
Genetic Screen Reveals the Role of Purine Metabolism in Staphylococcus aureus Persistence to Rifampicin


Abstract
:1. Introduction
2. Results
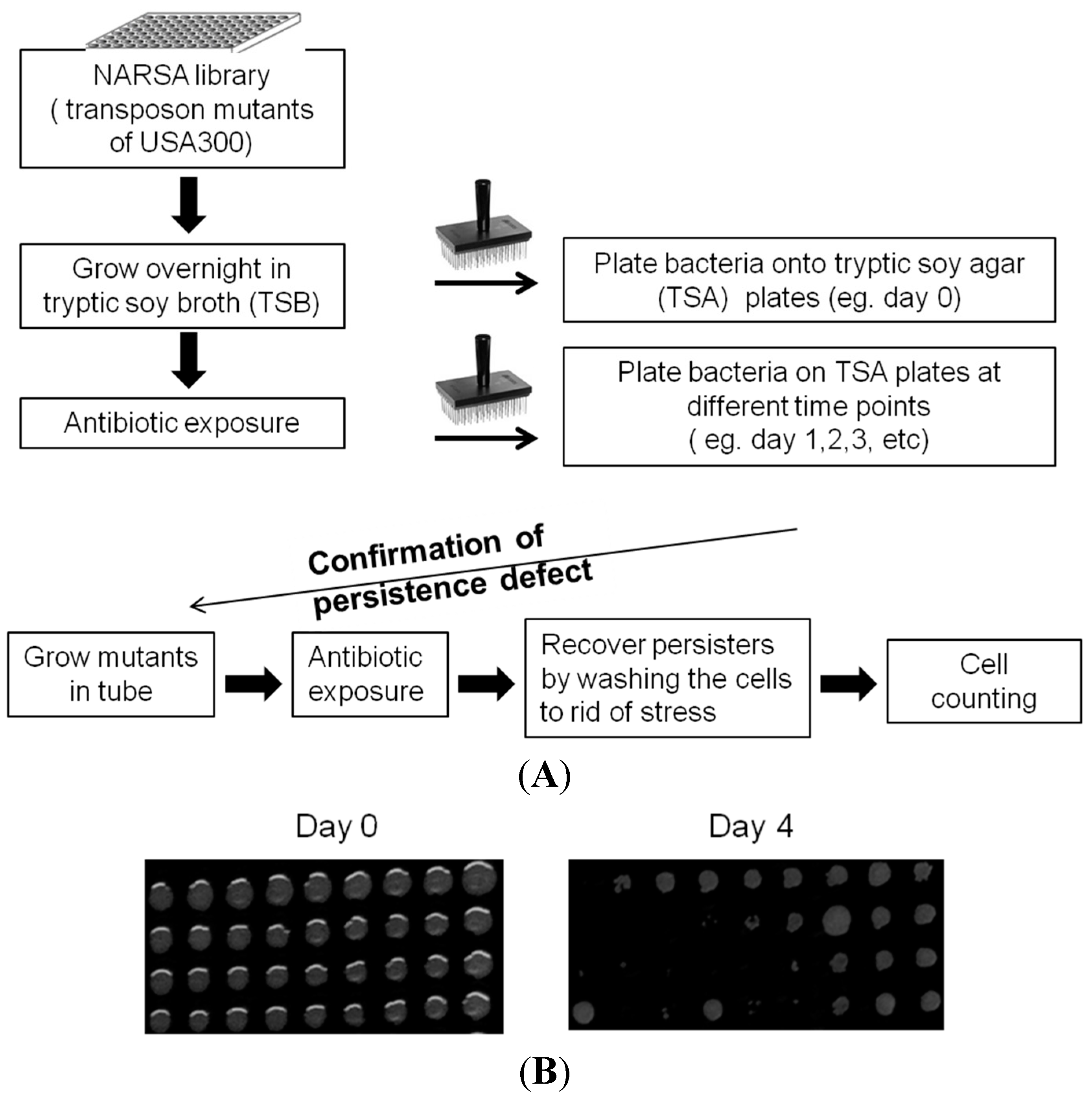
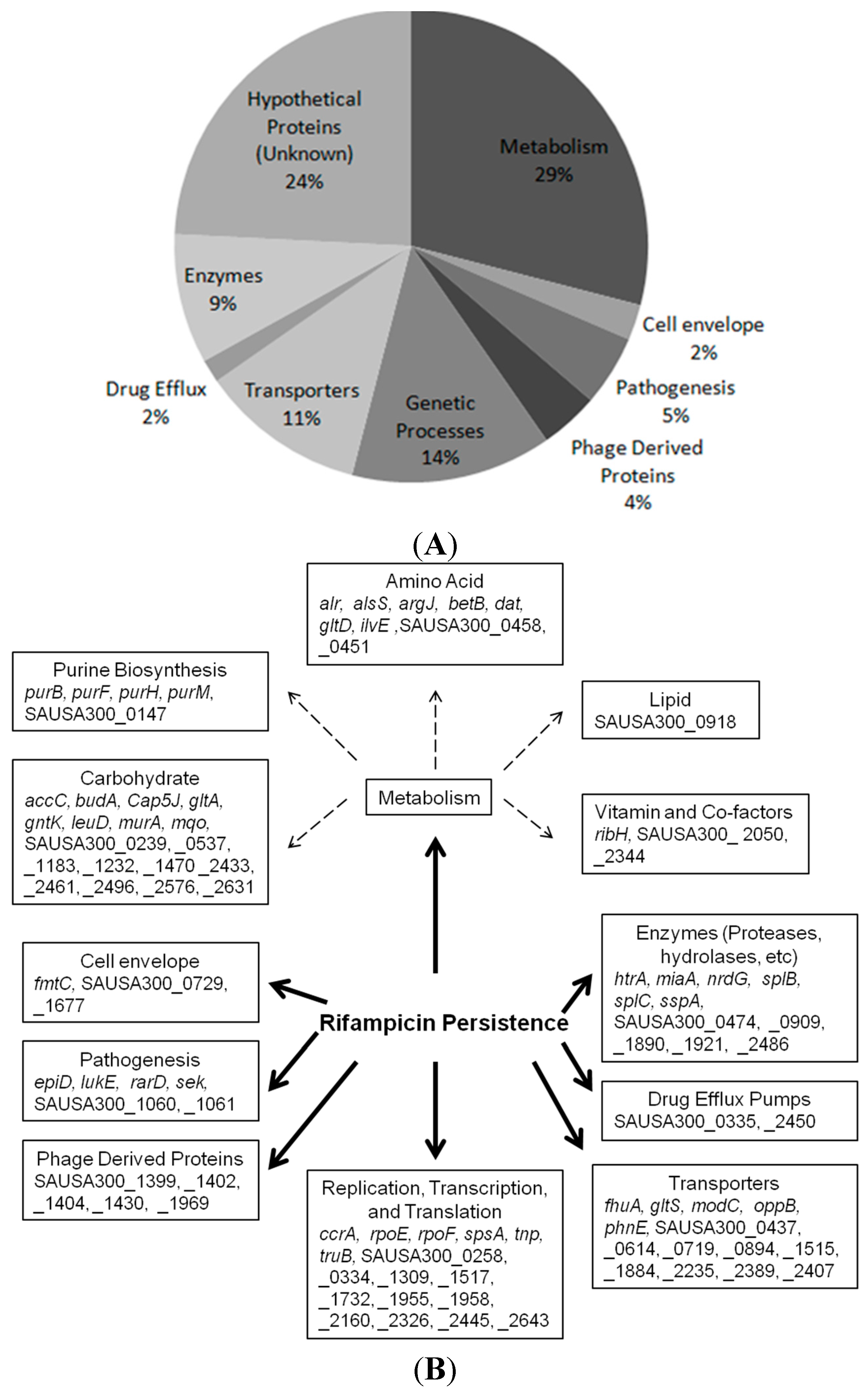
2.1. Identification of New Persister Genes
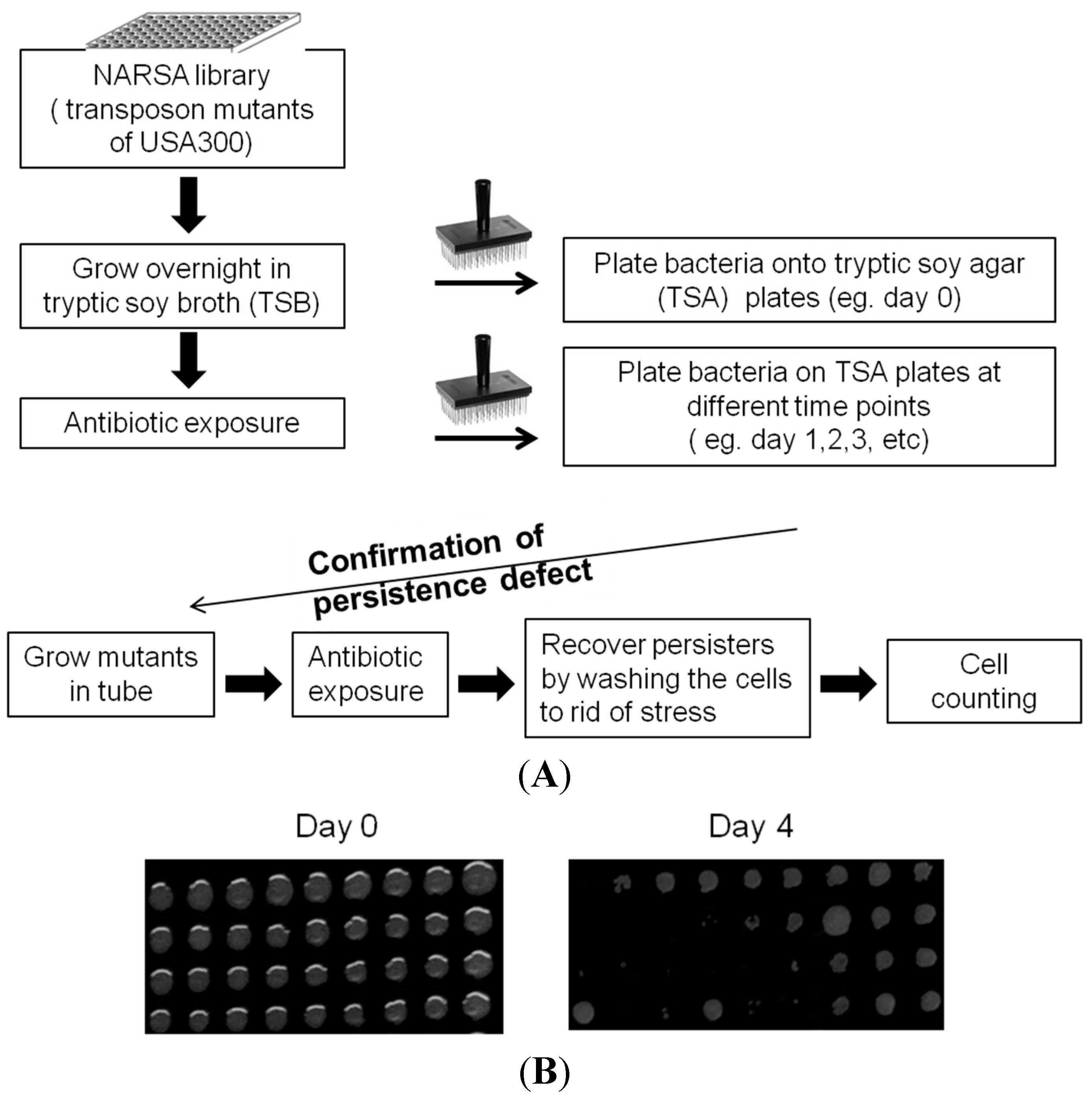
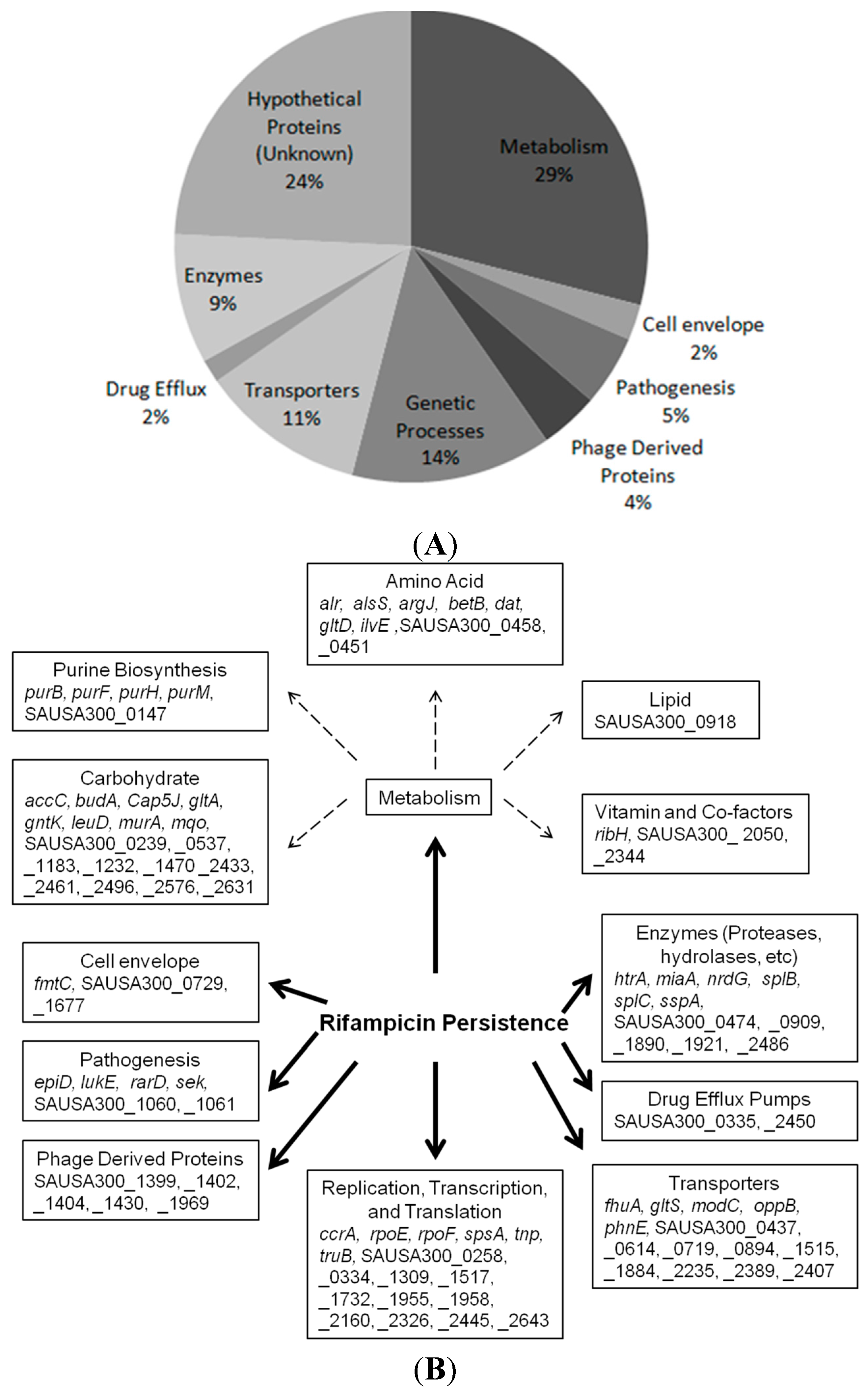
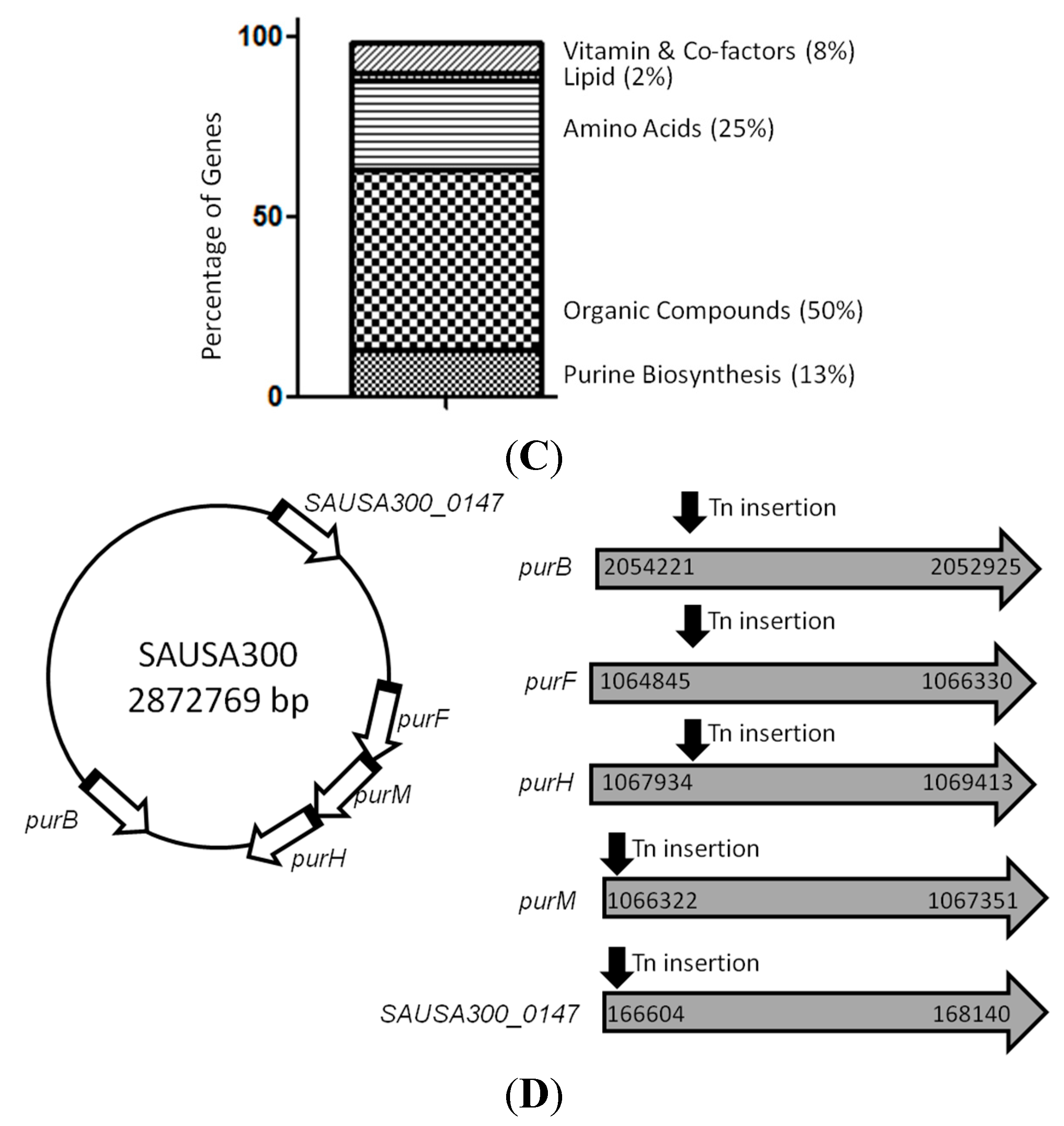
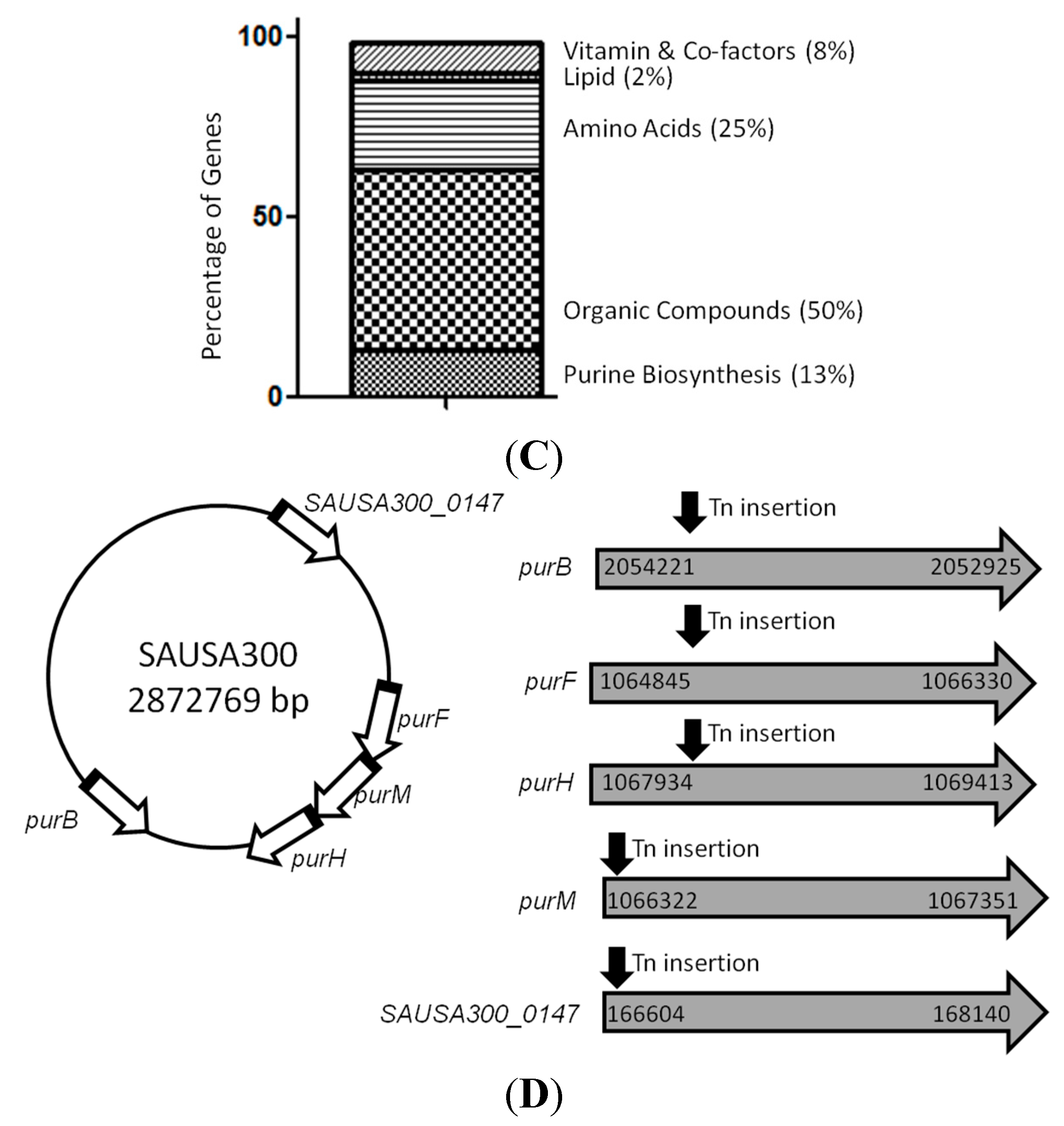
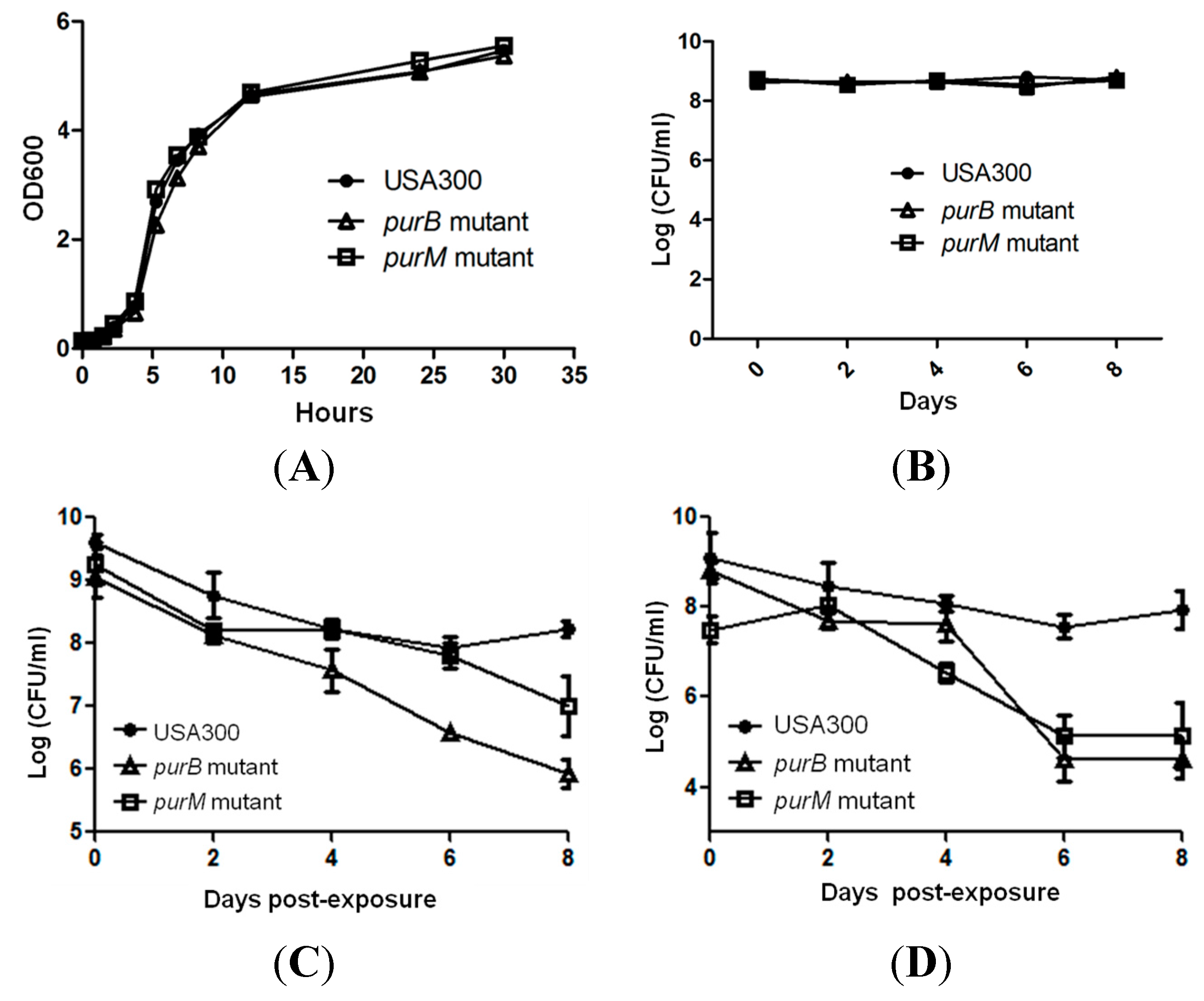
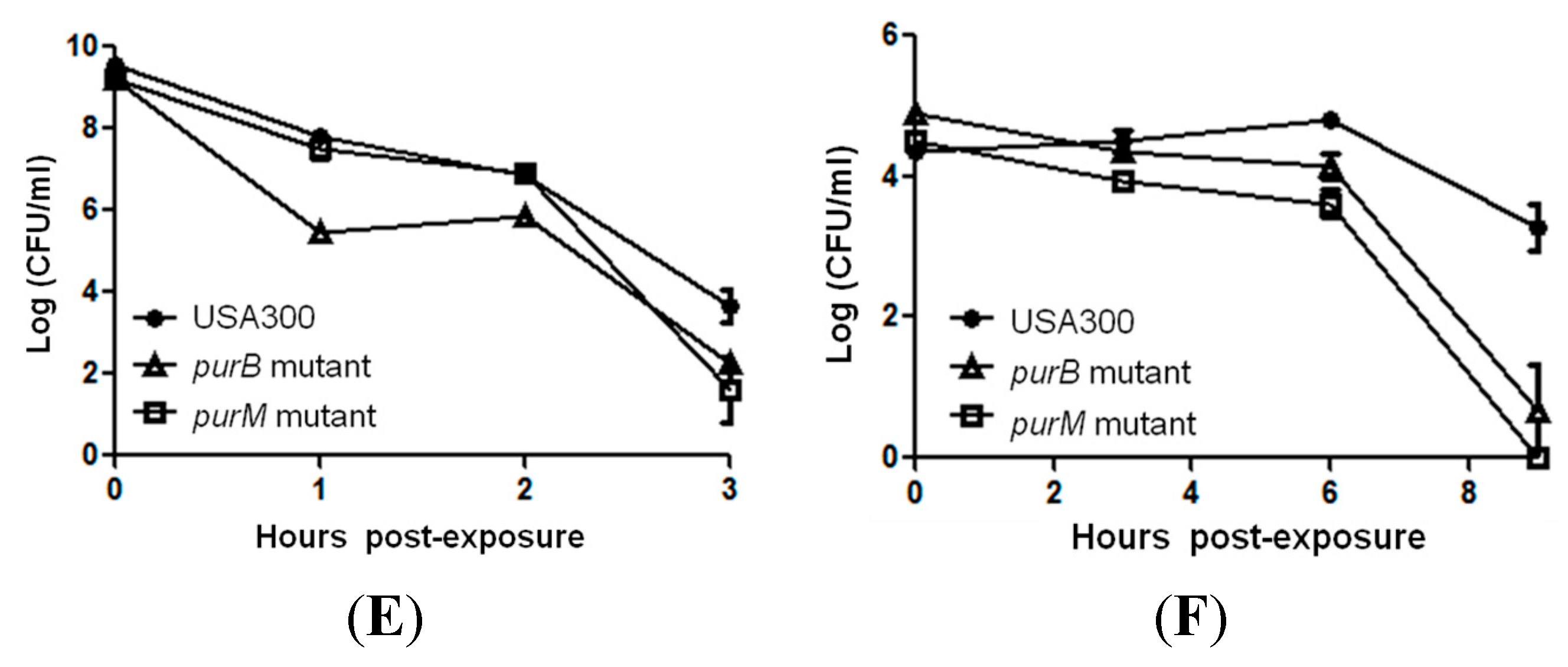
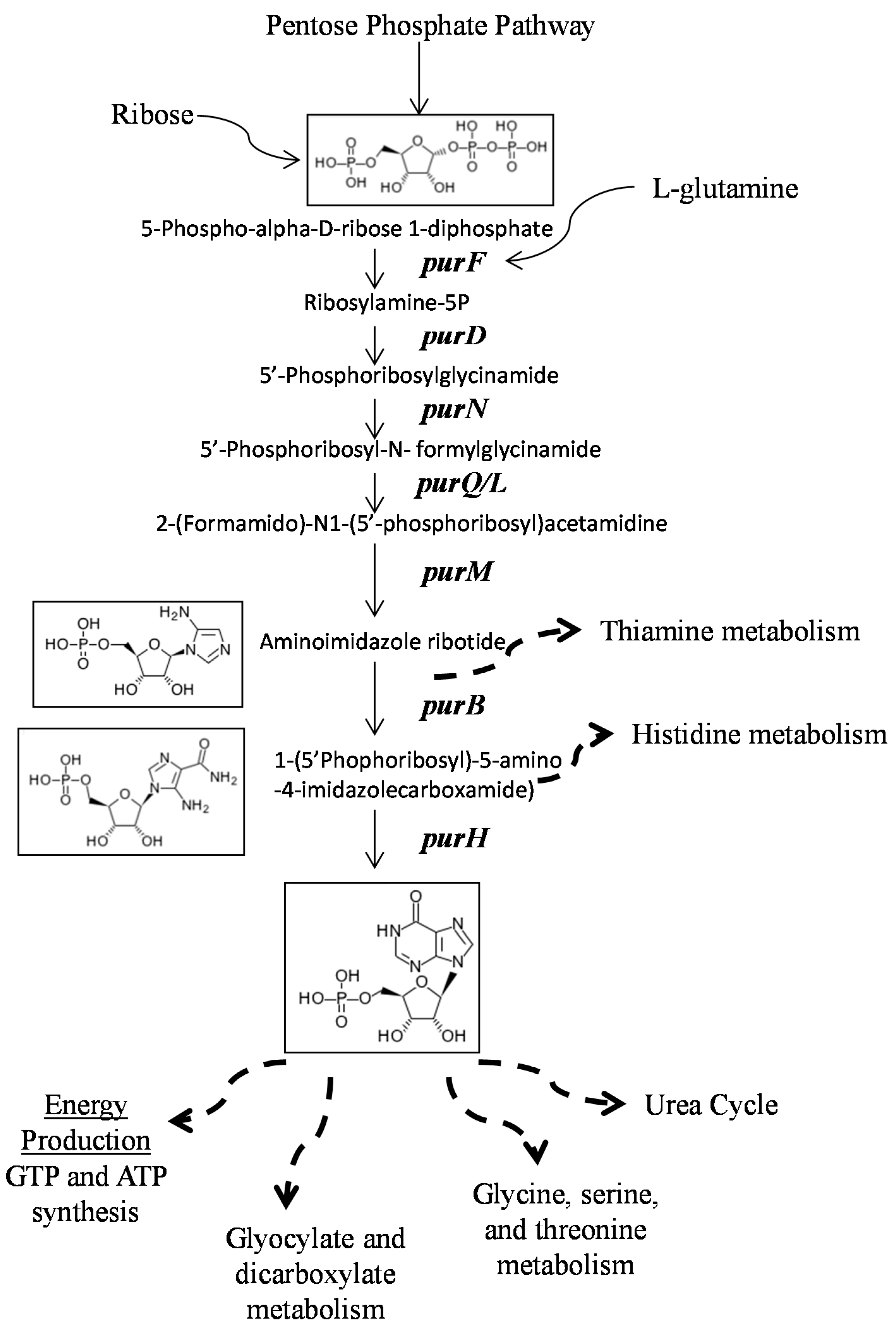
2.2. Mutants Involved in Regulating Purine Biosynthesis are More Susceptible to a Variety of Stresses Including Antibiotics, Heat and Low pH

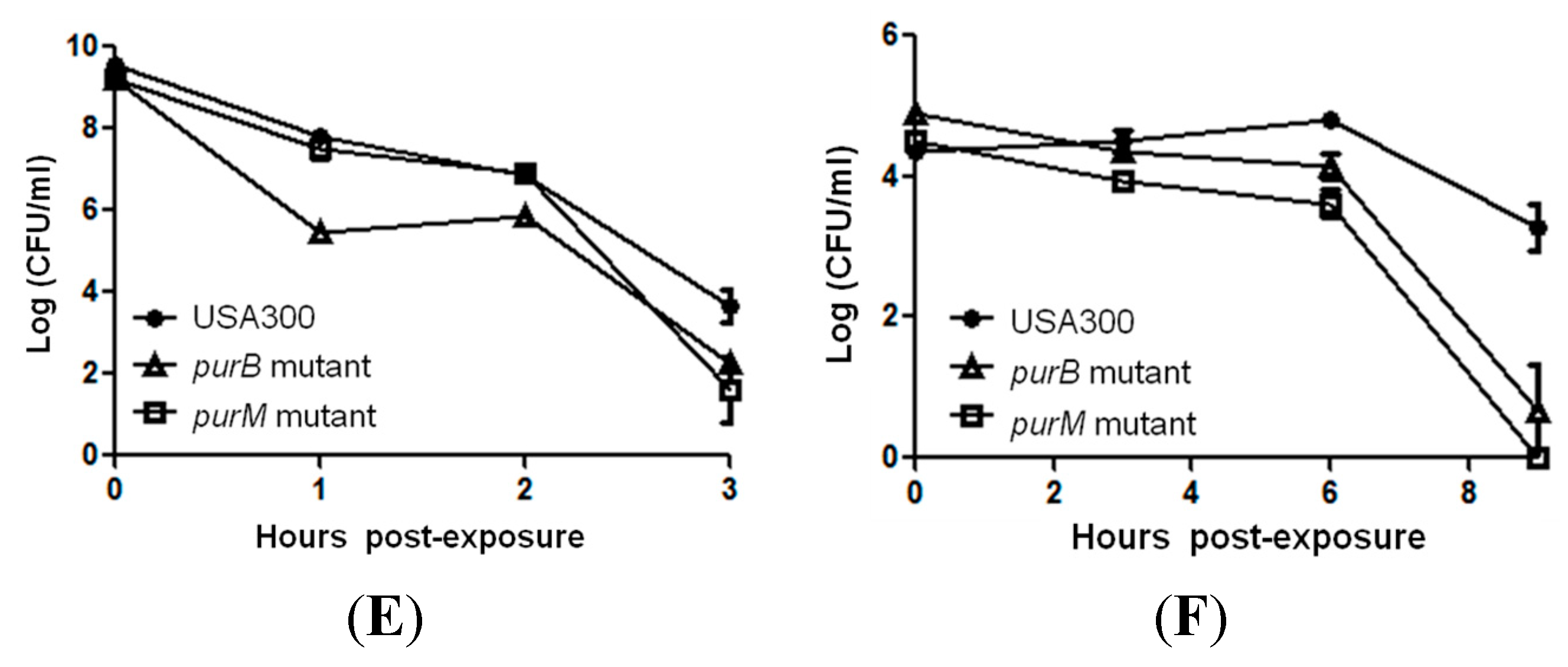
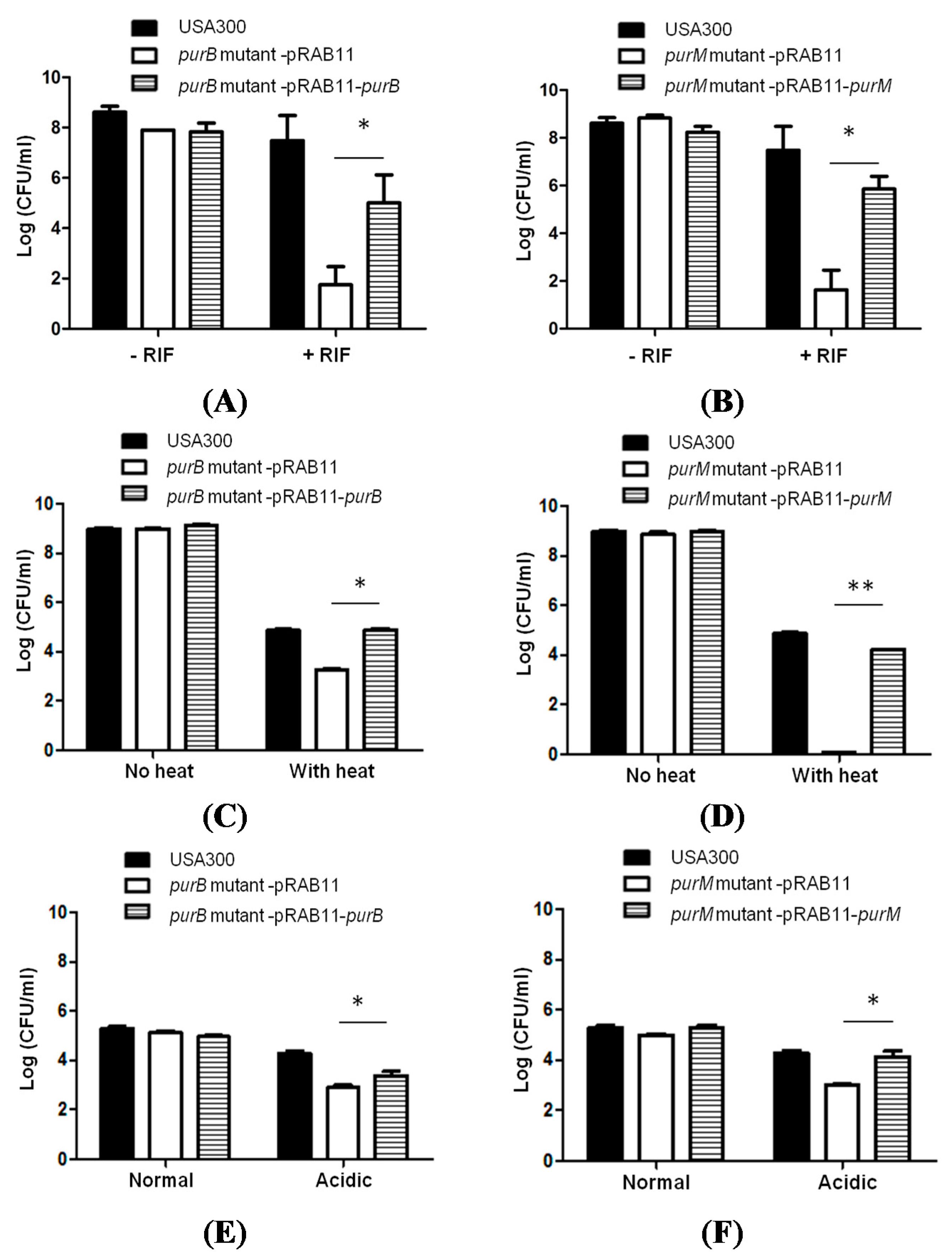
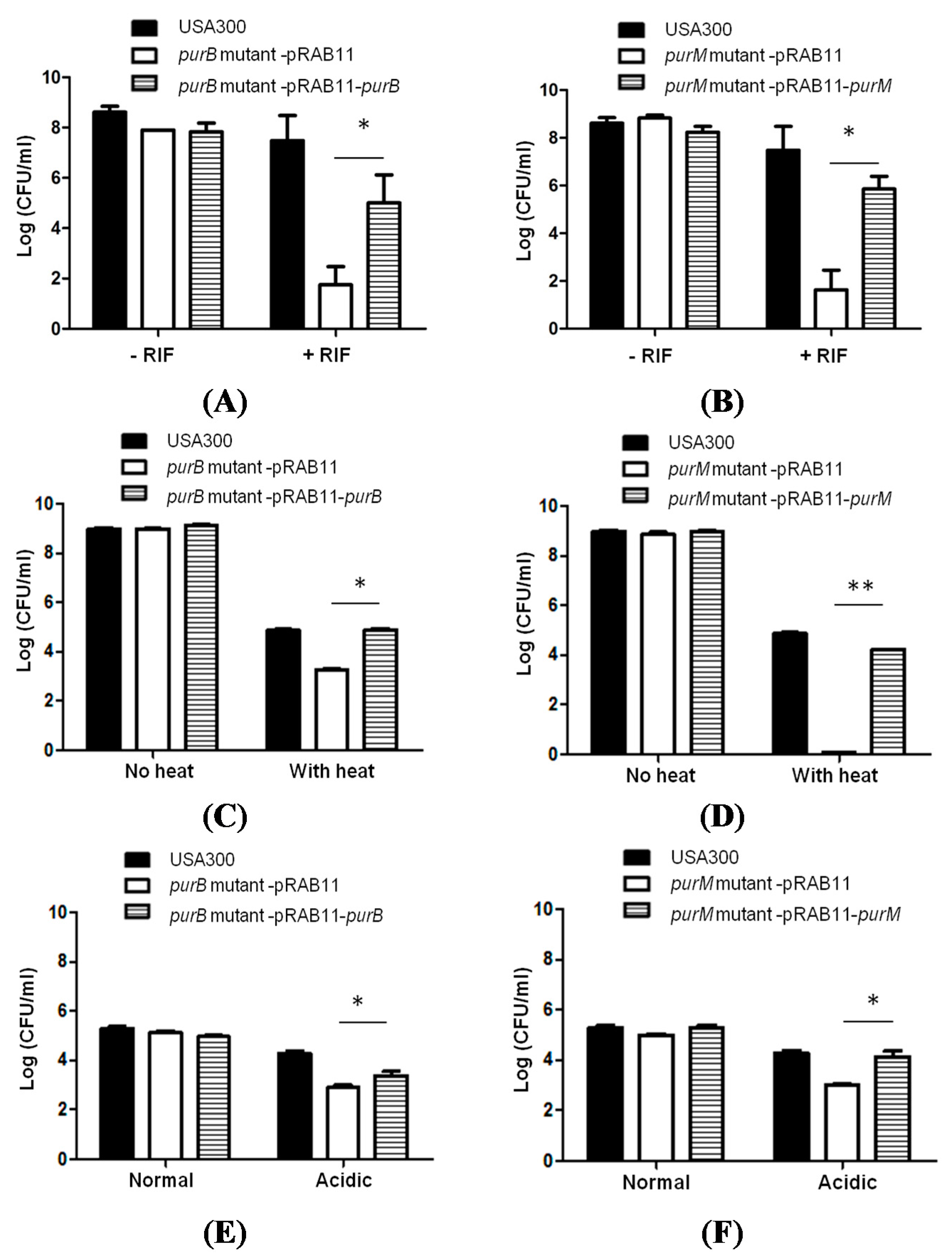
2.3. Complementation Studies to Confirm the Role of Purine Pathways in Persistence

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequence | Source or Reference |
|---|---|---|
| purBF | 5′-GCAAGATCTATGATTGAACGCTATTCTAG-3′ | This study |
| purBR | 5′-ACGGAATTCTTATGCTAATCCAGCGCGTTCG-3′ | This study |
| purMF | 5′-GCTAGATCTATGTCTAAAGCATATGAACAATC-3′ | This study |
| purMR | 5′-ACGGAATTCTTATACCCCCAACAATTCAAT-3′ | This study |
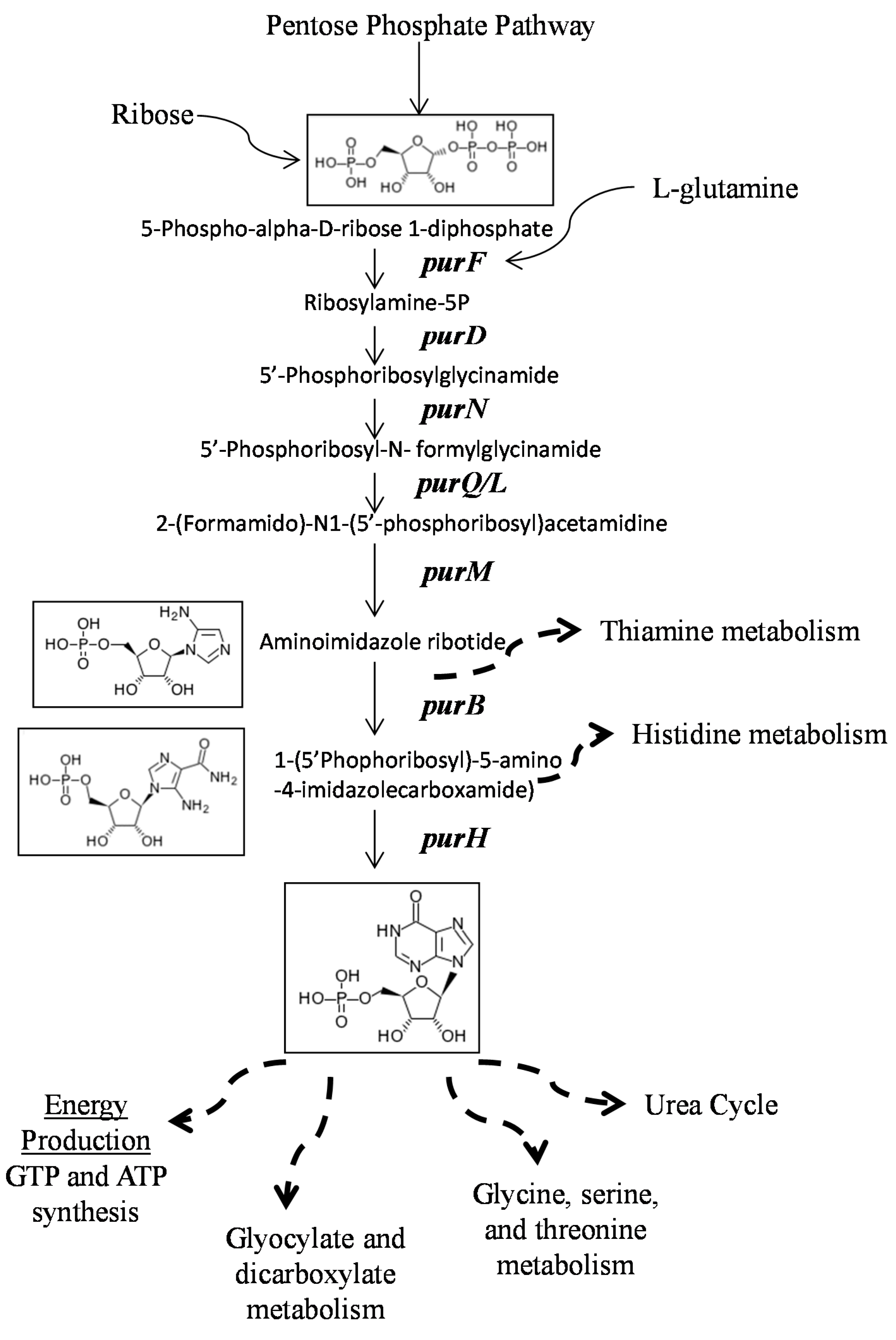
3. Discussion
4. Experimental Section
4.1. Culture Media, Antibiotics, and Chemicals
4.2. Library Screens to Identify Mutants with Defect in Persistence
4.3. Susceptibility of Mutants to Various Antibiotics and Stresses
4.4. Complementation of S. aureus Mutants
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vandenesch, F.; Naimi, T.; Enright, M.C.; Lina, G.; Nimmo, G.R.; Heffernan, H.; Liassine, N.; Bes, M.; Greenland, T.; Reverdy, M.E.; et al. Community-acquired methicillin-resistant Staphylococcus aureus carrying Panton-Valentine leukocidin genes: Worldwide emergence. Emerg. Infect. Dis. 2003, 9, 978–984. [Google Scholar] [CrossRef] [PubMed]
- Chambers, H.F.; Deleo, F.R. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol. 2009, 7, 629–641. [Google Scholar] [CrossRef] [PubMed]
- McDougal, L.K.; Steward, C.D.; Killgore, G.E.; Chaitram, J.M.; McAllister, S.K.; Tenover, F.C. Pulsed-field gel electrophoresis typing of oxacillin-resistant Staphylococcus aureus isolates from the United States: Establishing a national database. J. Clin. Microbiol. 2003, 41, 5113–5120. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.W.; Moran, G.J. Update on emerging infections: News from the Centers for Disease Control and Prevention. Methicillin-resistant Staphylococcus aureus infections among competitive sports participants—Colorado, Indiana, Pennsylvania, and Los Angeles County, 2000–2003. Ann. Emerg. Med. 2004, 43, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Tenover, F.C.; Goering, R.V. Methicillin-resistant Staphylococcus aureus strain USA300: Origin and epidemiology. J. Antimicrob. Chemother. 2009, 64, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Conlon, B.P. Staphylococcus aureus chronic and relapsing infections: Evidence of a role for persister cells: An investigation of persister cells, their formation and their role in S. aureus disease. Bioessays 2014, 36, 991–996. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. Persisters, Persistent Infections and the Yin-Yang Model. Emerg. Microbes Infect. 2014, 3, 10. [Google Scholar] [CrossRef] [PubMed]
- Proctor, R.A.; Kriegeskorte, A.; Kahl, B.C.; Becker, K.; Loffler, B.; Peters, G. Staphylococcus aureus Small Colony Variants (SCVs): A road map for the metabolic pathways involved in persistent infections. Front. Cell. Infect. Microbiol. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Von Eiff, C.; McNamara, P.; Becker, K.; Bates, D.; Lei, X.H.; Ziman, M.; Bochner, B.R.; Peters, G.; Proctor, R.A. Phenotype microarray profiling of Staphylococcus aureus menD and hemB mutants with the small-colony-variant phenotype. J. Bacteriol. 2006, 188, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Clasener, H. Pathogenicity of the L-phase of bacteria. Annu. Rev. Microbiol. 1972, 26, 55–84. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; He, L.; Shi, W.; Xu, X.; Wang, S.; Zhnag, S.; Zhang, Y. Glycerol Uptake is Important for L-Form Formation and Persistence in Staphylococcus aureus. PLoS ONE 2014, 9, e108325. [Google Scholar] [CrossRef] [PubMed]
- Bose, J.L.; Fey, P.D.; Bayles, K.W. Genetic tools to enhance the study of gene function and regulation in Staphylococcus aureus. Appl. Environ. Microbiol. 2013, 79, 2218–2224. [Google Scholar] [CrossRef] [PubMed]
- Korzeniowski, O.; Sande, M.A. Combination antimicrobial therapy for Staphylococcus aureus endocarditis in patients addicted to parenteral drugs and in nonaddicts: A prospective study. Ann. Intern. Med. 1982, 97, 496–503. [Google Scholar] [CrossRef] [PubMed]
- So, T.Y.; Farrington, E. Community-acquired methicillin-resistant Staphylococcus aureus infection in the pediatric population. J. Pediatr. Health Care 2008, 22, 211–217; quiz 218–220. [Google Scholar] [CrossRef] [PubMed]
- Diep, B.A.; Gill, S.R.; Chang, R.F.; Phan, T.H.; Chen, J.H.; Davidson, M.G.; Lin, F.; Lin, J.; Carleton, H.A.; Mongodin, E.F.; et al. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet 2006, 367, 731–739. [Google Scholar] [CrossRef]
- Yee, R.; Cui, P.; Shi, W.; Feng, J.; Zhang, W.; Zhang, Y. Role of arginine biosynthesis pathway in Staphylococcus aureus persistence. PLoS One 2015. under review. [Google Scholar]
- Li, Y.; Zhang, Y. PhoU is a persistence switch involved in persister formation and tolerance to multiple antibiotics and stresses in Escherichia coli. Antimicrob. Agents Chemother. 2007, 51, 2092–2099. [Google Scholar] [CrossRef] [PubMed]
- Helle, L.; Kull, M.; Mayer, S.; Marincola, G.; Zelder, M.E.; Goerke, C.; Wolz, C.; Bertram, R. Vectors for improved Tet repressor-dependent gradual gene induction or silencing in Staphylococcus aureus. Microbiology 2011, 157, 3314–3323. [Google Scholar] [CrossRef] [PubMed]
- Bigger, J.W. Treatment of staphylococcal infections with penicillin by intermittent sterilisation. Lancet 1944, 244, 497–500. [Google Scholar] [CrossRef]
- Ge, X.; Kitten, T.; Chen, Z.; Lee, S.P.; Munro, C.L.; Xu, P. Identification of Streptococcus sanguinis genes required for biofilm formation and examination of their role in endocarditis virulence. Infect. Immun. 2008, 76, 2551–2559. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Jang, H.A.; Won, Y.J.; Kikuchi, Y.; Han, S.H.; Kim, C.H.; Nikoh, N.; Fukatsu, T.; Lee, B.L. Purine biosynthesis-deficient Burkholderia mutants are incapable of symbiotic accommodation in the stinkbug. ISME J. 2014, 8, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Keer, J.; Smeulders, M.J.; Williams, H.D. A purF mutant of Mycobacterium smegmatis has impaired survival during oxygen-starved stationary phase. Microbiology 2001, 147, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Mongodin, E.; Finan, J.; Climo, M.W.; Rosato, A.; Gill, S.; Archer, G.L. Microarray transcription analysis of clinical Staphylococcus aureus isolates resistant to vancomycin. J. Bacteriol. 2003, 185, 4638–4643. [Google Scholar] [CrossRef] [PubMed]
- Glover, W.A.; Yang, Y.; Zhang, Y. Insights into the molecular basis of L-form formation and survival in Escherichia coli. PLoS ONE 2009, 4, e7316. [Google Scholar] [CrossRef] [PubMed]
- Kriel, A.; Bittner, A.N.; Kim, S.H.; Liu, K.; Tehranchi, A.K.; Zou, W.Y.; Rendon, S.; Chen, R.; Tu, B.P.; Wang, J.D. Direct regulation of GTP homeostasis by (p)ppGpp: A critical component of viability and stress resistance. Mol. Cell 2012, 48, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Potrykus, K.; Murphy, H.; Philippe, N.; Cashel, M. ppGpp is the major source of growth rate control in E. coli. Environ. Microbiol. 2011, 13, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Khakimova, M.; Ahlgren, H.G.; Harrison, J.J.; English, A.M.; Nguyen, D. The stringent response controls catalases in Pseudomonas aeruginosa and is required for hydrogen peroxide and antibiotic tolerance. J. Bacteriol. 2013, 195, 2011–2020. [Google Scholar] [CrossRef] [PubMed]
- Thurlow, L.R.; Joshi, G.S.; Clark, J.R.; Spontak, J.S.; Neely, C.J.; Maile, R.; Richardson, A.R. Functional modularity of the arginine catabolic mobile element contributes to the success of USA300 methicillin-resistant Staphylococcus aureus. Cell Host Microbe 2013, 13, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Diep, B.A.; Stone, G.G.; Basuino, L.; Graber, C.J.; Miller, A.; des Etages, S.A.; Jones, A.; Palazzolo-Ballance, A.M.; Perdreau-Remington, F.; Sensabaugh, G.F.; et al. The arginine catabolic mobile element and staphylococcal chromosomal cassette mec linkage: Convergence of virulence and resistance in the USA300 clone of methicillin-resistant Staphylococcus aureus. J. Infect. Dis. 2008, 197, 1523–1530. [Google Scholar] [CrossRef] [PubMed]
- Scherr, T.D.; Roux, C.M.; Hanke, M.L.; Angle, A.; Dunman, P.D.; Kielian, T. Global transcriptome analysis of Staphylococcus aureus biofilms in response to innate immune cells. Infect. Immun. 2013, 81, 4363–4376. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Chisholm, A.D. Methods for skin wounding and assays for wound responses in C. elegans. J. Vis. Exp. 2014. [Google Scholar] [CrossRef] [PubMed]
- Leduc, D.; Gallaud, J.; Stingl, K.; de Reuse, H. Coupled amino acid deamidase-transport systems essential for Helicobacter pylori colonization. Infect. Immun. 2010, 78, 2782–2792. [Google Scholar] [CrossRef] [PubMed]
- Dunning, D.W.; McCall, L.W.; Powell, W.F., Jr.; Arscott, W.T.; McConocha, E.M.; McClurg, C.J.; Goodman, S.D.; Spatafora, G.A. SloR modulation of the Streptococcus mutans acid tolerance response involves the GcrR response regulator as an essential intermediary. Microbiology 2008, 154, 1132–1143. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, G.; Ronca, R.; Cannio, R.; Rossi, M.; Bartolucci, S. MarR-like transcriptional regulator involved in detoxification of aromatic compounds in Sulfolobus solfataricus. J. Bacteriol. 2007, 189, 7351–7360. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.D.; Bubeck Wardenburg, J.; Gardner, D.J.; Long, D.; Whitney, A.R.; Braughton, K.R.; Schneewind, O.; DeLeo, F.R. Targeting of alpha-hemolysin by active or passive immunization decreases severity of USA300 skin infection in a mouse model. J. Infect. Dis. 2010, 202, 1050–1058. [Google Scholar] [CrossRef] [PubMed]
- Rauch, S.; DeDent, A.C.; Kim, H.K.; Bubeck Wardenburg, J.; Missiakas, D.M.; Schneewind, O. Abscess formation and alpha-hemolysin induced toxicity in a mouse model of Staphylococcus aureus peritoneal infection. Infect. Immun. 2012, 80, 3721–3732. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.S.; Dodson, K.W.; Hultgren, S.J. A murine model of urinary tract infection. Nat. Protoc. 2009, 4, 1230–1243. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yee, R.; Cui, P.; Shi, W.; Feng, J.; Zhang, Y. Genetic Screen Reveals the Role of Purine Metabolism in Staphylococcus aureus Persistence to Rifampicin. Antibiotics 2015, 4, 627-642. https://doi.org/10.3390/antibiotics4040627
Yee R, Cui P, Shi W, Feng J, Zhang Y. Genetic Screen Reveals the Role of Purine Metabolism in Staphylococcus aureus Persistence to Rifampicin. Antibiotics. 2015; 4(4):627-642. https://doi.org/10.3390/antibiotics4040627
Chicago/Turabian StyleYee, Rebecca, Peng Cui, Wanliang Shi, Jie Feng, and Ying Zhang. 2015. "Genetic Screen Reveals the Role of Purine Metabolism in Staphylococcus aureus Persistence to Rifampicin" Antibiotics 4, no. 4: 627-642. https://doi.org/10.3390/antibiotics4040627