
Small Molecule Docking Supports Broad and Narrow Spectrum Potential for the Inhibition of the Novel Antibiotic Target Bacterial Pth1



Abstract
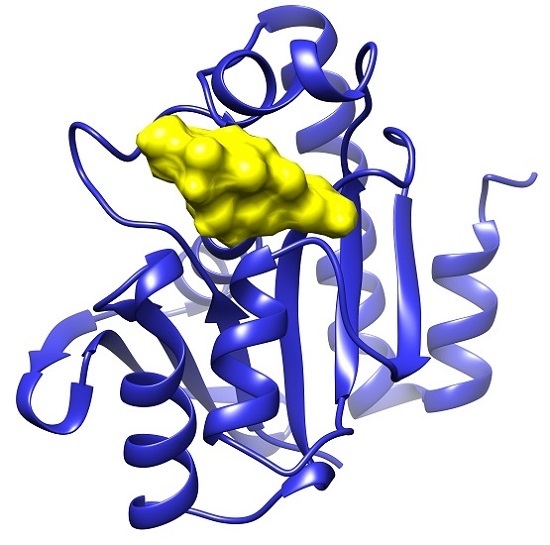
:
1. Introduction
2. Results and Discussion
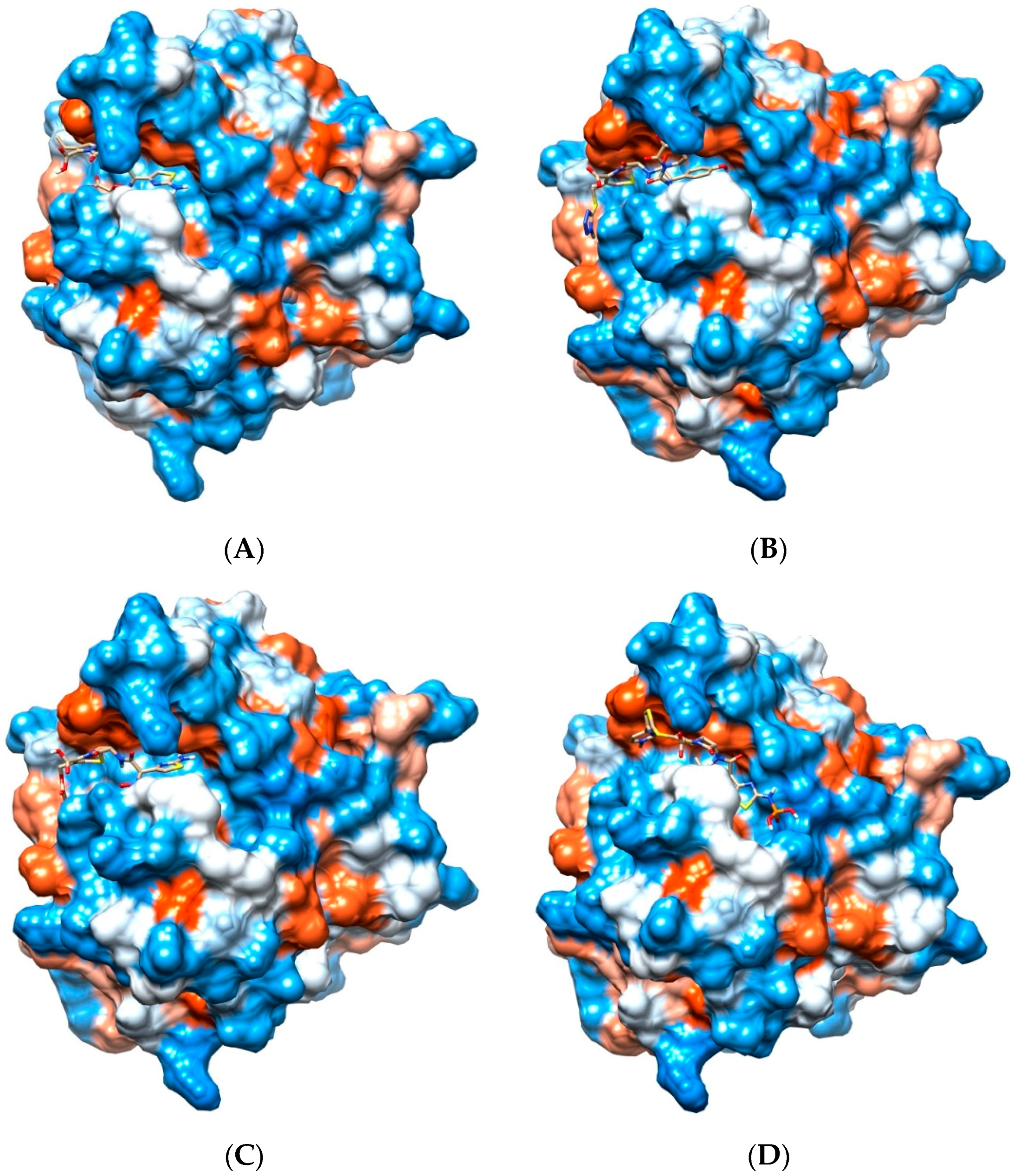
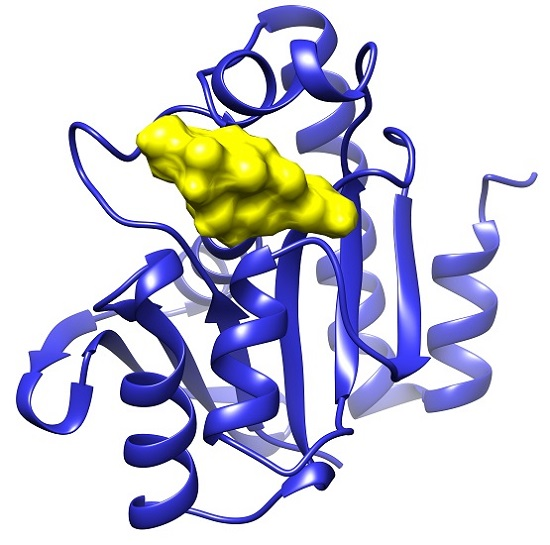
2.1. Docking Results
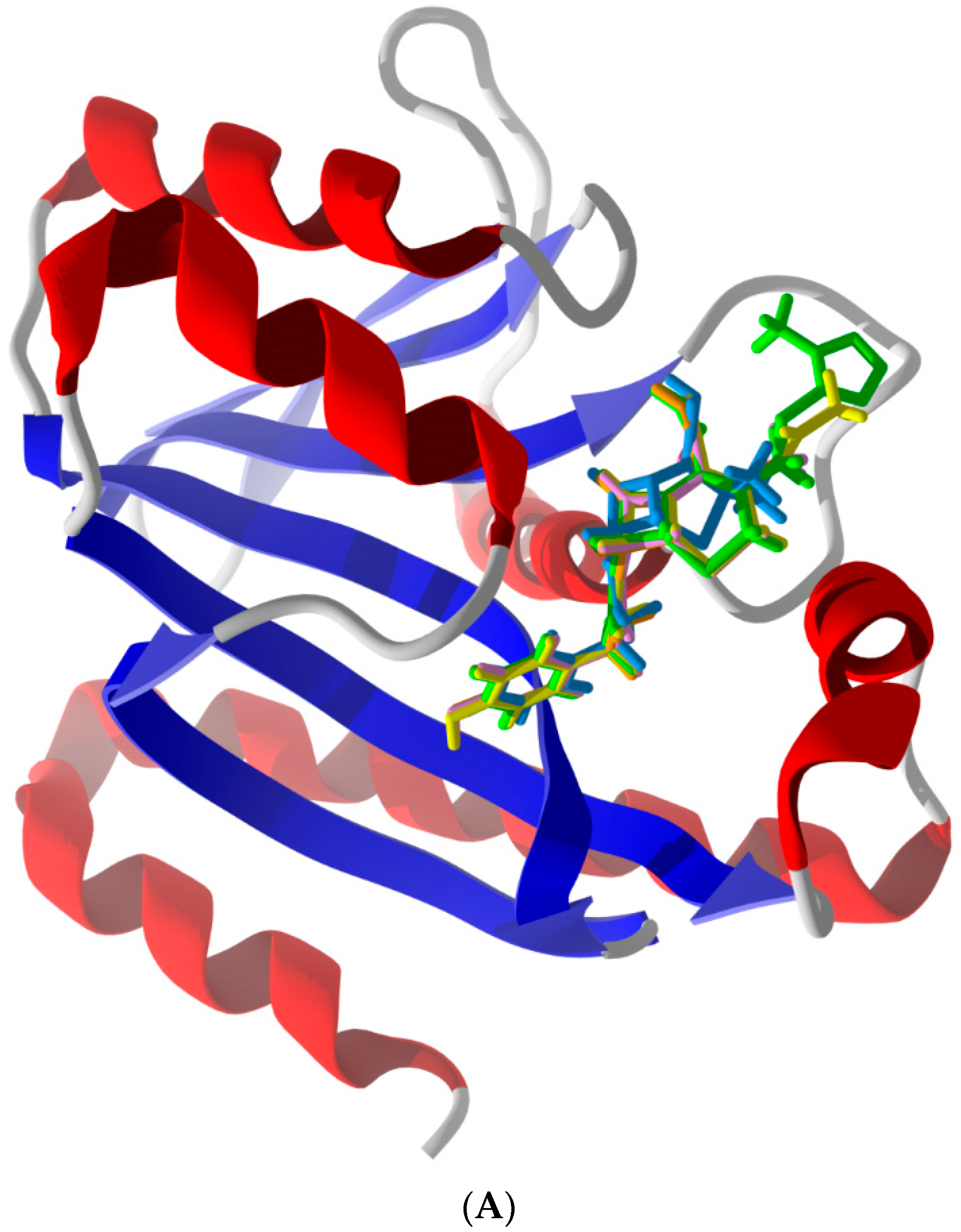
2.2. Common Favorable Pth1 Binders
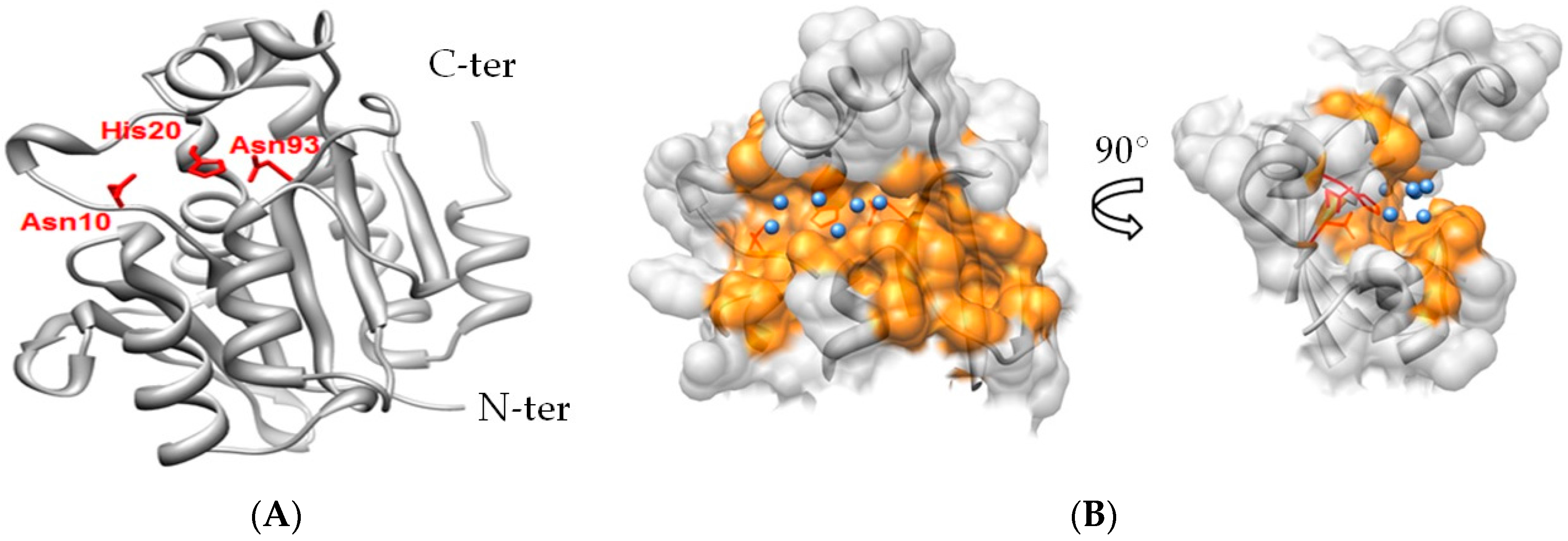
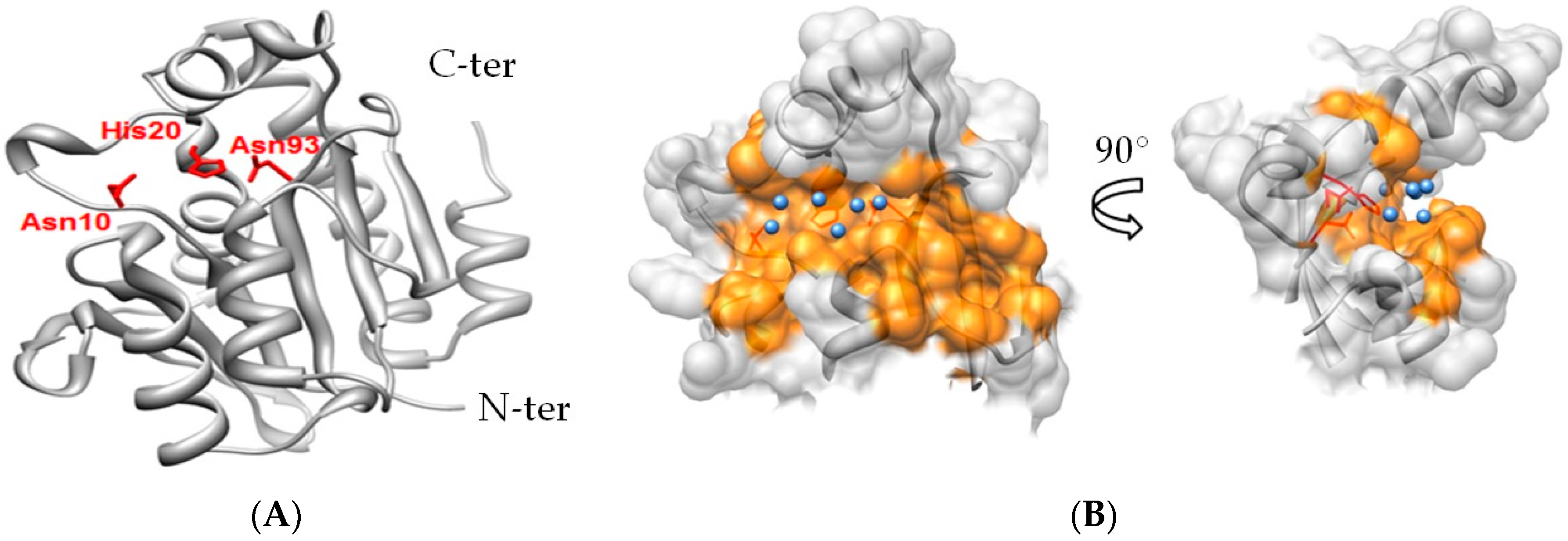
2.3. Notable Hydrogen Bonding
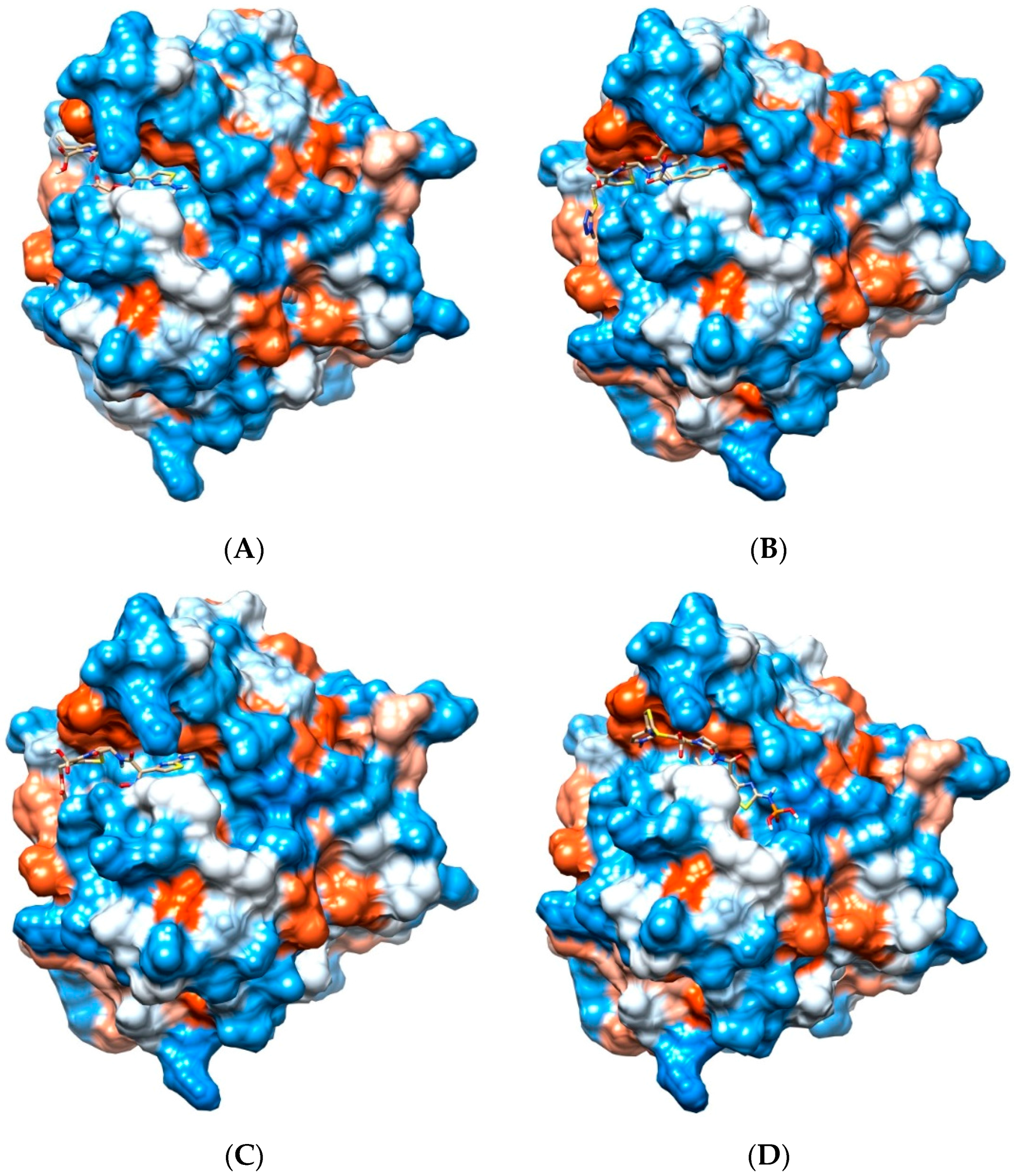
2.4. Potential for Pth1 Ligand Selectivity
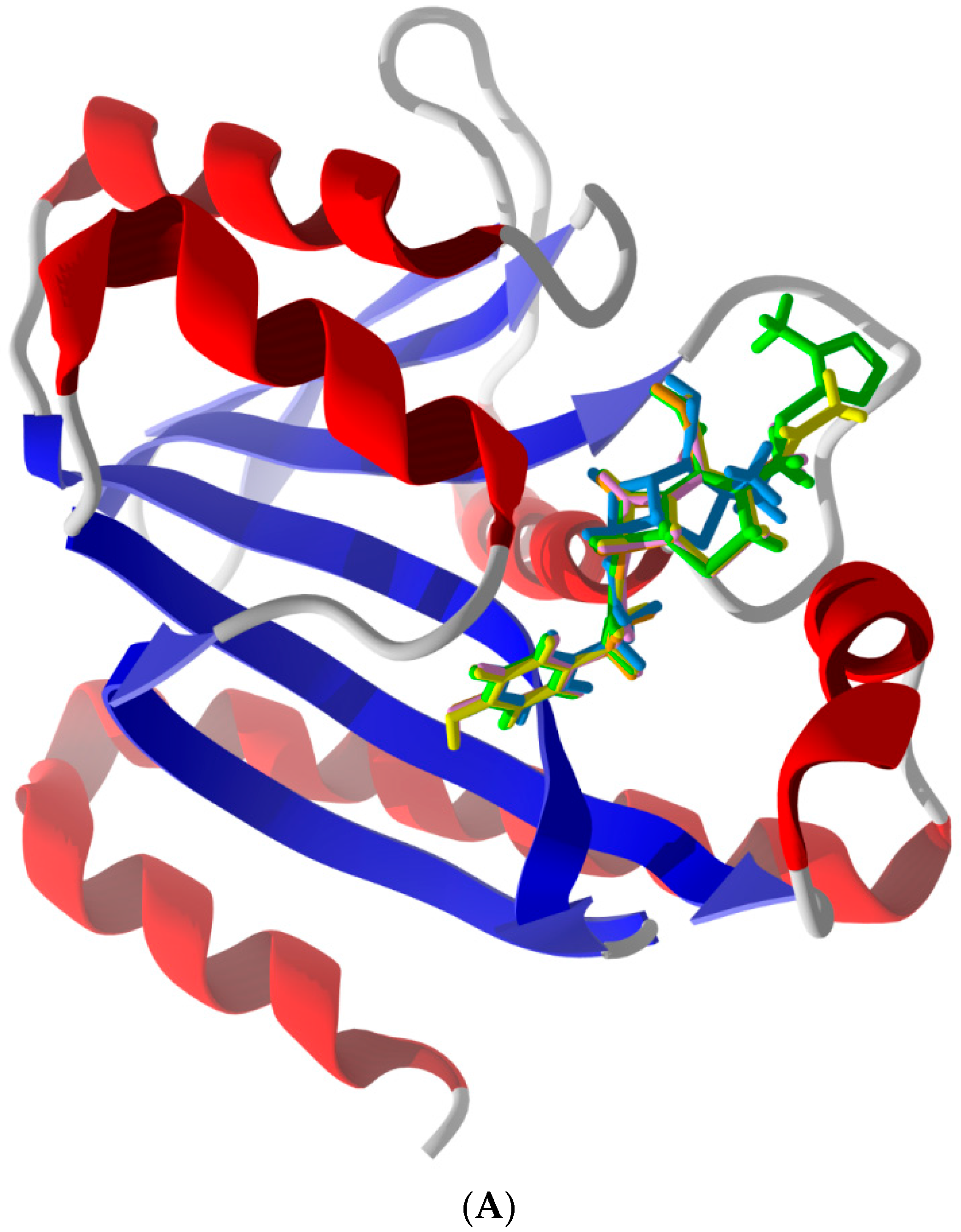
2.5. Other Compounds of Note
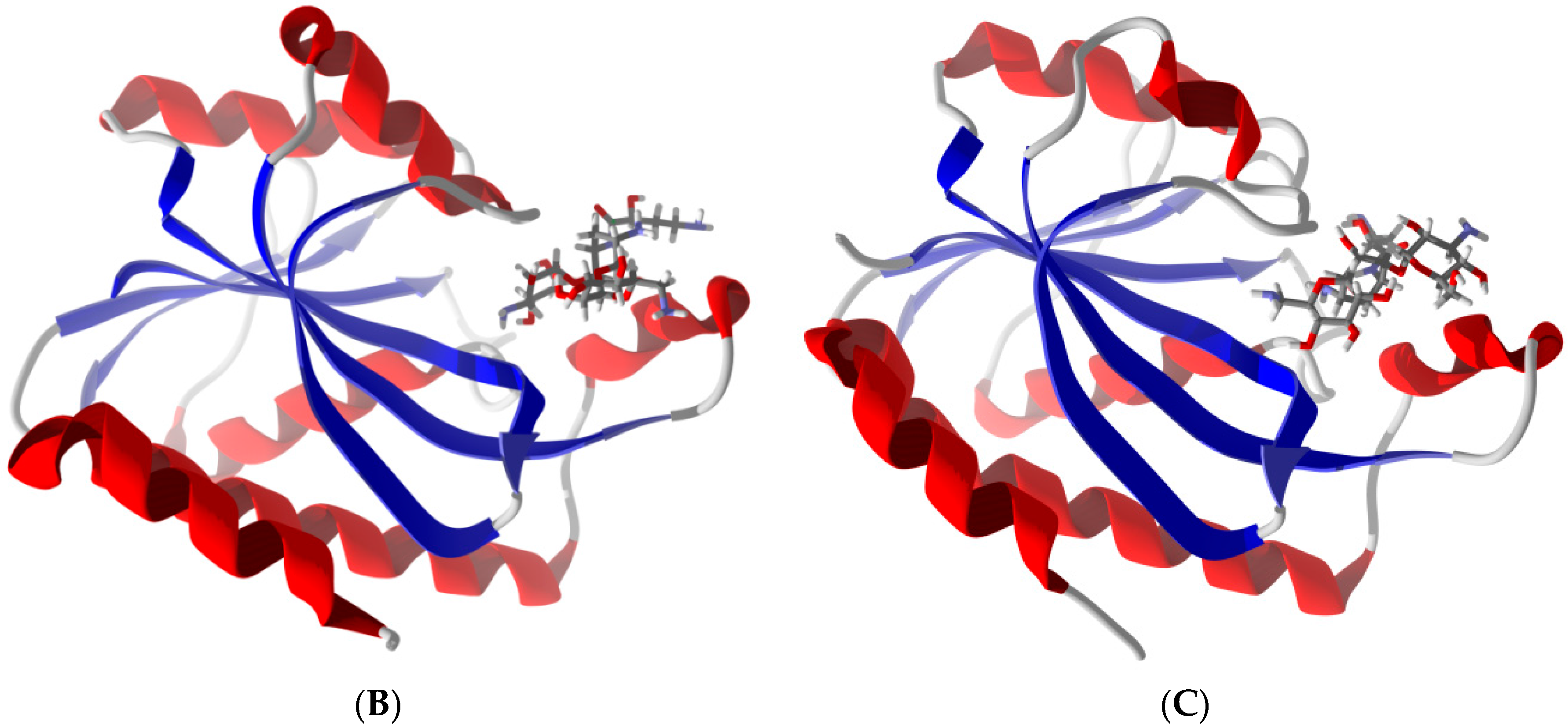
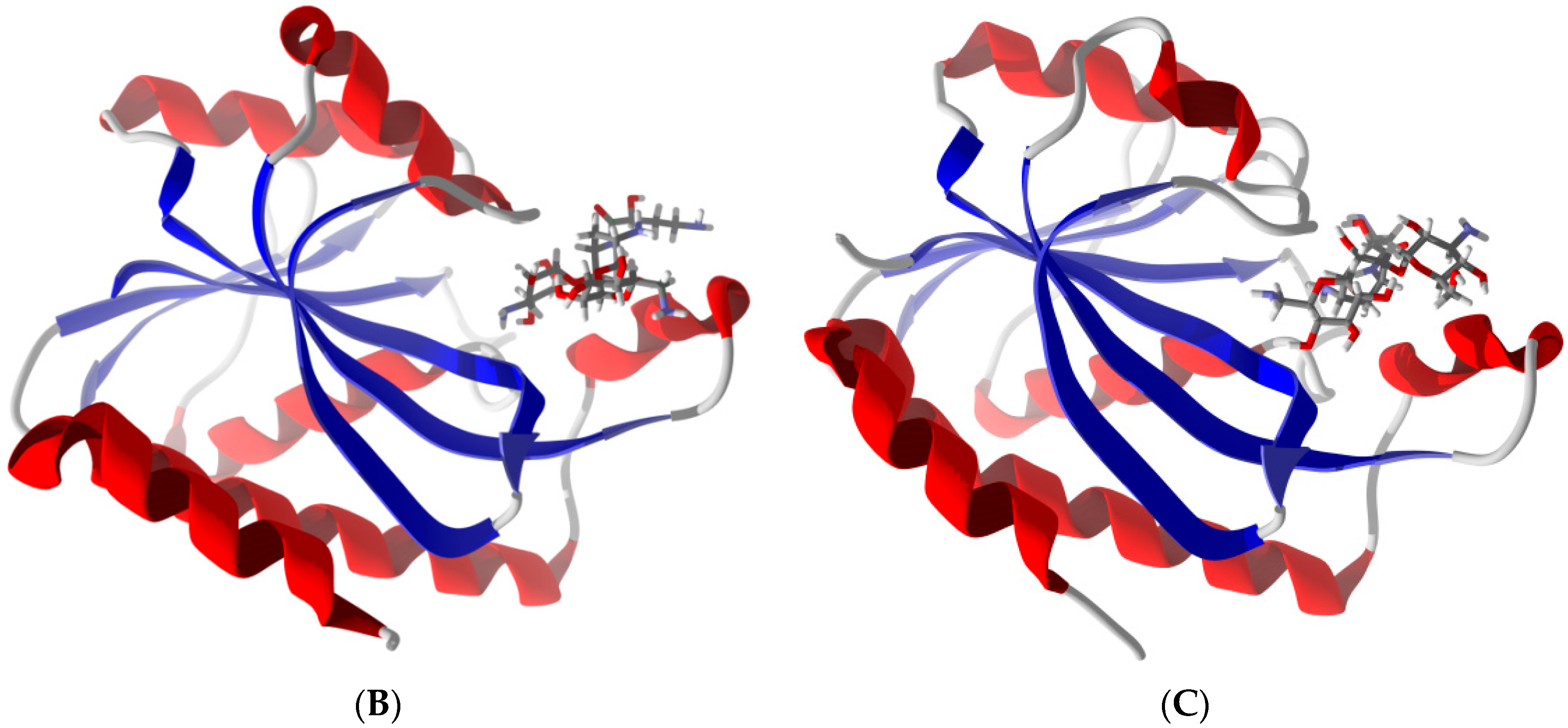
2.6. Differentiation of Pth1 from Pth2
3. Materials and Methods
3.1. Computational Methods
3.2. Ligand Dataset
3.3. Protein Structure Data Set
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Pth | Peptidyl-tRNA hydrolase |
| Pth1 | bacterial peptidyl-tRNA hydrolase |
References
- Heurgué-Hamard, V.; Karimi, R.; Mora, L.; MacDougall, J.; Leboeuf, C.; Grentzmann, G.; Ehrenberg, M.; Buckingham, R.H. Ribosome release factor RF4 and termination factor RF3 are involved in dissociation of peptidyl-tRNA from the ribosome. EMBO J. 1998, 17, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Vera, L.R.; Hernández-Ramón, E.; Pérez-Zamorano, B.; Guarneros, G. The rate of peptidyl-tRNA dissociation from the ribosome during minigene expression depends on the nature of the last decoding interaction. J. Biol. Chem. 2003, 278, 26065–26070. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Sánchez, J.; Valadez, J.G.; Herrera, J.V.; Ontiveros, C.; Guarneros, G. λ bar minigene-mediated inhibition of protein synthesis involves accumulation of peptidyl-tRNA and starvation for tRNA. EMBO J. 1998, 17, 3758–3765. [Google Scholar] [CrossRef] [PubMed]
- Tenson, T.; Herrera, J.V.; Kloss, P.; Guarneros, G.; Mankin, A.S. Inhibition of translation and cell growth by minigene expression. J. Bacteriol. 1999, 181, 1617–1622. [Google Scholar] [PubMed]
- Hames, M.; McFeeters, H.; Holloway, W.; Stanley, C.; Urban, V.; McFeeters, R. Small Molecule Binding, Docking, and Characterization of the Interaction between Pth1 and Peptidyl-tRNA. Int. J. Mol Sci. 2013, 14, 22741–22752. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.; Crow, P.; White, J. The site of hydrolysis by rabbit reticulocyte peptidyl-tRNA hydrolase is the 3’-AMP terminus of susceptible tRNA substrates. J. Biol. Chem. 1992, 267, 2080–2086. [Google Scholar] [PubMed]
- Powers, R.; Mirkovic, N.; Goldsmith-Fischman, S.; Acton, T.B.; Chiang, Y.; Huang, Y.J.; Ma, L.; Rajan, P.K.; Cort, J.R.; Kennedy, M.A.; et al. Solution structure of Archaeglobus fulgidis peptidyl-tRNA hydrolase (Pth2) provides evidence for an extensive conserved family of Pth2 enzymes in archea, bacteria, and eukaryotes. Protein Sci. 2005, 14, 2849–2861. [Google Scholar] [CrossRef] [PubMed]
- Menez, J.B.R.; de Zamaroczy, M.; Campelli, C.K. Peptidyl-tRNA hydrolase in Bacillus subtilis, encoded by spoVC, is essential to vegetative growth, whereas the homologous enzyme in Saccharomyces cerevisiae is dispensable. Mol. Microbiol. 2002, 45, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Sandoval, G.; Ambrogelly, A.; Rinehart, J.; Wei, D.; Cruz-Vera, L.R.; Graham, D.E.; Stetter, K.O.; Guarneros, G.; Söll, D. Orthologs of a novel archaeal and of the bacterial peptidyl-tRNA hydrolase are nonessential in yeast. Proc. Natl. Acad. Sci. USA 2002, 99, 16707–16712. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.S.; Varshney, U. A physiological connection between tmRNA and peptidyl-tRNA hydrolase functions in Escherichia coli. Nucleic Acids Res. 2004, 32, 6028–6037. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, E.; Mechulam, Y.; Fromant, M.; Plateau, P.; Blanquet, S. Crystal structure at 1.2 Å resolution and active site mapping of Escherichia coli peptidyl-tRNA hydrolase. EMBO J. 1997, 16, 4760–4769. [Google Scholar] [CrossRef] [PubMed]
- Pulavarti, S.V.; Jain, A.; Pathak, P.P.; Mahmood, A.; Arora, A. Solution structure and dynamics of peptidyl-tRNA hydrolase from Mycobacterium tuberculosis H37Rv. J. Mol. Biol. 2008, 378, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, M.; Roy, S.; Singh, N.; Sangeetha, R.; Varshney, U.; Vijayan, M. Structural Plasticity and Enzyme Action: Crystal Structures of Mycobacterium tuberculosis Peptidyl-tRNA Hydrolase. J. Mol. Biol. 2007, 372, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.C.; McFeeters, H.; Coates, L.; McFeeters, R.L. Recombinant production, crystallization and X-ray crystallographic structure determination of the peptidyl-tRNA hydrolase of Pseudomonas aeruginosa. Acta Crystallogr. Sect. F: Struct. Biol. Cryst. Commun. 2012, 68, 1472–1476. [Google Scholar] [CrossRef] [PubMed]
- Clarke, T.E.; Romanov, V.; Lam, R.; Gothe, S.A.; Peddi, S.R.; Razumova, E.B.; Lipman, R.S.; Branstrom, A.A.; Chirgadze, N.Y. Structure of Francisella tularensis peptidyl-tRNA hydrolase. Acta Crystallogr. Sect. F: Struct. Biol. Cryst. Commun. 2011, 67, 446–449. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, N.; Yadav, R.; Kumar, R.P.; Sharma, S.; Arora, A.; Singh, T.P. Crystal structure of peptidyl-tRNA hydrolase from mycobacterium smegmatis reveals novel features related to enzyme dynamics. Int. J. Biochem. Mol. Biol. 2012, 3, 58–69. [Google Scholar] [PubMed]
- Kaushik, S.; Singh, N.; Yamini, S.; Singh, A.; Sinha, M.; Arora, A.; Kaur, P.; Sharma, S.; Singh, T.P. The Mode of Inhibitor Binding to Peptidyl-tRNA Hydrolase: Binding Studies and Structure Determination of Unbound and Bound Peptidyl-tRNA Hydrolase from Acinetobacter baumannii. PLoS ONE 2013, 8, e67547. [Google Scholar] [CrossRef] [PubMed]
- Vandavasi, V.; Taylor-Creel, K.; McFeeters, R.L.; Coates, L.; McFeeters, H. Recombinant production, crystallization and X-ray crystallographic structure determination of peptidyl-tRNA hydrolase from Salmonella typhimurium. Acta Crystallogr. Sect. F: Struct. Biol. Cryst. Commun. 2014, 70, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Song, Y.; Niu, L.; Teng, M.; Li, X. Crystal structure of Staphylococcus aureus peptidyl-tRNA hydrolase at a 2.25 A resolution. Acta Biochim. Biophys. Sin. 2015, 47, 1005–1010. [Google Scholar] [PubMed]
- Goodall, J.J.; Chen, G.J.; Page, M.G. Essential role of histidine 20 in the catalytic mechanism of Escherichia coli peptidyl-tRNA hydrolase. Biochemistry 2004, 43, 4583–4591. [Google Scholar] [CrossRef] [PubMed]
- McFeeters, R.L. Recent Antimicrobial Developments Targeting Peptidyl-tRNA Hydrolases. JSM Biotechnol. Biomed. Eng. 2013, 1, 1006. [Google Scholar]
- Giorgi, L.; Bontems, F.; Fromant, M.; Aubard, C.; Blanquet, S.; Plateau, P. RNA-binding site of Escherichia coli peptidyl-tRNA hydrolase. J. Biol. Chem. 2011, 286, 39585–39594. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, L.; Plateau, P.; O’Mahony, G.; Aubard, C.; Fromant, M.; Thureau, A.; Grotli, M.; Blanquet, S.; Bontems, F. NMR-Based Substrate Analog Docking to Escherichia coli Peptidyl-tRNA Hydrolase. J. Mol. Biol. 2011, 412, 619–633. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Murakami, R.; Mochizuki, M.; Qi, H.; Shimizu, Y.; Miura, K.; Ueda, T.; Uchiumi, T. Structural basis for the substrate recognition and catalysis of peptidyl-tRNA hydrolase. Nucleic Acids Res. 2012, 40, 10521–10531. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.M.; McFeeters, H.; Ogungbe, I.V.; Cruz-Vera, L.R.; Setzer, W.N.; Jackes, B.R.; McFeeters, R.L. Peptidyl-tRNA Hydrolase Screening Combined with Molecular Docking Reveals the Antibiotic Potential of Syzygium johnsonii Bark Extract. Nat. Prod. Commun. 2011, 6, 1421–1424. [Google Scholar] [PubMed]
- McFeeters, H.; Gilbert, M.J.; Thompson, R.M.; Setzer, W.N.; Cruz-Vera, L.R.; McFeeters, R.L. Inhibition of essential bacterial peptidyl-tRNA hydrolase activity by tropical plant extracts. Nat. Prod. Commun. 2012, 7, 1107–1110. [Google Scholar] [PubMed]
- McFeeters, R.L. Naural Products Demonstrate Broad and Narrow Spectrum Pth1 Inhibition. Personal communication. Unsubmitted.
- Molegro Virtual Docker v 5.0; Molegro ApS: Aarhus, Denmark, 2011.
- Thomsen, R.; Christensen, M.H. MolDock: A new technique for high-accuracy molecular docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef] [PubMed]
- Spartan ’10 for Windows, v 1.1; Wavefunction, Inc.: Irvine, CA, USA, 2011.
- Baugh, L.; Gallagher, L.A.; Patrapuvich, R.; Clifton, M.C.; Gardberg, A.S.; Edwards, T.E.; Armour, B.; Begley, D.W.; Dieterich, S.H.; Dranow, D.M.; et al. Combining functional and structural genomics to sample the essential Burkholderia structome. PLoS ONE 2013, 8, e53851. [Google Scholar] [CrossRef] [PubMed]
- Fromant, M.; Schmitt, E.; Mechulam, Y.; Lazennec, C.; Plateau, P.; Blanquet, S. Crystal structure at 1.8 A resolution and identification of active site residues of Sulfolobus solfataricus peptidyl-tRNA hydrolase. Biochemistry 2005, 44, 4294–4301. [Google Scholar] [CrossRef] [PubMed]
- De Pereda, J.M.; Waas, W.F.; Jan, Y.; Ruoslahti, E.; Schimmel, P.; Pascual, J. Crystal structure of a human peptidyl-tRNA hydrolase reveals a new fold and suggests basis for a bifunctional activity. J. Biol. Chem. 2004, 279, 8111–8115. [Google Scholar] [CrossRef] [PubMed]
- McFeeters, H.; McFeeters, R.L. Current Methods for Analysis of Enzymatic Peptidyl-tRNA Hydrolysis. J. Anal. Bioanal. Tech. 2014, 5, 215. [Google Scholar] [CrossRef]
- Holloway, W.B.; McFeeters, H.; Powell, A.M.; Nidadavolu, G.S.; McFeeters, R.L. A Highly Adaptable Method for Quantification of Peptidyl-tRNA Hydrolase Activity. J. Anal. Bioanal. Tech. 2015, 6, 244. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Pth1 | Pth2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Eco 1 | Sty | Bth | Ftu | Par | Mtb | Msm | Aba | Hs | Ss | Af | |
| 2PTH | 4P7B | 3V2I | 3NEA | 4FYJ | 2Z2I | 3KJZ | 4FOP | 1Q7S | 1XTY | 3ERJ | |
| Amikacin | −80.3 | −77.1 | −81.1 | −60.2 | −82.8 | −91.7 | −67.5 | −23.7 | −90.1 | −94.1 | −81.1 |
| Amoxicillin | −88.1 | −87.4 | −89.7 | −34.8 | −76.7 | −75.2 | −75.9 | −95.3 | −87.9 | −82.8 | −78.7 |
| Ampicillin | −93.4 | −81.9 | −84.4 | −81.4 | −74.4 | −71.2 | −77.9 | −86.6 | −85.6 | −92.6 | −64.9 |
| Anisomycin | −90.9 | −95.6 | −87.6 | −80.7 | −84.3 | −63.7 | −63.4 | −93.7 | −68.3 | −69.9 | −76.7 |
| Azlocillin | −114.6 | −119.7 | −106.4 | −85.4 | −112 | −101.5 | −90 | −52.8 | −40.5 | −120.2 | nd |
| Aztreonam | −104.8 | −92.2 | −98.5 | −105.2 | −76.2 | −82.6 | −86.8 | −23.4 | −73 | −91.9 | −94.6 |
| Blasticidin S | −89.5 | −99 | −94.5 | −92.5 | −80.8 | −76.4 | −77.6 | −86.4 | −82.6 | −94.5 | −70.2 |
| Capreomycin | −114.3 | −95.6 | −75.3 | −71.4 | −90.6 | −83.5 | −79 | −103.5 | −90.4 | −108 | −81.1 |
| Carbenicillin | −101.9 | −88.5 | −87.3 | −26.4 | −91.3 | −76.7 | −77.1 | nd | nd | −94.6 | −65.9 |
| Cefaclor | −102.7 | −84.8 | −91.3 | −76.3 | −56.5 | −71.6 | −69.4 | −97.7 | −102.7 | −84 | −74.4 |
| Cefadroxil | −102.7 | −96.4 | −90.6 | −85.7 | −84.8 | −73.7 | −81 | −112.1 | −120.8 | −85.5 | −73.2 |
| Cefamandole | −110.6 | −92 | −38.2 | −97.6 | −73 | −94.5 | −91.6 | −96.6 | −95.3 | −96.6 | −86.5 |
| Cefazolin | −118.3 | −115.5 | −93.7 | −99.4 | −68.5 | −93.6 | −104 | −53.7 | −97.1 | −98.7 | −90.8 |
| Cefdinir | nd | nd | nd | nd | nd | nd | nd | −99.2 | −90.9 | nd | nd |
| Cefditoren | −132 | −107.6 | −100.4 | −81.8 | −108.3 | −98.6 | −89 | −100.9 | −94.7 | −117.7 | −89.1 |
| Cefepime | −105.7 | −99.1 | −96.3 | −100.4 | −96.3 | −85 | −91.2 | −87.1 | −122.5 | −106.9 | −96.8 |
| Cefixime | −115.2 | −110 | −106.8 | −106.7 | −98 | −98.1 | −94.8 | −118.2 | −102.4 | −108.1 | −96.2 |
| Cefoperazone | −123.2 | −115.3 | −92 | −109.8 | −93.8 | −99.4 | −79.6 | −116.8 | −100.6 | −101.1 | −109.1 |
| Cefotaxime | −105.8 | −98.1 | −103.6 | −121.5 | −93 | −91.3 | −92.2 | −87.6 | −89.6 | −100.4 | −101.7 |
| Cefoxitin | −103.4 | −88 | −85.5 | −105.9 | −51.3 | −83.3 | −83.7 | −90.9 | −82.1 | −97.2 | −78.1 |
| Cefpodoxime | −105.2 | −93.7 | −110 | −110.3 | −92.2 | −86.9 | −84.6 | −91.7 | −82.3 | −95 | −97.5 |
| Cefprozil | −106.5 | −90.3 | −94.2 | −92.8 | −104.1 | −83.4 | −84.3 | −90.4 | −97.4 | −85.3 | −70.5 |
| Ceftaroline fosamil | −118.6 | −117.4 | −112.3 | −92 | −96.9 | −114.9 | −98.6 | −82.3 | −81.5 | −134.1 | −110.5 |
| Ceftazidime | −108.3 | −94.3 | −93.7 | −107.9 | −104.8 | −83.9 | −86.3 | −81.4 | −75.6 | −110.1 | −103.1 |
| Ceftibuten | −104.8 | −108.7 | −97.2 | −100.5 | −100.8 | −87.6 | −89.3 | −66.1 | −86.6 | −101.1 | −103.7 |
| Ceftizoxime | −103.9 | −100.4 | −101.9 | −96.1 | −73.3 | −85.7 | −86.9 | −95.9 | −84.1 | −97.4 | −90.5 |
| Cephalexin | −103.1 | −80.4 | −89.4 | −82.1 | −85 | −76.8 | −80.6 | −66.5 | −74.9 | −84.5 | −81.1 |
| Cephalothin | −111.4 | −96.2 | −95.8 | −80.7 | −103.8 | −87.5 | −84.8 | −98 | −76.9 | −92.2 | −87.7 |
| Chloramphenicol | −92.1 | −91.4 | −91 | −83.4 | −63.3 | −66.1 | −65.3 | −69.3 | −68.9 | −80.5 | −84.9 |
| Ciprofloxacin | −87.4 | −78.4 | −66.8 | −98.3 | −84.1 | −69.7 | −64.3 | −46.9 | −43.5 | −89.7 | −75.2 |
| Clindamycin | −85.1 | −86.1 | −85.1 | −83.5 | −88.2 | −83.3 | −74.7 | −85 | −86.2 | −81.4 | −78.6 |
| Clofazimine | −99.3 | −75.8 | −99.3 | −78.7 | −90.6 | −90.1 | −66.2 | −71.5 | −70.9 | −74.9 | −72.5 |
| Cloxacillin | −95.8 | −84.3 | −47.9 | −94 | −97.5 | −91.2 | −82.5 | −88 | −41.6 | −88.4 | −25.6 |
| Dicloxacillin | −81.6 | −77.7 | −85.8 | −93.9 | −99 | −89.8 | −79.8 | −71.3 | −50.6 | −95.2 | −64.6 |
| Doripenem | −104.6 | −113.8 | −105.9 | −107.2 | −83.4 | −92.4 | −81 | −105.4 | −82.2 | −94.1 | −70.4 |
| Doxycycline | −61.4 | −57.9 | nd | −77 | −62.8 | −66.5 | −51.3 | −103.1 | −89.5 | −78.4 | −59.5 |
| Enoxacin | −83.6 | −74.5 | −66.4 | −95.1 | −73.9 | −64.4 | −60.8 | −96.4 | −81.3 | −84.7 | −88.5 |
| Ertapenem | −137.7 | −128.9 | −106.3 | −111.7 | −123.3 | −102 | −88.3 | −18.9 | −25 | −107.2 | −74.7 |
| Flucloxacillin | −90.9 | −82.8 | −88.3 | −93.3 | −98.9 | −90.4 | −77.4 | −83.6 | −72.9 | −88.6 | −81.4 |
| Fosfomycin | −43.7 | −48.5 | −42.7 | −43.1 | −43.8 | −31.6 | −40.7 | −105.6 | −85.7 | −38.8 | −37.5 |
| Furazolidone | −90.6 | −95.7 | −84.9 | −76 | −82.2 | −68.7 | −66.6 | −78.8 | −76.9 | −72.8 | −84.3 |
| Gatifloxacin | −59.4 | −81.5 | −53.4 | −98.9 | −89.2 | −65.9 | −61.8 | −82.3 | −78.8 | −86.2 | −27.8 |
| Geldanamycin | −75.5 | −60.8 | −67.8 | −70 | −74.2 | −75.4 | −59.4 | −62.8 | −99.9 | −74.6 | −57.2 |
| Gentamicin C | −109.5 | −64 | −86.2 | −87.7 | −69.4 | −75.1 | −53.7 | −64.3 | −51.3 | −81.1 | −67.8 |
| Herbimycin | −91.5 | −47.1 | −63.7 | −55.2 | −58.2 | −65.4 | −62.8 | −95.8 | −91.4 | −63.6 | −62.9 |
| Imipenem | −128 | −105.7 | −95.7 | −83.7 | −90.8 | −80.8 | −90.3 | −116.5 | −116 | −81 | −33 |
| Kanamycin | −52.7 | −102.7 | −81.4 | −103.1 | −89.6 | −83.4 | −71.6 | −74.4 | −89.7 | −99.5 | −73.4 |
| Levofloxacin | −76.2 | −75.4 | −79.8 | −65.5 | −81.1 | −58.3 | −58 | −37.8 | −78.3 | −73.2 | −29.9 |
| Lincomycin | −96 | −88.1 | −85.8 | −80.6 | −83.9 | −79.3 | −68.9 | −85.1 | −86.8 | −87.1 | −62.2 |
| Linezolid | −87.6 | −98.9 | −91.3 | −79.3 | −98.9 | −78.6 | −80.1 | −77.9 | −87.5 | −92.5 | −69.1 |
| Lomefloxacin | −102.1 | −96.6 | −64.5 | −73.9 | −84.5 | −73.2 | −67.1 | −78.4 | −66.5 | −80.5 | −92.4 |
| Loracarbef | −75 | −76 | −86.4 | −74 | −84.3 | −74.3 | −78.1 | −86.1 | −68.9 | −82.2 | −70.8 |
| Mafenide | −64.1 | −67.5 | −58.4 | −71.4 | −66.3 | −49.4 | −52.6 | −97.2 | −95.6 | −53.6 | −56.5 |
| Meropenem | −86.2 | −106.8 | −109.4 | −94.5 | −35.6 | −86.9 | −75.3 | −111.4 | −112.9 | −91.1 | −76.6 |
| Methicillin | −92.1 | −82.9 | −76.1 | −79.8 | −76 | −69.9 | −67.3 | −100.6 | −95.2 | −96.6 | −47.1 |
| Metronidazole | −87.4 | −64.5 | −66.2 | −64.2 | −66 | −53.3 | −48.8 | −80.3 | −93.1 | −52.2 | −48.8 |
| Mezlocillin | −76.1 | −123.3 | −100.9 | −102 | −128.9 | −91.6 | −95.8 | −88.1 | −78.9 | −97.9 | −76.2 |
| Minocycline | −94.8 | −64.2 | nd | −49.8 | −68.4 | −66.2 | −63.8 | −83.4 | −66.1 | −72.8 | −91.1 |
| Moxifloxacin | −92.2 | −82.1 | −85.5 | −99.3 | −90 | −68.8 | −65.3 | −65.9 | −47.6 | −70.9 | −26.2 |
| Nalidixic acid | −111.9 | −73.6 | −57.6 | −78.6 | −68.6 | −49.5 | −58 | −76.1 | −91.1 | −61.6 | −78.3 |
| Neomycin | −87 | −77.4 | −64.3 | −67.8 | −86.1 | −81.8 | −67.7 | −77.5 | −57.7 | −96 | −95.7 |
| Netilmicin | −93.5 | −68 | −74.7 | −76.4 | −71.5 | −80.4 | −53.7 | −94.9 | −72.4 | −77.1 | −30.9 |
| Norfloxacin | −77 | −73.8 | −67.8 | −72.7 | −54.7 | −68.5 | −61.9 | −88.5 | −85 | −86 | −93.2 |
| Ofloxacin | −53 | −64.3 | −36.4 | −75.8 | −70.3 | −69.9 | −58.5 | −102.7 | −82.5 | −68 | −92.7 |
| Oxytetracycline | −99.4 | −51.7 | −57.8 | −78.4 | −64.9 | −57.5 | −55.1 | −87.4 | −73.1 | −81.9 | −60.2 |
| Penicillin G | −106.6 | −98.5 | −86 | −76.1 | −90.7 | −73.2 | −78.6 | −50.4 | −67.1 | −92.1 | −72.4 |
| Penicillin V | −54.1 | −95.5 | −68.3 | −83.2 | −87.8 | −80.2 | −72.7 | −81 | −100 | −87.7 | −44.6 |
| Platensimycin | −121.8 | −93.3 | −103.5 | −78.1 | −36.5 | −79.5 | −78.3 | −25.3 | −77.9 | −85.3 | −82 |
| Puromycin | −53.8 | −96.6 | −109.6 | −93.2 | −111.5 | −99.4 | −81.1 | −79.2 | −69.1 | −105.7 | −89.1 |
| Sparsomycin | −91.5 | −104.1 | −90.6 | −89.1 | −101.5 | −75.9 | −93.1 | −79.6 | −81.2 | −81.5 | −60.6 |
| Streptogramin A | −93.3 | −73.2 | −84.5 | −69.7 | −78.7 | −99.4 | −68.9 | −104.9 | −102.4 | −87.3 | −76.4 |
| Streptomycin | −69.8 | −91.2 | −97.6 | −84.7 | −78.6 | −77.8 | −74.9 | −71.5 | −61.1 | −89.5 | −63.3 |
| Sulfacetamide | −86.1 | −70.9 | −66.3 | −68.1 | −72.7 | −55.8 | −52.6 | −35.3 | nd | −59.8 | −67.5 |
| Sulfadiazine | −93.5 | −85.6 | −74.4 | −67.3 | −73.5 | −63.3 | −58.1 | −57.4 | −77 | −71.3 | −74.7 |
| Sulfamethizole | −87.5 | −99.1 | −80.1 | −81.1 | −93.4 | −67.3 | −67 | −65.4 | −64.4 | −71.5 | −76.4 |
| Sulfamethoxazole | −114.8 | −99.9 | −80.9 | −78.6 | −95.5 | −67.4 | −72.2 | −74.5 | −73.2 | −71 | −63.7 |
| Sulfasalazine | −95.1 | −107.7 | −107 | −71.2 | −110.9 | −82.3 | −75.9 | −73.9 | −88 | −85.7 | −84.7 |
| Sulfisoxazole | −78.8 | −102.4 | −80.1 | −77.1 | −89 | −71.7 | −70.3 | −64.2 | −79.8 | −75.3 | −64.2 |
| Tetracycline | −103.1 | −51.1 | −63.2 | −78.3 | −52.3 | −58.2 | −57.2 | −88.9 | −82.9 | −60.6 | −58.4 |
| Thiamphenicol | −65.2 | −91.1 | −89.4 | −85.5 | −88.9 | −67.3 | −65.2 | −75.7 | −58.4 | −77.7 | −41.5 |
| Tigecycline | −69.7 | −63.7 | −79.4 | −63.7 | −78.4 | −89.6 | −74.2 | nd | −93.8 | −85 | nd |
| Tinidazole | −86 | −62.3 | −71.9 | −76.3 | −16.4 | −62.4 | −58.3 | −81.5 | −73.8 | −68.6 | −51.5 |
| Tobramycin | −91.5 | −79.9 | −77.3 | −78.4 | −75.5 | −77.9 | −64.5 | −55.8 | −47.8 | −95.8 | −65.7 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferguson, P.P.; Holloway, W.B.; Setzer, W.N.; McFeeters, H.; McFeeters, R.L. Small Molecule Docking Supports Broad and Narrow Spectrum Potential for the Inhibition of the Novel Antibiotic Target Bacterial Pth1. Antibiotics 2016, 5, 16. https://doi.org/10.3390/antibiotics5020016
Ferguson PP, Holloway WB, Setzer WN, McFeeters H, McFeeters RL. Small Molecule Docking Supports Broad and Narrow Spectrum Potential for the Inhibition of the Novel Antibiotic Target Bacterial Pth1. Antibiotics. 2016; 5(2):16. https://doi.org/10.3390/antibiotics5020016
Chicago/Turabian StyleFerguson, Paul P., W. Blake Holloway, William N. Setzer, Hana McFeeters, and Robert L. McFeeters. 2016. "Small Molecule Docking Supports Broad and Narrow Spectrum Potential for the Inhibition of the Novel Antibiotic Target Bacterial Pth1" Antibiotics 5, no. 2: 16. https://doi.org/10.3390/antibiotics5020016
APA StyleFerguson, P. P., Holloway, W. B., Setzer, W. N., McFeeters, H., & McFeeters, R. L. (2016). Small Molecule Docking Supports Broad and Narrow Spectrum Potential for the Inhibition of the Novel Antibiotic Target Bacterial Pth1. Antibiotics, 5(2), 16. https://doi.org/10.3390/antibiotics5020016