Host Antimicrobial Peptides in Bacterial Homeostasis and Pathogenesis of Disease

Abstract
:
1. Introduction
2. Modulation of AMPs during Viral Infection Facilitates Bacterial Disease
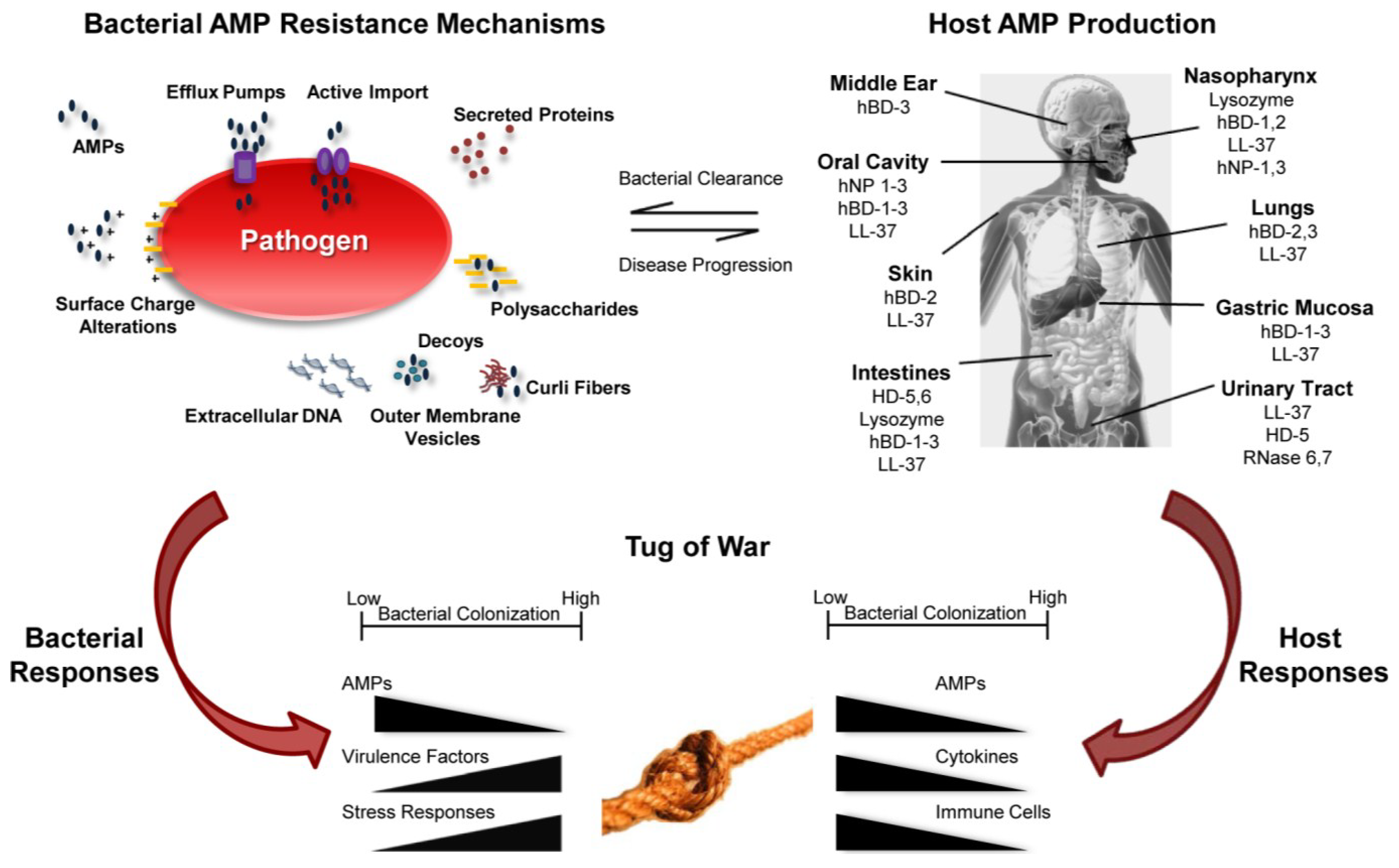
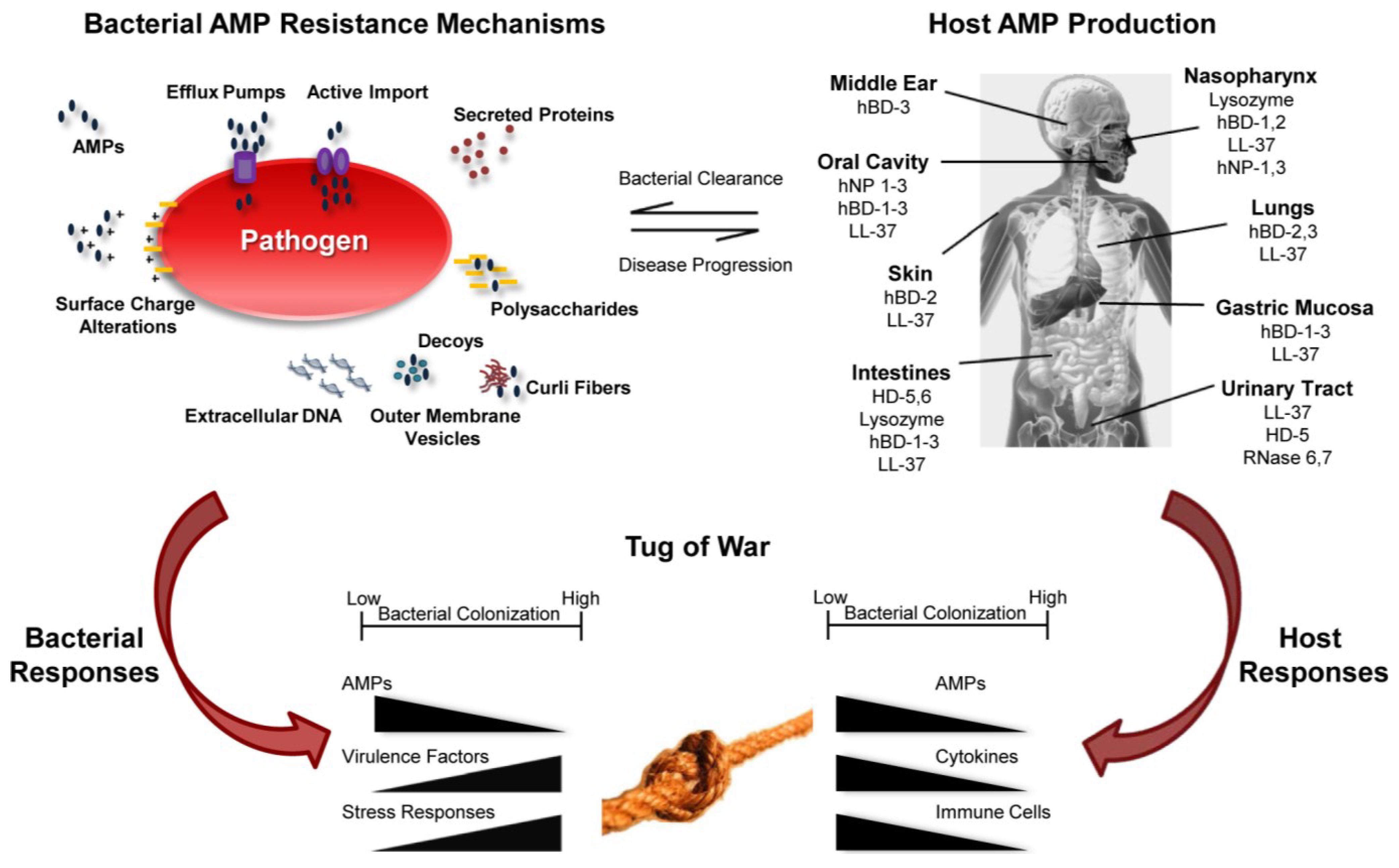
3. Bacterial Mechanisms of AMP Resistance
3.1. Surface Charge Alterations
3.2. Efflux Pumps
3.3. Import of AMPs into the Cytoplasm for Degradation
3.4. Secreted Proteins that Reduce AMP Activity
3.5. Decoys That Sequester AMPs
4. Colonization and Host Microenvironmental Factors That Influence AMP Activity
4.1. AMP Activity of the Skin
4.2. Bacterial Homeostasis of the Nasopharynx
4.3. AMPs and Lung Disease
4.4. AMPs and Periodontal Disease: Oral Cavity
4.5. AMPs and the Gastric Mucosa
4.6. Homeostasis and Diseases of the Intestines
4.7. Urinary Tract Infections
5. Host-Pathogen Tug of War
5.1. AMPs Influence Bacterial Gene Expression
5.2. AMPs Influence Disease Progression

{kind=link}
{kind=link}
| Disease Site | Animal Model | Host AMP Neutralized | Bacteria | Gene | Source |
|---|---|---|---|---|---|
| Disseminated | Drosophila, Mouse | --- | S. aureus | dltA | [39,40] |
| Mouse | --- | Francisella tularensis | naxD | [34] | |
| Mouse | --- | S. Typhimurium | pmrF | [37] | |
| Mouse | --- | Y. pseudotuberculosis | pmrF | [137] | |
| Mouse | --- | GAS | Ralp3, lsa | [150] | |
| Rabbit | --- | S. aureus | lytS | [140] | |
| Intestine | Mouse | CRAMP | E. coli | --- | [119] |
| --- | S. aureus | dltA | [38] | ||
| α-defensins | --- | --- | [114] | ||
| CRAMP | C. rodentium | --- | [113] | ||
| α-defensins | C. trachomatis | --- | [115] | ||
| CRAMP, α-defensins | S. Typhimurium | phoPQ and pmrAB | [148] | ||
| --- | V. cholerae | msbB | [27] | ||
| Kidney | Mouse | --- | S. aureus | apsS | [141] |
| Lung | Mouse | CRAMP | K. pneumoniae, P. aeruginosa | --- | [89] |
| Middle Ear | Chinchilla | CβD-1 | NTHI | sapA | [50] |
| Nasopharynx | Chinchilla | CβD-1 | NTHI | --- | [21] |
| Skin | Human | --- | NTHI | sapBC | [51] |
| Mouse | CRAMP | GAS | --- | [78] | |
| Stomach | C. elegans | Spp1 | S. typhimurium | phoPQ | [149] |
| Urogenital Tract | Mouse, Human | --- | N. gonorrhoeae | lptA | [35,36] |
| Mouse | CRAMP | E. coli | --- | [121] |
5.3. Using What We Know About AMPs for Potential Therapeutic Use
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hoffmann, J.A.; Kafatos, F.C.; Janeway, C.A.; Ezekowitz, R.A. Phylogenetic perspectives in innate immunity. Science 1999, 284, 1313–1318. [Google Scholar]
- Finlay, B.B.; Hancock, R.E. Can innate immunity be enhanced to treat microbial infections? Nat. Rev. Microbiol. 2004, 2, 497–504. [Google Scholar]
- Hancock, R.E.; Chapple, D.S. Peptide antibiotics. Antimicrob. Agents Chemother. 1999, 43, 1317–1323. [Google Scholar] [PubMed]
- Peschel, A. How do bacteria resist human antimicrobial peptides? Trends Microbiol. 2002, 10, 179–186. [Google Scholar] [CrossRef] [PubMed]
- White, S.H.; Wimley, W.C.; Selsted, M.E. Structure, function, and membrane integration of defensins. Curr. Opin. Struct. Biol. 1995, 5, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Mason, K.M.; Bruggeman, M.E.; Munson, R.S.; Bakaletz, L.O. The non-typeable Haemophilus influenzae Sap transporter provides a mechanism of antimicrobial peptide resistance and SapD-dependent potassium acquisition. Mol. Microbiol. 2006, 62, 1357–1372. [Google Scholar] [CrossRef]
- Gries, C.M.; Bose, J.L.; Nuxoll, A.S.; Fey, P.D.; Bayles, K.W. The Ktr potassium transport system in Staphylococcus aureus and its role in cell physiology, antimicrobial resistance and pathogenesis. Mol. Microbiol. 2013, 89, 760–773. [Google Scholar] [CrossRef]
- Boyton, R.J.; Openshaw, P.J. Pulmonary defences to acute respiratory infection. Br. Med. Bull. 2002, 61, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Krijgsveld, J.; Zaat, S.A.; Meeldijk, J.; van Veelen, P.A.; Fang, G.; Poolman, B.; Brandt, E.; Ehlert, J.E.; Kuijpers, A.J.; Engbers, G.H.; et al. Thrombocidins, microbicidal proteins from human blood platelets, are C-terminal deletion products of CXC chemokines. J. Biol. Chem. 2000, 275, 20374–20381. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, R.I.; Ganz, T. Cathelicidins: A family of endogenous antimicrobial peptides. Curr. Opin. Hematol. 2002, 9, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, F.; Pufe, T.; Conradi, L.; Varoga, D.; Tsokos, M.; Papendieck, J.; Petersen, W. Antimicrobial peptides are expressed and produced in healthy and inflamed human synovial membranes. J. Pathol. 2002, 198, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Raj, P.A.; Dentino, A.R. Current status of defensins and their role in innate and adaptive immunity. FEMS Microbiol. Lett. 2002, 206, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Guilhelmelli, F.; Vilela, N.; Albuquerque, P.; Derengowski Lda, S.; Silva-Pereira, I.; Kyaw, C.M. Antibiotic development challenges: The various mechanisms of action of antimicrobial peptides and of bacterial resistance. Front. Microbiol. 2013, 4, e353. [Google Scholar]
- Tecle, T.; Tripathi, S.; Hartshorn, K.L. Review: Defensins and cathelicidins in lung immunity. Innate Immun. 2010, 16, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Bernard, J.J.; Gallo, R.L. Protecting the boundary: The sentinel role of host defense peptides in the skin. Cell Mol. Life Sci. 2011, 68, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Salzman, N.H.; Underwood, M.A.; Bevins, C.L. Paneth cells, defensins, and the commensal microbiota: A hypothesis on intimate interplay at the intestinal mucosa. Semin. Immunol. 2007, 19, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, B.R.; de Freitas, V.A.; Nascimento-Neto, L.G.; Carneiro, V.A.; Arruda, F.V.; de Aguiar, A.S.; Cavada, B.S.; Teixeira, E.H. Antimicrobial peptide control of pathogenic microorganisms of the oral cavity: A review of the literature. Peptides 2012, 36, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Gorr, S.U. Antimicrobial peptides of the oral cavity. Periodontology 2009, 51, 152–180. [Google Scholar] [CrossRef]
- Ali, A.S.; Townes, C.L.; Hall, J.; Pickard, R.S. Maintaining a sterile urinary tract: The role of antimicrobial peptides. J. Urol. 2009, 182, 21–28. [Google Scholar] [CrossRef] [PubMed]
- McGillivary, G.; Ray, W.C.; Bevins, C.L.; Munson, R.S., Jr.; Bakaletz, L.O. A member of the cathelicidin family of antimicrobial peptides is produced in the upper airway of the chinchilla and its mRNA expression is altered by common viral and bacterial co-pathogens of otitis media. Mol. Immunol. 2007, 44, 2446–2458. [Google Scholar] [CrossRef]
- McGillivary, G.; Mason, K.M.; Jurcisek, J.A.; Peeples, M.E.; Bakaletz, L.O. Respiratory syncytial virus-induced dysregulation of expression of a mucosal beta-defensin augments colonization of the upper airway by non-typeable Haemophilus influenzae. Cell Microbiol. 2009, 11, 1399–1408. [Google Scholar] [CrossRef] [PubMed]
- Mallia, P.; Footitt, J.; Sotero, R.; Jepson, A.; Contoli, M.; Trujillo-Torralbo, M.B.; Kebadze, T.; Aniscenko, J.; Oleszkiewicz, G.; Gray, K.; et al. Rhinovirus infection induces degradation of antimicrobial peptides and secondary bacterial infection in chronic obstructive pulmonary disease. Am. J. Respir Crit. Care Med. 2012, 186, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.M.; McHugh, K.J.; Mandalapu, S.; Clay, M.E.; Lee, B.; Scheller, E.V.; Enelow, R.I.; Chan, Y.R.; Kolls, J.K.; Alcorn, J.F. Influenza A virus exacerbates Staphylococcus aureus pneumonia in mice by attenuating antimicrobial peptide production. J. Infect. Dis. 2014, 209, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Lysenko, E.S.; Gould, J.; Bals, R.; Wilson, J.M.; Weiser, J.N. Bacterial phosphorylcholine decreases susceptibility to the antimicrobial peptide LL-37/hCAP18 expressed in the upper respiratory tract. Infect. Immun. 2000, 68, 1664–1671. [Google Scholar] [CrossRef]
- Taneja, N.K.; Ganguly, T.; Bakaletz, L.O.; Nelson, K.J.; Dubey, P.; Poole, L.B.; Deora, R. D-Alanine modification of a protease-susceptible outer membrane component by the Bordetella pertussis dra locus promotes resistance to antimicrobial peptides and polymorphonuclear leukocyte-mediated killing. J. Bacteriol. 2013, 195, 5102–5111. [Google Scholar] [CrossRef] [PubMed]
- Starner, T.D.; Swords, W.E.; Apicella, M.A.; McCray, P.B., Jr. Susceptibility of nontypeable Haemophilus influenzae to human beta-defensins is influenced by lipooligosaccharide acylation. Infect. Immun 2002, 70, 5287–5289. [Google Scholar] [CrossRef] [PubMed]
- Matson, J.S.; Yoo, H.J.; Hakansson, K.; Dirita, V.J. Polymyxin B resistance in El Tor Vibrio cholerae requires lipid acylation catalyzed by MsbB. J. Bacteriol. 2010, 192, 2044–2052. [Google Scholar] [CrossRef] [PubMed]
- Ouhara, K.; Komatsuzawa, H.; Kawai, T.; Nishi, H.; Fujiwara, T.; Fujiue, Y.; Kuwabara, M.; Sayama, K.; Hashimoto, K.; Sugai, M. Increased resistance to cationic antimicrobial peptide LL-37 in methicillin-resistant strains of Staphylococcus aureus. J. Antimicrob. Chemother. 2008, 61, 1266–1269. [Google Scholar] [CrossRef] [PubMed]
- Fabretti, F.; Theilacker, C.; Baldassarri, L.; Kaczynski, Z.; Kropec, A.; Holst, O.; Huebner, J. Alanine esters of enterococcal lipoteichoic acid play a role in biofilm formation and resistance to antimicrobial peptides. Infect. Immun. 2006, 74, 4164–4171. [Google Scholar] [CrossRef] [PubMed]
- Kristian, S.A.; Datta, V.; Weidenmaier, C.; Kansal, R.; Fedtke, I.; Peschel, A.; Gallo, R.L.; Nizet, V. D-Alanylation of teichoic acids promotes group a streptococcus antimicrobial peptide resistance, neutrophil survival, and epithelial cell invasion. J. Bacteriol. 2005, 187, 6719–6725. [Google Scholar] [CrossRef] [PubMed]
- Peschel, A.; Otto, M.; Jack, R.W.; Kalbacher, H.; Jung, G.; Gotz, F. Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J. Biol. Chem. 1999, 274, 8405–8410. [Google Scholar] [CrossRef] [PubMed]
- Herbert, S.; Bera, A.; Nerz, C.; Kraus, D.; Peschel, A.; Goerke, C.; Meehl, M.; Cheung, A.; Gotz, F. Molecular basis of resistance to muramidase and cationic antimicrobial peptide activity of lysozyme in staphylococci. PLoS Pathog. 2007, 3, e102. [Google Scholar] [CrossRef] [PubMed]
- Morey, P.; Viadas, C.; Euba, B.; Hood, D.W.; Barberan, M.; Gil, C.; Grillo, M.J.; Bengoechea, J.A.; Garmendia, J. Relative contributions of lipooligosaccharide inner and outer core modifications to nontypeable Haemophilus influenzae pathogenesis. Infect. Immun. 2013, 81, 4100–4111. [Google Scholar] [CrossRef] [PubMed]
- Llewellyn, A.C.; Zhao, J.; Song, F.; Parvathareddy, J.; Xu, Q.; Napier, B.A.; Laroui, H.; Merlin, D.; Bina, J.E.; Cotter, P.A.; et al. NaxD is a deacetylase required for lipid A modification and Francisella pathogenesis. Mol. Microbiol. 2012, 86, 611–627. [Google Scholar] [CrossRef] [PubMed]
- Packiam, M.; Yedery, R.D.; Begum, A.A.; Carlson, R.W.; Ganguly, J.; Sempowski, G.D.; Ventevogel, M.S.; Shafer, W.M.; Jerse, A.E. Phosphoethanolamine decoration of Neisseria gonorrhoeae lipid A plays a dual immunostimulatory and protective role during experimental genital tract infection. Infect. Immun. 2014, 82, 2170–2179. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, M.M.; Anderson, J.E.; Balthazar, J.T.; Kandler, J.L.; Carlson, R.W.; Ganguly, J.; Begum, A.A.; Duncan, J.A.; Lin, J.T.; Sparling, P.F.; et al. Lipid Aʼs structure mediates Neisseria gonorrhoeae fitness during experimental infection of mice and men. MBio 2013, 4. [Google Scholar] [CrossRef]
- Gunn, J.S.; Ryan, S.S.; van Velkinburgh, J.C.; Ernst, R.K.; Miller, S.I. Genetic and functional analysis of a PmrA-PmrB-regulated locus necessary for lipopolysaccharide modification, antimicrobial peptide resistance, and oral virulence of Salmonella enterica serovar typhimurium. Infect. Immun. 2000, 68, 6139–6146. [Google Scholar] [PubMed]
- Walter, J.; Loach, D.M.; Alqumber, M.; Rockel, C.; Hermann, C.; Pfitzenmaier, M.; Tannock, G.W. D-Alanyl ester depletion of teichoic acids in Lactobacillus reuteri 100-23 results in impaired colonization of the mouse gastrointestinal tract. Environ. Microbiol 2007, 9, 1750–1760. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.V.; Kristian, S.A.; Weidenmaier, C.; Faigle, M.; van Kessel, K.P.; van Strijp, J.A.; Gotz, F.; Neumeister, B.; Peschel, A. Staphylococcus aureus strains lacking D-alanine modifications of teichoic acids are highly susceptible to human neutrophil killing and are virulence attenuated in mice. J. Infect. Dis. 2002, 186, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Tabuchi, Y.; Shiratsuchi, A.; Kurokawa, K.; Gong, J.H.; Sekimizu, K.; Lee, B.L.; Nakanishi, Y. Inhibitory role for D-alanylation of wall teichoic acid in activation of insect Toll pathway by peptidoglycan of Staphylococcus aureus. J. Immunol. 2010, 185, 2424–2431. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.J.; Kuo, T.Y.; Lin, C.C.; Chow, L.P.; Chen, W.J. Proteomic identification of membrane proteins regulating antimicrobial peptide resistance in Vibrio parahaemolyticus. J. Appl. Microbiol. 2010, 108, 1398–1407. [Google Scholar] [CrossRef]
- Shafer, W.M.; Qu, X.; Waring, A.J.; Lehrer, R.I. Modulation of Neisseria gonorrhoeae susceptibility to vertebrate antibacterial peptides due to a member of the resistance/nodulation/division efflux pump family. Proc. Natl. Acad. Sci. USA 1998, 95, 1829–1833. [Google Scholar] [CrossRef]
- Tzeng, Y.L.; Ambrose, K.D.; Zughaier, S.; Zhou, X.; Miller, Y.K.; Shafer, W.M.; Stephens, D.S. Cationic antimicrobial peptide resistance in Neisseria meningitidis. J. Bacteriol. 2005, 187, 5387–5396. [Google Scholar] [CrossRef] [PubMed]
- Zahner, D.; Zhou, X.; Chancey, S.T.; Pohl, J.; Shafer, W.M.; Stephens, D.S. Human antimicrobial peptide LL-37 induces MefE/Mel-mediated macrolide resistance in Streptococcus pneumoniae. Antimicrob. Agents Chemother. 2010, 54, 3516–3519. [Google Scholar] [CrossRef] [PubMed]
- Rinker, S.D.; Trombley, M.P.; Gu, X.; Fortney, K.R.; Bauer, M.E. Deletion of mtrC in Haemophilus ducreyi increases sensitivity to human antimicrobial peptides and activates the CpxRA regulon. Infect. Immun. 2011, 79, 2324–2334. [Google Scholar] [CrossRef] [PubMed]
- Rieg, S.; Huth, A.; Kalbacher, H.; Kern, W.V. Resistance against antimicrobial peptides is independent of Escherichia coli AcrAB, Pseudomonas aeruginosa MexAB and Staphylococcus aureus NorA efflux pumps. Int. J. Antimicrob. Agents 2009, 33, 174–176. [Google Scholar] [CrossRef] [PubMed]
- Parra-Lopez, C.; Baer, M.T.; Groisman, E.A. Molecular genetic analysis of a locus required for resistance to antimicrobial peptides in Salmonella typhimurium. EMBO J. 1993, 12, 4053–4062. [Google Scholar] [PubMed]
- Mason, K.M.; Munson, R.S., Jr.; Bakaletz, L.O. Nontypeable Haemophilus influenzae gene expression induced in vivo in a chinchilla model of otitis media. Infect. Immun. 2003, 71, 3454–3462. [Google Scholar] [CrossRef] [PubMed]
- Mason, K.M.; Munson, R.S., Jr.; Bakaletz, L.O. A mutation in the sap operon attenuates survival of nontypeable Haemophilus influenzae in a chinchilla model of otitis media. Infect. Immun. 2005, 73, 599–608. [Google Scholar] [CrossRef]
- Shelton, C.L.; Raffel, F.K.; Beatty, W.L.; Johnson, S.M.; Mason, K.M. Sap transporter mediated import and subsequent degradation of antimicrobial peptides in Haemophilus. PLoS Pathog. 2011, 7, e1002360. [Google Scholar] [CrossRef] [PubMed]
- Rinker, S.D.; Gu, X.; Fortney, K.R.; Zwickl, B.W.; Katz, B.P.; Janowicz, D.M.; Spinola, S.M.; Bauer, M.E. Permeases of the sap transporter are required for cathelicidin resistance and virulence of Haemophilus ducreyi in humans. J. Infect. Dis. 2012, 206, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Runti, G.; Lopez Ruiz Mdel, C.; Stoilova, T.; Hussain, R.; Jennions, M.; Choudhury, H.G.; Benincasa, M.; Gennaro, R.; Beis, K.; Scocchi, M. Functional characterization of SbmA, a bacterial inner membrane transporter required for importing the antimicrobial peptide Bac7(1-35). J. Bacteriol. 2013, 195, 5343–5351. [Google Scholar] [CrossRef] [PubMed]
- Schmidtchen, A.; Frick, I.M.; Andersson, E.; Tapper, H.; Bjorck, L. Proteinases of common pathogenic bacteria degrade and inactivate the antibacterial peptide LL-37. Mol. Microbiol. 2002, 46, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Villaruz, A.E.; Li, M.; Cha, D.J.; Sturdevant, D.E.; Otto, M. The human anionic antimicrobial peptide dermcidin induces proteolytic defence mechanisms in staphylococci. Mol. Microbiol. 2007, 63, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Puklo, M.; Guentsch, A.; Hiemstra, P.S.; Eick, S.; Potempa, J. Analysis of neutrophil-derived antimicrobial peptides in gingival crevicular fluid suggests importance of cathelicidin LL-37 in the innate immune response against periodontogenic bacteria. Oral Microbiol. Immunol. 2008, 23, 328–335. [Google Scholar] [CrossRef] [PubMed]
- McCrudden, M.T.; Orr, D.F.; Yu, Y.; Coulter, W.A.; Manning, G.; Irwin, C.R.; Lundy, F.T. LL-37 in periodontal health and disease and its susceptibility to degradation by proteinases present in gingival crevicular fluid. J. Clin. Periodontol. 2013, 40, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Hollands, A.; Gonzalez, D.; Leire, E.; Donald, C.; Gallo, R.L.; Sanderson-Smith, M.; Dorrestein, P.C.; Nizet, V. A bacterial pathogen co-opts host plasmin to resist killing by cathelicidin antimicrobial peptides. J. Biol. Chem. 2012, 287, 40891–40897. [Google Scholar] [CrossRef]
- Braff, M.H.; Jones, A.L.; Skerrett, S.J.; Rubens, C.E. Staphylococcus aureus exploits cathelicidin antimicrobial peptides produced during early pneumonia to promote staphylokinase-dependent fibrinolysis. J. Infect. Dis. 2007, 195, 1365–1372. [Google Scholar] [CrossRef]
- Bokarewa, M.; Tarkowski, A. Human alpha-defensins neutralize fibrinolytic activity exerted by staphylokinase. Thromb. Haemost. 2004, 91, 991–999. [Google Scholar] [PubMed]
- Herasimenka, Y.; Benincasa, M.; Mattiuzzo, M.; Cescutti, P.; Gennaro, R.; Rizzo, R. Interaction of antimicrobial peptides with bacterial polysaccharides from lung pathogens. Peptides 2005, 26, 1127–1132. [Google Scholar] [CrossRef] [PubMed]
- Llobet, E.; Tomas, J.M.; Bengoechea, J.A. Capsule polysaccharide is a bacterial decoy for antimicrobial peptides. Microbiology 2008, 154, 3877–3886. [Google Scholar] [CrossRef] [PubMed]
- Islam, D.; Bandholtz, L.; Nilsson, J.; Wigzell, H.; Christensson, B.; Agerberth, B.; Gudmundsson, G. Downregulation of bactericidal peptides in enteric infections: A novel immune escape mechanism with bacterial DNA as a potential regulator. Nat. Med. 2001, 7, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Kai-Larsen, Y.; Luthje, P.; Chromek, M.; Peters, V.; Wang, X.; Holm, A.; Kadas, L.; Hedlund, K.O.; Johansson, J.; Chapman, M.R.; et al. Uropathogenic Escherichia coli modulates immune responses and its curli fimbriae interact with the antimicrobial peptide LL-37. PLoS Pathog. 2010, 6, e1001010. [Google Scholar] [CrossRef] [PubMed]
- Vuong, C.; Voyich, J.M.; Fischer, E.R.; Braughton, K.R.; Whitney, A.R.; DeLeo, F.R.; Otto, M. Polysaccharide intercellular adhesin (PIA) protects Staphylococcus epidermidis against major components of the human innate immune system. Cell Microbiol. 2004, 6, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.; Horsman, S.R.; Charron-Mazenod, L.; Turnbull, A.L.; Mulcahy, H.; Surette, M.G.; Lewenza, S. Extracellular DNA-induced antimicrobial peptide resistance in Salmonella enterica serovar Typhimurium. BMC Microbiol. 2013, 13, e115. [Google Scholar] [CrossRef]
- Lewenza, S. Extracellular DNA-induced antimicrobial peptide resistance mechanisms in Pseudomonas aeruginosa. Front. Microbiol. 2013, 4, e21. [Google Scholar] [CrossRef]
- Jones, E.A.; McGillivary, G.; Bakaletz, L.O. Extracellular DNA within a nontypeable Haemophilus influenzae-induced biofilm binds human beta defensin-3 and reduces its antimicrobial activity. J. Innate Immun. 2013, 5, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Duperthuy, M.; Sjostrom, A.E.; Sabharwal, D.; Damghani, F.; Uhlin, B.E.; Wai, S.N. Role of the Vibrio cholerae matrix protein Bap1 in cross-resistance to antimicrobial peptides. PLoS Pathog. 2013, 9, e1003620. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.T.; Stenger, S.; Li, H.; Wenzel, L.; Tan, B.H.; Krutzik, S.R.; Ochoa, M.T.; Schauber, J.; Wu, K.; Meinken, C.; et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 2006, 311, 1770–1773. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhao, L.; Ma, L.; Lv, C.; Ding, Y.; Xia, T.; Wang, J.; Dou, X. Vitamin D status and expression of vitamin D receptor and LL-37 in patients with spontaneous bacterial peritonitis. Dig. Dis. Sci. 2012, 57, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Love, J.F.; Tran-Winkler, H.J.; Wessels, M.R. Vitamin D and the human antimicrobial peptide LL-37 enhance group a streptococcus resistance to killing by human cells. MBio 2012, 3. [Google Scholar] [CrossRef]
- Reines, M.; Llobet, E.; Llompart, C.M.; Moranta, D.; Perez-Gutierrez, C.; Bengoechea, J.A. Molecular basis of Yersinia enterocolitica temperature-dependent resistance to antimicrobial peptides. J. Bacteriol. 2012, 194, 3173–3188. [Google Scholar] [CrossRef] [PubMed]
- Perez, J.C.; Groisman, E.A. Acid pH activation of the PmrA/PmrB two-component regulatory system of Salmonella enterica. Mol. Microbiol. 2007, 63, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Prost, L.R.; Daley, M.E.; Bader, M.W.; Klevit, R.E.; Miller, S.I. The PhoQ histidine kinases of Salmonella and Pseudomonas spp. are structurally and functionally different: Evidence that pH and antimicrobial peptide sensing contribute to mammalian pathogenesis. Mol. Microbiol. 2008, 69, 503–519. [Google Scholar] [CrossRef] [PubMed]
- Thomassin, J.L.; Brannon, J.R.; Gibbs, B.F.; Gruenheid, S.; Le Moual, H. OmpT outer membrane proteases of enterohemorrhagic and enteropathogenic Escherichia coli contribute differently to the degradation of human LL-37. Infect. Immun. 2012, 80, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Frick, I.M.; Nordin, S.L.; Baumgarten, M.; Morgelin, M.; Sorensen, O.E.; Olin, A.I.; Egesten, A. Constitutive and inflammation-dependent antimicrobial peptides produced by epithelium are differentially processed and inactivated by the commensal Finegoldia magna and the pathogen Streptococcus pyogenes. J. Immunol. 2011, 187, 4300–4309. [Google Scholar] [CrossRef] [PubMed]
- Nagy, I.; Pivarcsi, A.; Kis, K.; Koreck, A.; Bodai, L.; McDowell, A.; Seltmann, H.; Patrick, S.; Zouboulis, C.C.; Kemeny, L. Propionibacterium acnes and lipopolysaccharide induce the expression of antimicrobial peptides and proinflammatory cytokines/chemokines in human sebocytes. Microbes Infect. 2006, 8, 2195–2205. [Google Scholar] [CrossRef] [PubMed]
- Nizet, V.; Ohtake, T.; Lauth, X.; Trowbridge, J.; Rudisill, J.; Dorschner, R.A.; Pestonjamasp, V.; Piraino, J.; Huttner, K.; Gallo, R.L. Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature 2001, 414, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Kim, J.M.; Jeong, S.K.; Jeon, J.E.; Yoon, H.J.; Jeong, M.K.; Lee, S.H. Protease-activated receptor-2 mediates the expression of inflammatory cytokines, antimicrobial peptides, and matrix metalloproteinases in keratinocytes in response to Propionibacterium acnes. Arch. Dermatol. Res. 2010, 302, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Bechara, F.G.; Sand, M.; Skrygan, M.; Kreuter, A.; Altmeyer, P.; Gambichler, T. Acne inversa: Evaluating antimicrobial peptides and proteins. Ann. Dermatol. 2012, 24, 393–397. [Google Scholar] [PubMed]
- Dossel, J.; Meyer-Hoffert, U.; Schroder, J.M.; Gerstel, U. Pseudomonas aeruginosa-derived rhamnolipids subvert the host innate immune response through manipulation of the human beta-defensin-2 expression. Cell Microbiol. 2012, 14, 1364–1375. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Andalibi, A.; Webster, P.; Moon, S.K.; Teufert, K.; Kang, S.H.; Li, J.D.; Nagura, M.; Ganz, T.; Lim, D.J. Antimicrobial activity of innate immune molecules against Streptococcus pneumoniae, Moraxella catarrhalis and nontypeable Haemophilus influenzae. BMC Infect. Dis. 2004, 4, e12. [Google Scholar] [CrossRef]
- Jones, E.A.; Kananurak, A.; Bevins, C.L.; Hollox, E.J.; Bakaletz, L.O. Copy number variation of the beta defensin gene cluster on chromosome 8p influences the bacterial microbiota within the nasopharynx of otitis-prone children. PLoS One 2014, 9, e98269. [Google Scholar] [CrossRef] [PubMed]
- Habets, M.G.; Rozen, D.E.; Brockhurst, M.A. Variation in Streptococcus pneumoniae susceptibility to human antimicrobial peptides may mediate intraspecific competition. Proc. Biol. Sci. 2012, 279, 3803–3811. [Google Scholar] [CrossRef] [PubMed]
- Thienhaus, M.L.; Wohlers, J.; Podschun, R.; Hedderich, J.; Ambrosch, P.; Laudien, M. Antimicrobial peptides in nasal secretion and mucosa with respect to Staphylococcus aureus colonization in chronic rhinosinusitis with nasal polyps. Rhinology 2011, 49, 554–561. [Google Scholar] [PubMed]
- Cole, A.M.; Tahk, S.; Oren, A.; Yoshioka, D.; Kim, Y.H.; Park, A.; Ganz, T. Determinants of Staphylococcus aureus nasal carriage. Clin. Diagn. Lab. Immunol. 2001, 8, 1064–1069. [Google Scholar] [PubMed]
- Starner, T.D.; McCray, P.B., Jr. Pathogenesis of early lung disease in cystic fibrosis: A window of opportunity to eradicate bacteria. Ann. Intern. Med. 2005, 143, 816–822. [Google Scholar] [CrossRef] [PubMed]
- Scharf, S.; Zahlten, J.; Szymanski, K.; Hippenstiel, S.; Suttorp, N.; N’Guessan, P.D. Streptococcus pneumoniae induces human beta-defensin-2 and -3 in human lung epithelium. Exp. Lung Res. 2012, 38, 100–110. [Google Scholar] [CrossRef]
- Kovach, M.A.; Ballinger, M.N.; Newstead, M.W.; Zeng, X.; Bhan, U.; Yu, F.S.; Moore, B.B.; Gallo, R.L.; Standiford, T.J. Cathelicidin-related antimicrobial peptide is required for effective lung mucosal immunity in Gram-negative bacterial pneumonia. J. Immunol. 2012, 189, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.Y.; Xiao, W.; Zhu, M.X.; Yang, Z.H.; Pan, X.J.; Zhang, Y.; Sun, C.C.; Xing, Y. The effect of human antibacterial peptide LL-37 in the pathogenesis of chronic obstructive pulmonary disease. Respir. Med. 2012, 106, 1680–1689. [Google Scholar] [CrossRef] [PubMed]
- Valore, E.V.; Park, C.H.; Quayle, A.J.; Wiles, K.R.; McCray, P.B., Jr.; Ganz, T. Human beta-defensin-1: An antimicrobial peptide of urogenital tissues. J. Clin. Invest. 1998, 101, 1633–1642. [Google Scholar] [CrossRef] [PubMed]
- Morrison, G.; Kilanowski, F.; Davidson, D.; Dorin, J. Characterization of the mouse beta defensin 1, Defb1, mutant mouse model. Infect. Immun. 2002, 70, 3053–3060. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.J.; Anderson, G.M.; Stolzenberg, E.D.; Kari, U.P.; Zasloff, M.; Wilson, J.M. Human beta-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell 1997, 88, 553–560. [Google Scholar] [CrossRef]
- Scudiero, O.; Galdiero, S.; Cantisani, M.; di Noto, R.; Vitiello, M.; Galdiero, M.; Naclerio, G.; Cassiman, J.J.; Pedone, C.; Castaldo, G.; et al. Novel synthetic, salt-resistant analogs of human beta-defensins 1 and 3 endowed with enhanced antimicrobial activity. Antimicrob. Agents Chemother. 2010, 54, 2312–2322. [Google Scholar] [CrossRef] [PubMed]
- Bergsson, G.; Reeves, E.P.; McNally, P.; Chotirmall, S.H.; Greene, C.M.; Greally, P.; Murphy, P.; OʼNeill, S.J.; McElvaney, N.G. LL-37 complexation with glycosaminoglycans in cystic fibrosis lungs inhibits antimicrobial activity, which can be restored by hypertonic saline. J. Immunol. 2009, 183, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Bucki, R.; Byfield, F.J.; Janmey, P.A. Release of the antimicrobial peptide LL-37 from DNA/F-actin bundles in cystic fibrosis sputum. Eur. Respir. J. 2007, 29, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jiang, Y.; Sun, C.; Wang, Q.; Yang, Z.; Pan, X.; Zhu, M.; Xiao, W. The human cathelicidin LL-37 enhances airway mucus production in chronic obstructive pulmonary disease. Biochem. Biophys. Res. Commun. 2014, 443, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Limoli, D.H.; Rockel, A.B.; Host, K.M.; Jha, A.; Kopp, B.T.; Hollis, T.; Wozniak, D.J. Cationic antimicrobial peptides promote microbial mutagenesis and pathoadaptation in chronic infections. PLoS Pathog. 2014, 10, e1004083. [Google Scholar] [CrossRef] [PubMed]
- Putsep, K.; Carlsson, G.; Boman, H.G.; Andersson, M. Deficiency of antibacterial peptides in patients with morbus Kostmann: An observation study. Lancet 2002, 360, 1144–1149. [Google Scholar] [CrossRef]
- Loo, W.T.; Bai, L.J.; Fan, C.B.; Yue, Y.; Dou, Y.D.; Wang, M.; Liang, H.; Cheung, M.N.; Chow, L.; Li, J.L.; et al. Clinical application of human beta-defensin and CD14 gene polymorphism in evaluating the status of chronic inflammation. J. Transl. Med. 2012, 10, S9. [Google Scholar] [CrossRef] [PubMed]
- To, M.; Kamata, Y.; Saruta, J.; Shimizu, T.; Sato, T.; Kondo, Y.; Hayashi, T.; Hamada, N.; Tsukinoki, K. Induction of beta-defensin expression by Porphyromonas gingivalis-infected human Gingival graft transplanted in nu/nu mouse subdermis. Acta Histochem. Cytochem. 2013, 46, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Pingel, L.C.; Kohlgraf, K.G.; Hansen, C.J.; Eastman, C.G.; Dietrich, D.E.; Burnell, K.K.; Srikantha, R.N.; Xiao, X.; Belanger, M.; Progulske-Fox, A.; et al. Human beta-defensin 3 binds to hemagglutinin B (rHagB), a non-fimbrial adhesin from Porphyromonas gingivalis, and attenuates a pro-inflammatory cytokine response. Immunol. Cell Biol. 2008, 86, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Nuding, S.; Gersemann, M.; Hosaka, Y.; Konietzny, S.; Schaefer, C.; Beisner, J.; Schroeder, B.O.; Ostaff, M.J.; Saigenji, K.; Ott, G.; et al. Gastric antimicrobial peptides fail to eradicate Helicobacter pylori infection due to selective induction and resistance. PLoS One 2013, 8, e73867. [Google Scholar] [CrossRef] [PubMed]
- Bauer, B.; Wex, T.; Kuester, D.; Meyer, T.; Malfertheiner, P. Differential expression of human beta defensin 2 and 3 in gastric mucosa of Helicobacter pylori-infected individuals. Helicobacter 2013, 18, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.R.; Smith, K.; Letley, D.P.; Cook, K.W.; Memon, A.A.; Ingram, R.J.; Staples, E.; Backert, S.; Zaitoun, A.M.; Atherton, J.C.; et al. Helicobacter pylori downregulates expression of human beta-defensin 1 in the gastric mucosa in a type IV secretion-dependent fashion. Cell Microbiol. 2013, 15, 2080–2092. [Google Scholar] [CrossRef] [PubMed]
- Bajaj-Elliott, M.; Fedeli, P.; Smith, G.V.; Domizio, P.; Maher, L.; Ali, R.S.; Quinn, A.G.; Farthing, M.J. Modulation of host antimicrobial peptide (beta-defensins 1 and 2) expression during gastritis. Gut 2002, 51, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Kawauchi, K.; Yagihashi, A.; Tsuji, N.; Uehara, N.; Furuya, D.; Kobayashi, D.; Watanabe, N. Human beta-defensin-3 induction in H. pylori-infected gastric mucosal tissues. World J. Gastroenterol. 2006, 12, 5793–5797. [Google Scholar] [PubMed]
- Hase, K.; Murakami, M.; Iimura, M.; Cole, S.P.; Horibe, Y.; Ohtake, T.; Obonyo, M.; Gallo, R.L.; Eckmann, L.; Kagnoff, M.F. Expression of LL-37 by human gastric epithelial cells as a potential host defense mechanism against Helicobacter pylori. Gastroenterology 2003, 125, 1613–1625. [Google Scholar] [CrossRef] [PubMed]
- Bauer, B.; Pang, E.; Holland, C.; Kessler, M.; Bartfeld, S.; Meyer, T.F. The Helicobacter pylori virulence effector CagA abrogates human beta-defensin 3 expression via inactivation of EGFR signaling. Cell Host Microbe 2012, 11, 576–586. [Google Scholar] [CrossRef]
- Shirin, T.; Rahman, A.; Danielsson, A.; Uddin, T.; Bhuyian, T.R.; Sheikh, A.; Qadri, S.S.; Qadri, F.; Hammarstrom, M.L. Antimicrobial peptides in the duodenum at the acute and convalescent stages in patients with diarrhea due to Vibrio cholerae O1 or enterotoxigenic Escherichia coli infection. Microbes Infect. 2011, 13, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.M.; Lee, J.Y.; Yoo, D.; Sim, Y.S.; Kim, Y.J.; Oh, Y.K.; Kang, J.S.; Kim, S.; Kim, J.S.; Kim, J.M. Bacteroides fragilis enterotoxin induces human beta-defensin-2 expression in intestinal epithelial cells via a mitogen-activated protein kinase/I kappaB kinase/NF-kappaB-dependent pathway. Infect. Immun. 2010, 78, 2024–2033. [Google Scholar] [CrossRef] [PubMed]
- Madi, A.; Alnabhani, Z.; Leneveu, C.; Mijouin, L.; Feuilloley, M.; Connil, N. Pseudomonas fluorescens can induce and divert the human beta-defensin-2 secretion in intestinal epithelial cells to enhance its virulence. Arch. Microbiol. 2013, 195, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Iimura, M.; Gallo, R.L.; Hase, K.; Miyamoto, Y.; Eckmann, L.; Kagnoff, M.F. Cathelicidin mediates innate intestinal defense against colonization with epithelial adherent bacterial pathogens. J. Immunol. 2005, 174, 4901–4907. [Google Scholar] [CrossRef] [PubMed]
- Salzman, N.H.; Hung, K.; Haribhai, D.; Chu, H.; Karlsson-Sjoberg, J.; Amir, E.; Teggatz, P.; Barman, M.; Hayward, M.; Eastwood, D.; et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nat. Immunol. 2010, 11, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Schmidt, A.P.; Peterson, E.M.; Wilson, C.L.; de la Maza, L.M. Role of matrix metalloproteinase-7 in the modulation of a Chlamydia trachomatis infection. Immunology 2006, 117, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Underwood, M.A.; Kananurak, A.; Coursodon, C.F.; Adkins-Reick, C.K.; Chu, H.; Bennett, S.H.; Wehkamp, J.; Castillo, P.A.; Leonard, B.C.; Tancredi, D.J.; et al. Bifidobacterium bifidum in a rat model of necrotizing enterocolitis: Antimicrobial peptide and protein responses. Pediatr Res. 2012, 71, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Tai, E.K.; Wong, H.P.; Lam, E.K.; Wu, W.K.; Yu, L.; Koo, M.W.; Cho, C.H. Cathelicidin stimulates colonic mucus synthesis by up-regulating MUC1 and MUC2 expression through a mitogen-activated protein kinase pathway. J. Cell. Biochem. 2008, 104, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Tai, E.K.; Wu, W.K.; Wong, H.P.; Lam, E.K.; Yu, L.; Cho, C.H. A new role for cathelicidin in ulcerative colitis in mice. Exp. Biol. Med. 2007, 232, 799–808. [Google Scholar]
- Chromek, M.; Arvidsson, I.; Karpman, D. The antimicrobial peptide cathelicidin protects mice from Escherichia coli O157:H7-mediated disease. PLoS One 2012, 7, e46476. [Google Scholar] [CrossRef] [PubMed]
- Nuding, S.; Zabel, L.T.; Enders, C.; Porter, E.; Fellermann, K.; Wehkamp, J.; Mueller, H.A.; Stange, E.F. Antibacterial activity of human defensins on anaerobic intestinal bacterial species: A major role of HBD-3. Microbes Infect. 2009, 11, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Chromek, M.; Slamova, Z.; Bergman, P.; Kovacs, L.; Podracka, L.; Ehren, I.; Hokfelt, T.; Gudmundsson, G.H.; Gallo, R.L.; Agerberth, B.; et al. The antimicrobial peptide cathelicidin protects the urinary tract against invasive bacterial infection. Nat. Med. 2006, 12, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Townes, C.L.; Ali, A.; Robson, W.; Pickard, R.; Hall, J. Tolerance of bacteriuria after urinary diversion is linked to antimicrobial peptide activity. Urology 2011, 77, e1–e8. [Google Scholar] [CrossRef] [PubMed]
- Becknell, B.; Spencer, J.D.; Carpenter, A.R.; Chen, X.; Singh, A.; Ploeger, S.; Kline, J.; Ellsworth, P.; Li, B.; Proksch, E.; et al. Expression and antimicrobial function of beta-defensin 1 in the lower urinary tract. PLoS One 2013, 8, e77714. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.D.; Schwaderer, A.L.; Wang, H.; Bartz, J.; Kline, J.; Eichler, T.; DeSouza, K.R.; Sims-Lucas, S.; Baker, P.; Hains, D.S. Ribonuclease 7, an antimicrobial peptide upregulated during infection, contributes to microbial defense of the human urinary tract. Kidney Int. 2013, 83, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.D.; Schwaderer, A.L.; Becknell, B.; Watson, J.; Hains, D.S. The innate immune response during urinary tract infection and pyelonephritis. Pediatr. Nephrol. 2014, 29, 1139–1149. [Google Scholar] [CrossRef] [PubMed]
- Hong, R.W.; Shchepetov, M.; Weiser, J.N.; Axelsen, P.H. Transcriptional profile of the Escherichia coli response to the antimicrobial insect peptide cecropin A. Antimicrob. Agents Chemother. 2003, 47, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ernst, R.K.; Guina, T.; Miller, S.I. Salmonella typhimurium outer membrane remodeling: Role in resistance to host innate immunity. Microbes Infect. 2001, 3, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Garcia Vescovi, E.; Soncini, F.C.; Groisman, E.A. The role of the PhoP/PhoQ regulon in Salmonella virulence. Res. Microbiol. 1994, 145, 473–480. [Google Scholar]
- Bader, M.W.; Navarre, W.W.; Shiau, W.; Nikaido, H.; Frye, J.G.; McClelland, M.; Fang, F.C.; Miller, S.I. Regulation of Salmonella typhimurium virulence gene expression by cationic antimicrobial peptides. Mol. Microbiol. 2003, 50, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.K.; Brannon, M.K.; Stevens, L.; Johansen, H.K.; Selgrade, S.E.; Miller, S.I.; Hoiby, N.; Moskowitz, S.M. PhoQ mutations promote lipid A modification and polymyxin resistance of Pseudomonas aeruginosa found in colistin-treated cystic fibrosis patients. Antimicrob. Agents Chemother. 2011, 55, 5761–5769. [Google Scholar] [CrossRef] [PubMed]
- Alteri, C.J.; Lindner, J.R.; Reiss, D.J.; Smith, S.N.; Mobley, H.L. The broadly conserved regulator PhoP links pathogen virulence and membrane potential in Escherichia coli. Mol. Microbiol. 2011, 82, 145–163. [Google Scholar] [CrossRef] [PubMed]
- Llobet, E.; Campos, M.A.; Gimenez, P.; Moranta, D.; Bengoechea, J.A. Analysis of the networks controlling the antimicrobial-peptide-dependent induction of Klebsiella pneumoniae virulence factors. Infect. Immun. 2011, 79, 3718–3732. [Google Scholar] [CrossRef] [PubMed]
- Moss, J.E.; Fisher, P.E.; Vick, B.; Groisman, E.A.; Zychlinsky, A. The regulatory protein PhoP controls susceptibility to the host inflammatory response in Shigella flexneri. Cell Microbiol. 2000, 2, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Newcombe, J.; Jeynes, J.C.; Mendoza, E.; Hinds, J.; Marsden, G.L.; Stabler, R.A.; Marti, M.; McFadden, J.J. Phenotypic and transcriptional characterization of the meningococcal PhoPQ system, a magnesium-sensing two-component regulatory system that controls genes involved in remodeling the meningococcal cell surface. J. Bacteriol. 2005, 187, 4967–4975. [Google Scholar] [CrossRef] [PubMed]
- O'Loughlin, J.L.; Spinner, J.L.; Minnich, S.A.; Kobayashi, S.D. Yersinia pestis two-component gene regulatory systems promote survival in human neutrophils. Infect. Immun. 2010, 78, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Gunn, J.S. The Salmonella PmrAB regulon: Lipopolysaccharide modifications, antimicrobial peptide resistance and more. Trends Microbiol. 2008, 16, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Marceau, M.; Sebbane, F.; Ewann, F.; Collyn, F.; Lindner, B.; Campos, M.A.; Bengoechea, J.A.; Simonet, M. The pmrF polymyxin-resistance operon of Yersinia pseudotuberculosis is upregulated by the PhoP-PhoQ two-component system but not by PmrA-PmrB, and is not required for virulence. Microbiology 2004, 150, 3947–3957. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, L.; Jenssen, H.; Bains, M.; Wiegand, I.; Gooderham, W.J.; Hancock, R.E. The two-component system CprRS senses cationic peptides and triggers adaptive resistance in Pseudomonas aeruginosa independently of ParRS. Antimicrob. Agents Chemother. 2012, 56, 6212–6222. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, L.; Gooderham, W.J.; Bains, M.; McPhee, J.B.; Wiegand, I.; Hancock, R.E. Adaptive resistance to the “last hope” antibiotics polymyxin B and colistin in Pseudomonas aeruginosa is mediated by the novel two-component regulatory system ParR-ParS. Antimicrob. Agents Chemother. 2010, 54, 3372–3382. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.J.; Xiong, Y.Q.; Yeaman, M.R.; Bayles, K.W.; Abdelhady, W.; Bayer, A.S. Role of the LytSR two-component regulatory system in adaptation to cationic antimicrobial peptides in Staphylococcus aureus. Antimicrob. Agents Chemother. 2013, 57, 3875–3882. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Cha, D.J.; Lai, Y.; Villaruz, A.E.; Sturdevant, D.E.; Otto, M. The antimicrobial peptide-sensing system aps of Staphylococcus aureus. Mol. Microbiol. 2007, 66, 1136–1147. [Google Scholar] [CrossRef] [PubMed]
- Falord, M.; Mader, U.; Hiron, A.; Debarbouille, M.; Msadek, T. Investigation of the Staphylococcus aureus GraSR regulon reveals novel links to virulence, stress response and cell wall signal transduction pathways. PLoS One 2011, 6, e21323. [Google Scholar] [CrossRef] [PubMed]
- Tran-Winkler, H.J.; Love, J.F.; Gryllos, I.; Wessels, M.R. Signal transduction through CsrRS confers an invasive phenotype in group A Streptococcus. PLoS Pathog. 2011, 7, e1002361. [Google Scholar] [CrossRef] [PubMed]
- Gryllos, I.; Tran-Winkler, H.J.; Cheng, M.F.; Chung, H.; Bolcome, R., 3rd; Lu, W.; Lehrer, R.I.; Wessels, M.R. Induction of group A Streptococcus virulence by a human antimicrobial peptide. Proc. Natl. Acad. Sci. USA 2008, 105, 16755–16760. [Google Scholar] [CrossRef]
- Froehlich, B.J.; Bates, C.; Scott, J.R. Streptococcus pyogenes CovRS mediates growth in iron starvation and in the presence of the human cationic antimicrobial peptide LL-37. J. Bacteriol. 2009, 191, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Strempel, N.; Neidig, A.; Nusser, M.; Geffers, R.; Vieillard, J.; Lesouhaitier, O.; Brenner-Weiss, G.; Overhage, J. Human host defense peptide LL-37 stimulates virulence factor production and adaptive resistance in Pseudomonas aeruginosa. PLoS One 2013, 8, e82240. [Google Scholar] [CrossRef] [PubMed]
- Gooderham, W.J.; Bains, M.; McPhee, J.B.; Wiegand, I.; Hancock, R.E. Induction by cationic antimicrobial peptides and involvement in intrinsic polymyxin and antimicrobial peptide resistance, biofilm formation, and swarming motility of PsrA in Pseudomonas aeruginosa. J. Bacteriol. 2008, 190, 5624–5634. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.M.; Strandberg, K.L.; Conroy, M.; Gunn, J.S. Cationic antimicrobial peptides serve as activation signals for the Salmonella Typhimurium PhoPQ and PmrAB regulons in vitro and in vivo. Front. Cell. Infect. Microbiol. 2012, 2, e102. [Google Scholar]
- Alegado, R.A.; Tan, M.W. Resistance to antimicrobial peptides contributes to persistence of Salmonella typhimurium in the C. elegans intestine. Cell Microbiol. 2008, 10, 1259–1273. [Google Scholar] [CrossRef] [PubMed]
- Kwinn, L.A.; Khosravi, A.; Aziz, R.K.; Timmer, A.M.; Doran, K.S.; Kotb, M.; Nizet, V. Genetic characterization and virulence role of the RALP3/LSA locus upstream of the streptolysin s operon in invasive M1T1 Group A Streptococcus. J. Bacteriol. 2007, 189, 1322–1329. [Google Scholar] [CrossRef] [PubMed]
- Niyonsaba, F.; Ushio, H.; Nagaoka, I.; Okumura, K.; Ogawa, H. The human beta-defensins (-1, -2, -3, -4) and cathelicidin LL-37 induce IL-18 secretion through p38 and ERK MAPK activation in primary human keratinocytes. J. Immunol. 2005, 175, 1776–1784. [Google Scholar] [CrossRef]
- Becknell, B.; Eichler, T.E.; Beceiro, S.; Li, B.; Easterling, R.S.; Carpenter, A.R.; James, C.L.; McHugh, K.M.; Hains, D.S.; Partida-Sanchez, S.; et al. Ribonucleases 6 and 7 have antimicrobial function in the human and murine urinary tract. Kidney Int. 2014. [Google Scholar] [CrossRef]
- Eberhard, J.; Menzel, N.; Dommisch, H.; Winter, J.; Jepsen, S.; Mutters, R. The stage of native biofilm formation determines the gene expression of human beta-defensin-2, psoriasin, ribonuclease 7 and inflammatory mediators: A novel approach for stimulation of keratinocytes with in situ formed biofilms. Oral Microbiol. Immunol. 2008, 23, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Zughaier, S.M.; Svoboda, P.; Pohl, J.; Stephens, D.S.; Shafer, W.M. The human host defense peptide LL-37 interacts with Neisseria meningitidis capsular polysaccharides and inhibits inflammatory mediators release. PLoS One 2010, 5, e13627. [Google Scholar] [CrossRef] [PubMed]
- Hing, T.C.; Ho, S.; Shih, D.Q.; Ichikawa, R.; Cheng, M.; Chen, J.; Chen, X.; Law, I.; Najarian, R.; Kelly, C.P.; et al. The antimicrobial peptide cathelicidin modulates Clostridium difficile-associated colitis and toxin A-mediated enteritis in mice. Gut 2013, 62, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- Bedran, T.B.; Mayer, M.P.; Spolidorio, D.P.; Grenier, D. Synergistic anti-inflammatory activity of the antimicrobial peptides human beta-defensin-3 (hBD-3) and cathelicidin (LL-37) in a three-dimensional co-culture model of gingival epithelial cells and fibroblasts. PLoS One 2014, 9, e106766. [Google Scholar] [CrossRef] [PubMed]
- Hou, M.; Zhang, N.; Yang, J.; Meng, X.; Yang, R.; Li, J.; Sun, T. Antimicrobial peptide LL-37 and IDR-1 ameliorate MRSA pneumonia in vivo. Cell. Physiol. Biochem. 2013, 32, 614–623. [Google Scholar] [CrossRef]
- Ruan, Y.; Shen, T.; Wang, Y.; Hou, M.; Li, J.; Sun, T. Antimicrobial peptide LL-37 attenuates LTA induced inflammatory effect in macrophages. Int. Immunopharmacol. 2013, 15, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Capparelli, R.; de Chiara, F.; Nocerino, N.; Montella, R.C.; Iannaccone, M.; Fulgione, A.; Romanelli, A.; Avitabile, C.; Blaiotta, G.; Capuano, F. New perspectives for natural antimicrobial peptides: Application as antinflammatory drugs in a murine model. BMC Immunol. 2012, 13, e61. [Google Scholar] [CrossRef]
- Li, S.A.; Liu, J.; Xiang, Y.; Wang, Y.J.; Lee, W.H.; Zhang, Y. Therapeutic potential of the antimicrobial peptide OH-CATH30 for antibiotic-resistant Pseudomonas aeruginosa keratitis. Antimicrob. Agents Chemother. 2014, 58, 3144–3150. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, P.E.; McHugh, B.; Gwyer Findlay, E.; Mackellar, A.; Mackenzie, K.J.; Gallo, R.L.; Govan, J.R.; Simpson, A.J.; Davidson, D.J. Cathelicidin host defence peptide augments clearance of pulmonary Pseudomonas aeruginosa infection by its influence on neutrophil function in vivo. PLoS One 2014, 9, e99029. [Google Scholar] [CrossRef] [PubMed]
- Niyonsaba, F.; Ushio, H.; Hara, M.; Yokoi, H.; Tominaga, M.; Takamori, K.; Kajiwara, N.; Saito, H.; Nagaoka, I.; Ogawa, H.; et al. Antimicrobial peptides human beta-defensins and cathelicidin LL-37 induce the secretion of a pruritogenic cytokine IL-31 by human mast cells. J. Immunol. 2010, 184, 3526–3534. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.G.; Davidson, D.J.; Gold, M.R.; Bowdish, D.; Hancock, R.E. The human antimicrobial peptide LL-37 is a multifunctional modulator of innate immune responses. J. Immunol. 2002, 169, 3883–3891. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Mookherjee, N.; Wee, K.; Bowdish, D.M.; Pistolic, J.; Li, Y.; Rehaume, L.; Hancock, R.E. Host defense peptide LL-37, in synergy with inflammatory mediator IL-1beta, augments immune responses by multiple pathways. J. Immunol. 2007, 179, 7684–7691. [Google Scholar] [CrossRef] [PubMed]
- Kanthawong, S.; Bolscher, J.G.; Veerman, E.C.; van Marle, J.; de Soet, H.J.; Nazmi, K.; Wongratanacheewin, S.; Taweechaisupapong, S. Antimicrobial and antibiofilm activity of LL-37 and its truncated variants against Burkholderia pseudomallei. Int. J. Antimicrob. Agents 2012, 39, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Haisma, E.M.; de Breij, A.; Chan, H.; van Dissel, J.T.; Drijfhout, J.W.; Hiemstra, P.S.; El Ghalbzouri, A.; Nibbering, P.H. LL-37-derived peptides eradicate multidrug-resistant Staphylococcus aureus from thermally wounded human skin equivalents. Antimicrob. Agents Chemother. 2014, 58, 4411–4419. [Google Scholar] [CrossRef] [PubMed]
- Rivas-Santiago, B.; Rivas Santiago, C.E.; Castaneda-Delgado, J.E.; Leon-Contreras, J.C.; Hancock, R.E.; Hernandez-Pando, R. Activity of LL-37, CRAMP and antimicrobial peptide-derived compounds E2, E6 and CP26 against Mycobacterium tuberculosis. Int. J. Antimicrob. Agents 2013, 41, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Ostorhazi, E.; Voros, E.; Nemes-Nikodem, E.; Pinter, D.; Sillo, P.; Mayer, B.; Wade, J.D.; Otvos, L., Jr. Rapid systemic and local treatments with the antibacterial peptide dimer A3-APO and its monomeric metabolite eliminate bacteria and reduce inflammation in intradermal lesions infected with Propionibacterium acnes and meticillin-resistant Staphylococcus aureus. Int. J. Antimicrob. Agents 2013, 42, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.J.; Beck, K.; Fox, M.A.; Ulaeto, D.; Clark, G.C.; Gumbleton, M. Pegylation of antimicrobial peptides maintains the active peptide conformation, model membrane interactions, and antimicrobial activity while improving lung tissue biocompatibility following airway delivery. Antimicrob. Agents Chemother. 2012, 56, 3298–3308. [Google Scholar] [CrossRef] [PubMed]
- Koziel, J.; Bryzek, D.; Sroka, A.; Maresz, K.; Glowczyk, I.; Bielecka, E.; Kantyka, T.; Pyrc, K.; Svoboda, P.; Pohl, J.; et al. Citrullination alters immunomodulatory function of LL-37 essential for prevention of endotoxin-induced sepsis. J. Immunol. 2014, 192, 5363–5372. [Google Scholar] [CrossRef] [PubMed]
- Hertting, O.; Holm, A.; Luthje, P.; Brauner, H.; Dyrdak, R.; Jonasson, A.F.; Wiklund, P.; Chromek, M.; Brauner, A. Vitamin D induction of the human antimicrobial Peptide cathelicidin in the urinary bladder. PLoS One 2010, 5, e15580. [Google Scholar] [CrossRef] [PubMed]
- Muehleisen, B.; Bikle, D.D.; Aguilera, C.; Burton, D.W.; Sen, G.L.; Deftos, L.J.; Gallo, R.L. PTH/PTHrP and vitamin D control antimicrobial peptide expression and susceptibility to bacterial skin infection. Sci. Transl. Med. 2012, 4, 135ra66. [Google Scholar] [CrossRef] [PubMed]
- Travis, S.M.; Anderson, N.N.; Forsyth, W.R.; Espiritu, C.; Conway, B.D.; Greenberg, E.P.; McCray, P.B., Jr.; Lehrer, R.I.; Welsh, M.J.; Tack, B.F. Bactericidal activity of mammalian cathelicidin-derived peptides. Infect. Immun. 2000, 68, 2748–2755. [Google Scholar] [CrossRef] [PubMed]
- Nuding, S.; Frasch, T.; Schaller, M.; Stange, E.F.; Zabel, L.T. Synergistic effects of antimicrobial peptides and antibiotics against Clostridium difficile. Antimicrob. Agents Chemother. 2014, 58, 5719–5725. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Huang, Y.; Chen, M.; Li, G.; Chen, Y. Functional synergy of alpha-helical antimicrobial peptides and traditional antibiotics against Gram-negative and Gram-positive bacteria in vitro and in vivo. Eur. J. Clin. Microbiol. Infect. Dis. 2014. [Google Scholar] [PubMed]
- Hirt, H.; Gorr, S.U. Antimicrobial peptide GL13K is effective in reducing biofilms of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2013, 57, 4903–4910. [Google Scholar] [CrossRef] [PubMed]
- Pamp, S.J.; Gjermansen, M.; Johansen, H.K.; Tolker-Nielsen, T. Tolerance to the antimicrobial peptide colistin in Pseudomonas aeruginosa biofilms is linked to metabolically active cells, and depends on the pmr and mexAB-oprM genes. Mol. Microbiol. 2008, 68, 223–240. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heimlich, D.R.; Harrison, A.; Mason, K.M. Host Antimicrobial Peptides in Bacterial Homeostasis and Pathogenesis of Disease. Antibiotics 2014, 3, 645-676. https://doi.org/10.3390/antibiotics3040645
Heimlich DR, Harrison A, Mason KM. Host Antimicrobial Peptides in Bacterial Homeostasis and Pathogenesis of Disease. Antibiotics. 2014; 3(4):645-676. https://doi.org/10.3390/antibiotics3040645
Chicago/Turabian StyleHeimlich, Derek R., Alistair Harrison, and Kevin M. Mason. 2014. "Host Antimicrobial Peptides in Bacterial Homeostasis and Pathogenesis of Disease" Antibiotics 3, no. 4: 645-676. https://doi.org/10.3390/antibiotics3040645
APA StyleHeimlich, D. R., Harrison, A., & Mason, K. M. (2014). Host Antimicrobial Peptides in Bacterial Homeostasis and Pathogenesis of Disease. Antibiotics, 3(4), 645-676. https://doi.org/10.3390/antibiotics3040645