
Antibiotics and RNase P

{kind=link}
{kind=link}
Abstract
:1. Introduction
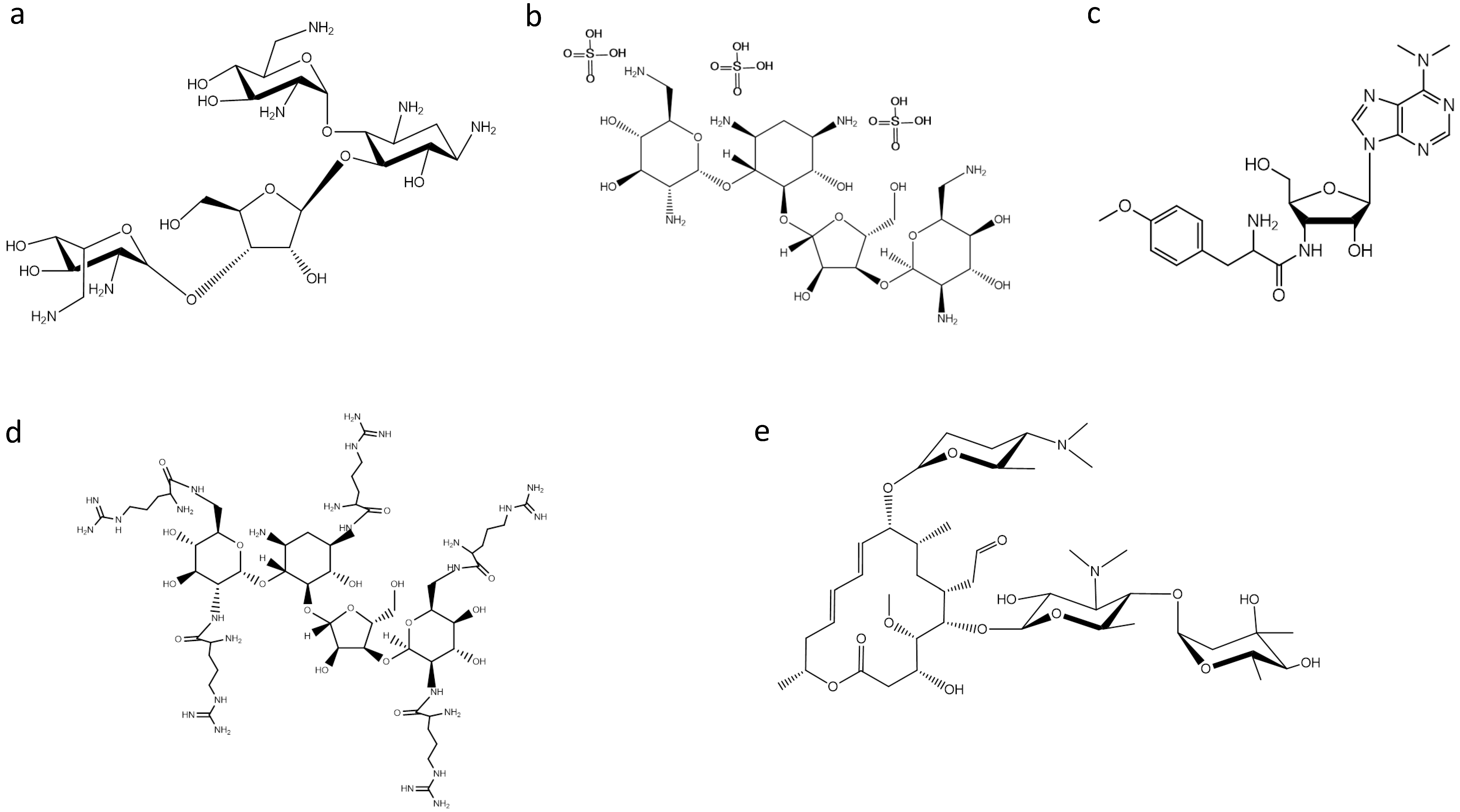
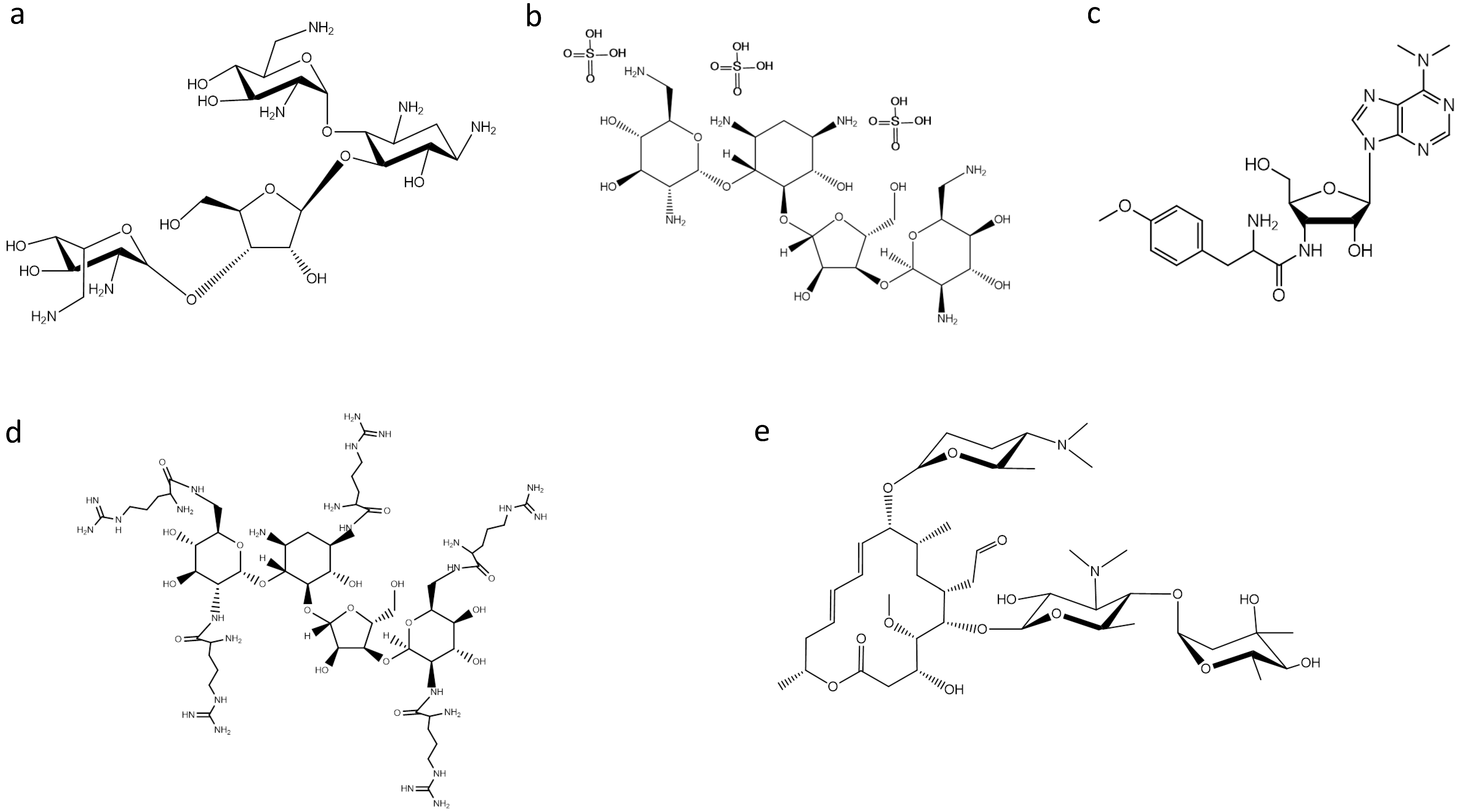
2. Ribosome-Targeting Antibiotics
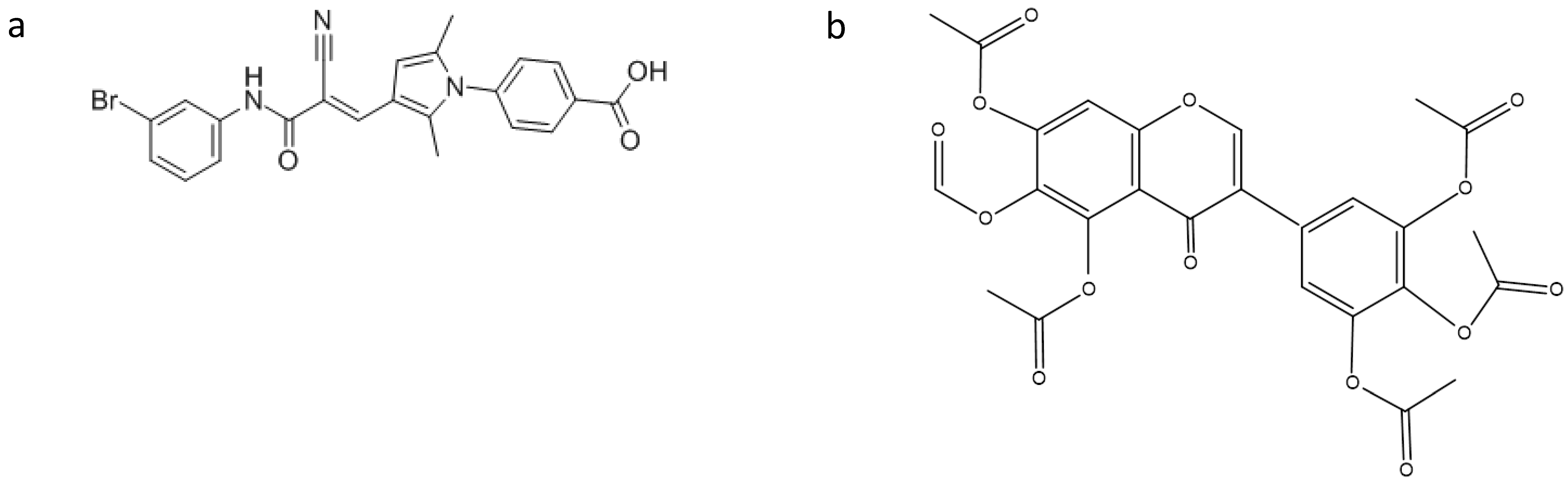
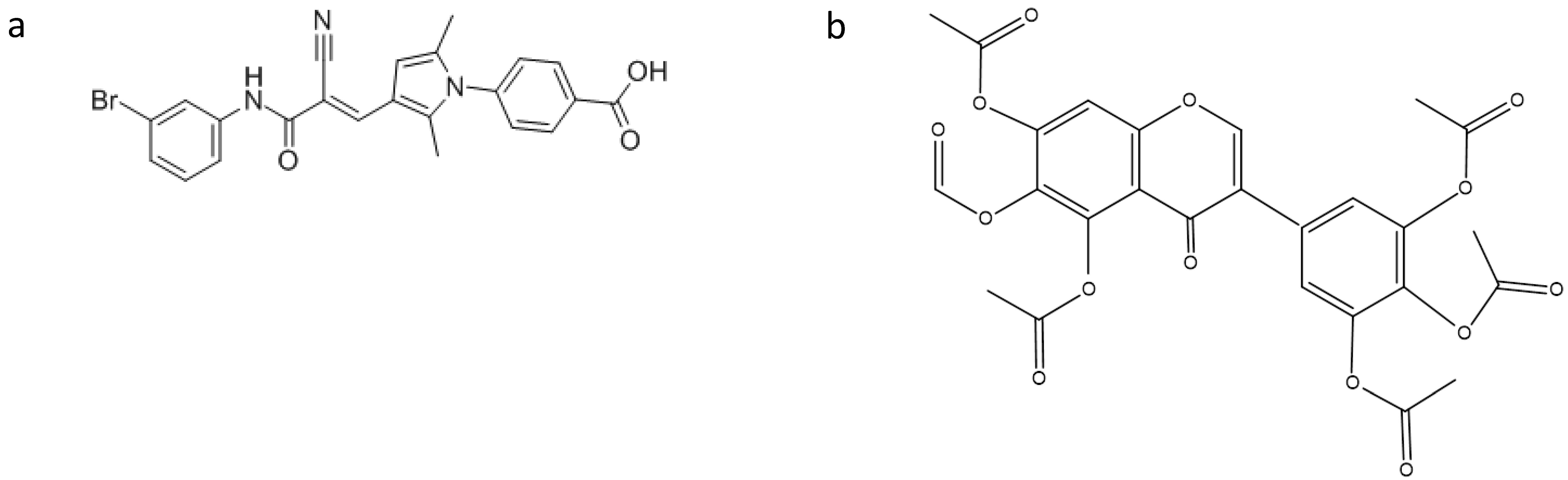
3. Searching for New Inhibitors
4. Conclusions
Conflicts of Interest
References
- Hartmann, E.; Hartmann, R.K. The enigma of ribonuclease P evolution. Trends Genet. 2003, 19, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Guerrier-Takada, C.; Gardiner, K.; Marsh, T.; Pace, N.; Altman, S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 1983, 35, 849–857. [Google Scholar] [CrossRef]
- Pannucci, J.A.; Haas, E.S.; Hall, T.A.; Harris, J.K.; Brown, J.W. RNase P RNAs from some Archaea are catalytically active. Proc. Natl. Acad. Sci. USA 1999, 6, 7803–7808. [Google Scholar] [CrossRef]
- Kikovska, E.; Svard, S.G.; Kirsebom, L.A. Eukaryotic RNase P RNA mediates cleavage in the absence of protein. Proc. Natl. Acad. Sci. USA 2007, 104, 2062–2067. [Google Scholar] [CrossRef] [PubMed]
- Holzmann, J.; Frank, P.; Löffler, E.; Bennett, K.L.; Gerner, C.; Rossmanith, W. RNase P without RNA: Identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell 2008, 135, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, B.; Gobert, A.; Giegé, P. PRORP proteins support RNase P activity in both organelles and the nucleus in Arabidopsis. Genes Dev. 2012, 26, 1022–1027. [Google Scholar] [CrossRef] [PubMed]
- Taschner, A.; Weber, C.; Buzet, A.; Hartmann, R.K.; Hartig, A.; Rossmanith, W. Nuclear RNase P of Trypanosoma brucei: A single protein in place of the multicomponent RNA-protein complex. Cell Rep. 2012, 2, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Toumpeki, C.; Stamatopoulou, V.; Bikou, M.; Grafanaki, K.; Kallia-Raftopoulou, S.; Papaioannou, D.; Stathopoulos, C.; Drainas, D. Targeting Ribonuclease P. In Antibiotics: Targets, Mechanisms and Resistance, 1st ed.; Gualerzi, C.O., Brandi, L., Fabbretti, A., Pon, C.L., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013; pp. 357–371. [Google Scholar]
- Steitz, T.A.; Moore, P.B. RNA, the first macromolecular catalyst: The ribosome is a ribozyme. Trends Biochem. Sci. 2003, 28, 411–418. [Google Scholar] [CrossRef]
- Vioque, A. Protein synthesis inhibitors and catalytic RNA. Effect of puromycin on tRNA precursor processing by the RNA component of Escherichia coli RNase P. FEBS Lett. 1989, 246, 137–139. [Google Scholar] [CrossRef]
- Mikkelsen, N.E.; Brannvall, M.; Virtanen, A.; Kirsebom, L.A. Inhibition of RNase P RNA cleavage by aminoglycosides. Proc. Natl. Acad. Sci. USA 1999, 96, 6155–6160. [Google Scholar] [CrossRef] [PubMed]
- Eubank, T.D.; Biswas, R.; Jovanovic, M.; Litovchick, A.; Lapidot, A.; Gopalan, V. Inhibition of bacterial RNase P by aminoglycoside-arginine conjugates. FEBS Lett. 2002, 511, 107–112. [Google Scholar] [CrossRef]
- Kawamoto, S.A.; Sudhahar, C.G.; Hatfield, C.L.; Sun, J.; Behrman, E.J.; Gopalan, V. Studies on the mechanism of inhibition of bacterial ribonuclease P by aminoglycoside derivatives. Nucleic Acids Res. 2008, 36, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.; Mellows, G. Interaction of pseudomonic acid A with Escherichia coli B isoleucyl-tRNA synthetase. Biochem. J. 1980, 191, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, C.; Brooks, L.; Beckley, A.; Colquhoun, J.; Dewhurst, S.; Dunman, P.M. Neomycin Sulfate Improves the Antimicrobial Activity of Mupirocin based Antibacterial Ointments. Antimicrob. Agents Chemother. 2015. [Google Scholar] [CrossRef] [PubMed]
- Toumpeki, C.; Vourekas, A.; Kalavrizioti, D.; Stamatopoulou, V.; Drainas, D. Activation of bacterial ribonuclease P by macrolides. Biochemistry 2008, 47, 4112–4118. [Google Scholar] [CrossRef] [PubMed]
- Olson, P.D.; Kuechenmeister, L.J.; Anderson, K.L.; Daily, S.; Beenken, K.E.; Roux, C.M.; Reniere, M.L.; Lewis, T.; Weiss, W.J.; Pulse, M.; et al. Small molecule inhibitors of Staphylococcus aureus RnpA alter cellular mRNA turnover, exhibit antimicrobial activity, and attenuate pathogenesis. PLoS Pathog. 2011, 7, e1001287. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chen, Y.; Fierke, C.A. A real-time fluorescence polarization activity assay to screen for inhibitors of bacterial ribonuclease P. Nucleic Acids Res. 2014, 42, e159. [Google Scholar] [CrossRef] [PubMed]
- Gopalan, V.; Vioque, A.; Altman, S. RNase P: Variations and uses. J. Biol. Chem. 2002, 277, 6759–6762. [Google Scholar] [CrossRef] [PubMed]
- Wesolowski, D.; Tae, H.S.; Gandotra, N.; Llopis, P.; Shen, N.; Altman, S. Basic peptide-morpholino oligomer conjugate that is very effective in killing bacteria by gene-specific and nonspecific modes. Proc. Natl. Acad. Sci. USA 2011, 108, 16582–16587. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, A.J.; Wesolowski, D.; Gandotra, N.; Stojadinovic, A.; Izadjoo, M.; Altman, S.; Kyriakides, T.R. A peptide-morpholino oligomer conjugate targeting Staphylococcus aureus gyrA mRNA improves healing in an infected mouse cutaneous wound model. Int. J. Pharm. 2013, 453, 651–655. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drainas, D. Antibiotics and RNase P. Antibiotics 2016, 5, 15. https://doi.org/10.3390/antibiotics5020015
Drainas D. Antibiotics and RNase P. Antibiotics. 2016; 5(2):15. https://doi.org/10.3390/antibiotics5020015
Chicago/Turabian StyleDrainas, Denis. 2016. "Antibiotics and RNase P" Antibiotics 5, no. 2: 15. https://doi.org/10.3390/antibiotics5020015
APA StyleDrainas, D. (2016). Antibiotics and RNase P. Antibiotics, 5(2), 15. https://doi.org/10.3390/antibiotics5020015