3.1 g | NT | 0.6 f | 0.2 f | 0.2 f | 0 | |
| VME | 2.1 | 0 f | 0 f | 5.8 f | 0 | 4.1 g | NT | 1.4 f | 1.2 f | 4.6 f | 0 | |
| Amikacin | C | NT | NT | NT | NT | 100 | 81.5 | 100 h | NT | NT | NT | NT |
| ME | NT | NT | NT | NT | 0 | 7.7 | 0 h | NT | NT | NT | NT | |
| VME | NT | NT | NT | NT | 0 | 10.8 | 0 h | NT | NT | NT | NT | |
| Gentamicin | C | 100 | 100 | 100 | 98.6 | 100 | NT | 100 | 100 | 99.6 | 99.8 | 100 |
| ME | 0 | 0 | 0 | 0 | 0 | NT | 0 | 0 | 0 | 0 | 0 | |
| VME | 0 | 0 | 0 | 1.4 | 0 | NT | 0 | 0 | 0.4 | 0.2 | 0 | |
| Tobramycin | C | NT | NT | NT | NT | NT | NT | 100 | NT | NT | NT | NT |
| ME | NT | NT | NT | NT | NT | NT | 0 | NT | NT | NT | NT | |
| VME | NT | NT | NT | NT | NT | NT | 0 | NT | NT | NT | NT | |
| Vancomycin | C | NR | NR | NR | NR | NR | NR | NT | 100 | 100 | 100 | NT |
| ME | NR | NR | NR | NR | NR | NR | NT | 0 | 0 | 0 | NT | |
| VME | NR | NR | NR | NR | NR | NR | NT | 0 | 0 | 0 | NT | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruppé, E.; Cherkaoui, A.; Lazarevic, V.; Emonet, S.; Schrenzel, J. Establishing Genotype-to-Phenotype Relationships in Bacteria Causing Hospital-Acquired Pneumonia: A Prelude to the Application of Clinical Metagenomics. Antibiotics 2017, 6, 30. https://doi.org/10.3390/antibiotics6040030
Ruppé E, Cherkaoui A, Lazarevic V, Emonet S, Schrenzel J. Establishing Genotype-to-Phenotype Relationships in Bacteria Causing Hospital-Acquired Pneumonia: A Prelude to the Application of Clinical Metagenomics. Antibiotics. 2017; 6(4):30. https://doi.org/10.3390/antibiotics6040030
Chicago/Turabian StyleRuppé, Etienne, Abdessalam Cherkaoui, Vladimir Lazarevic, Stéphane Emonet, and Jacques Schrenzel. 2017. "Establishing Genotype-to-Phenotype Relationships in Bacteria Causing Hospital-Acquired Pneumonia: A Prelude to the Application of Clinical Metagenomics" Antibiotics 6, no. 4: 30. https://doi.org/10.3390/antibiotics6040030
APA StyleRuppé, E., Cherkaoui, A., Lazarevic, V., Emonet, S., & Schrenzel, J. (2017). Establishing Genotype-to-Phenotype Relationships in Bacteria Causing Hospital-Acquired Pneumonia: A Prelude to the Application of Clinical Metagenomics. Antibiotics, 6(4), 30. https://doi.org/10.3390/antibiotics6040030