
Intravenous Clarithromycin in Critically Ill Adults: A Population Pharmacokinetic Study
 , , , , ,
, , , , ,  , , and
, , and
Abstract
1. Introduction
2. Results
2.1. Baseline Characteristics
2.2. Pharmacokinetic Analysis
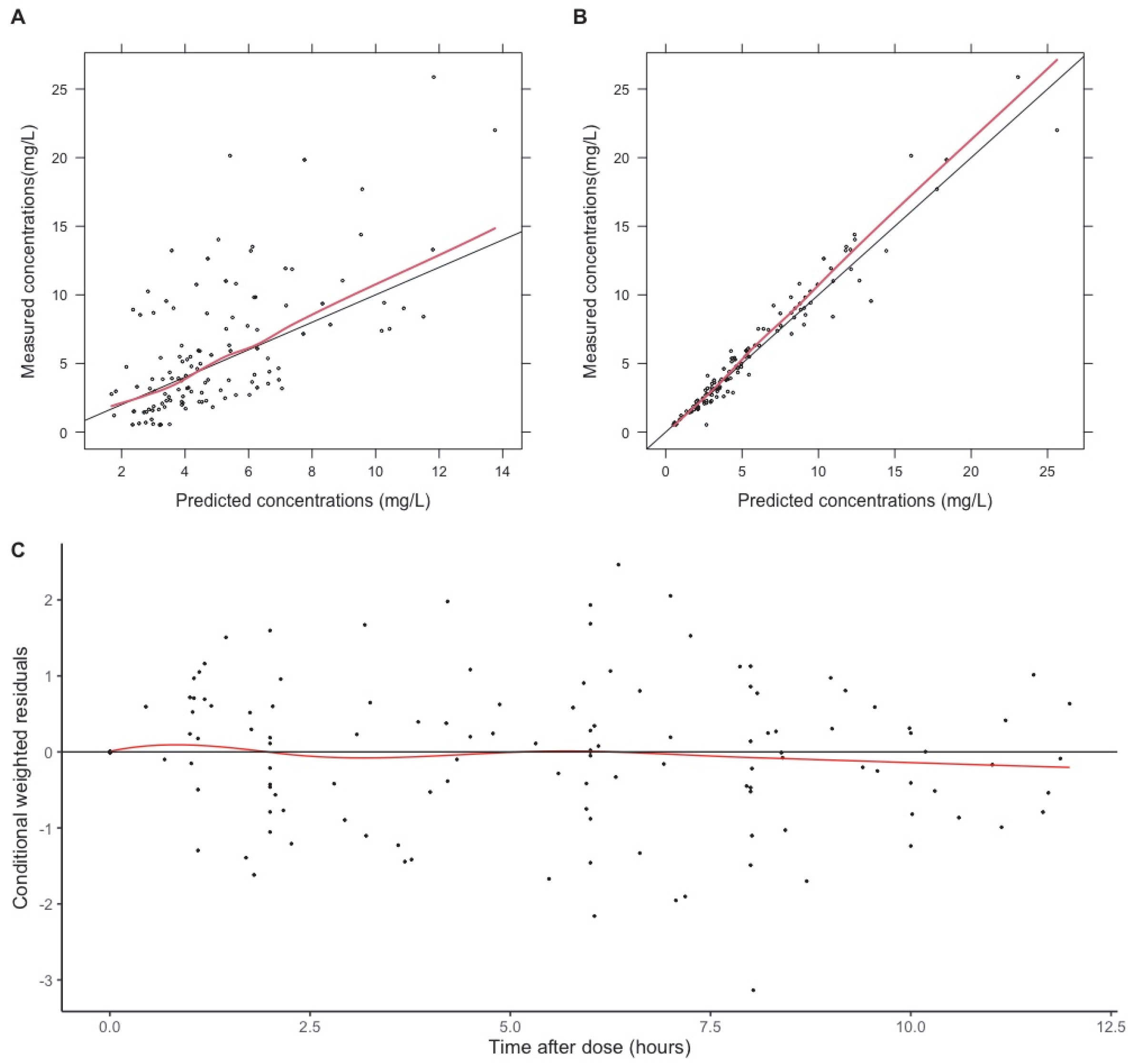
2.3. Evaluation Methods
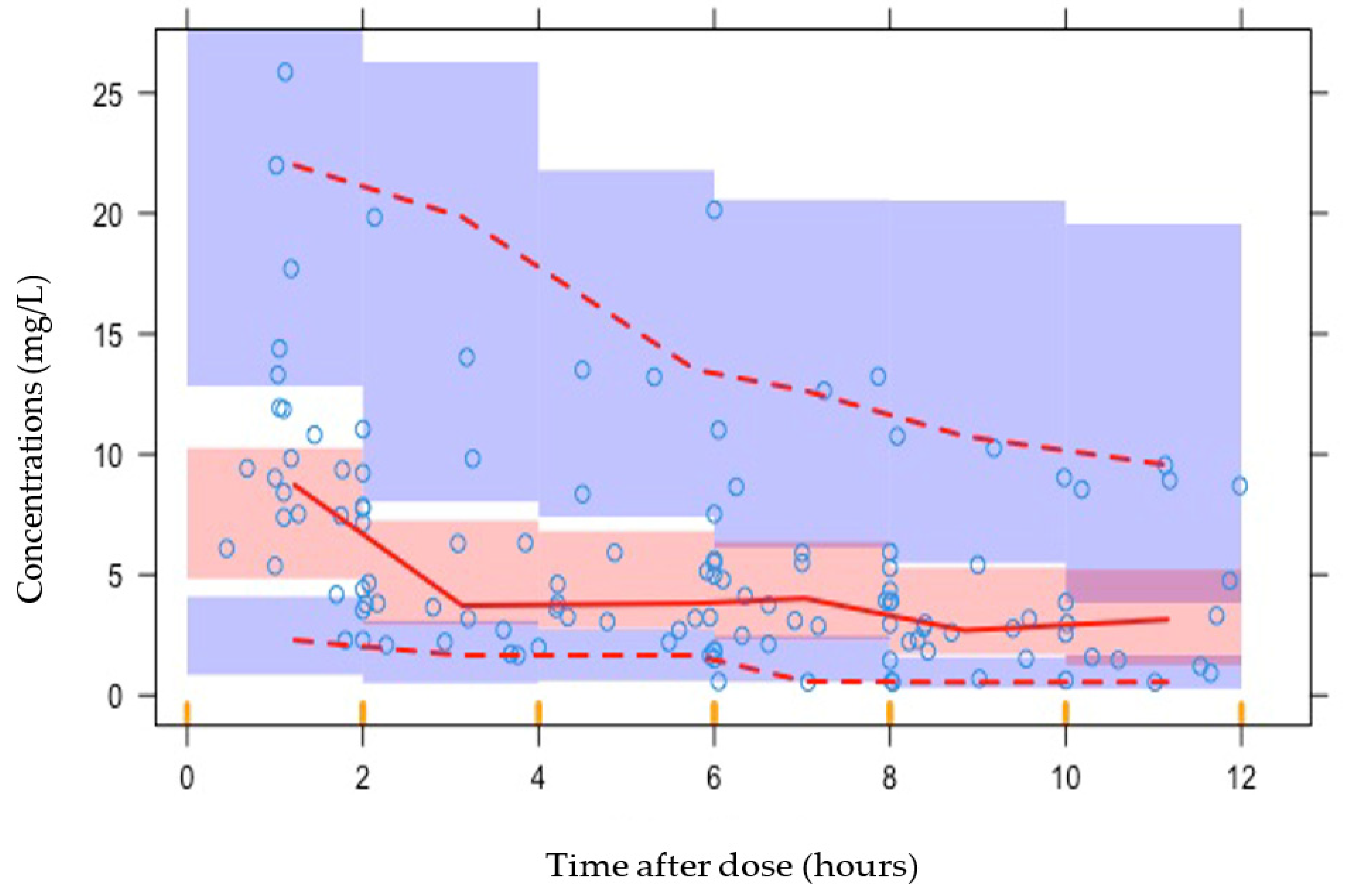
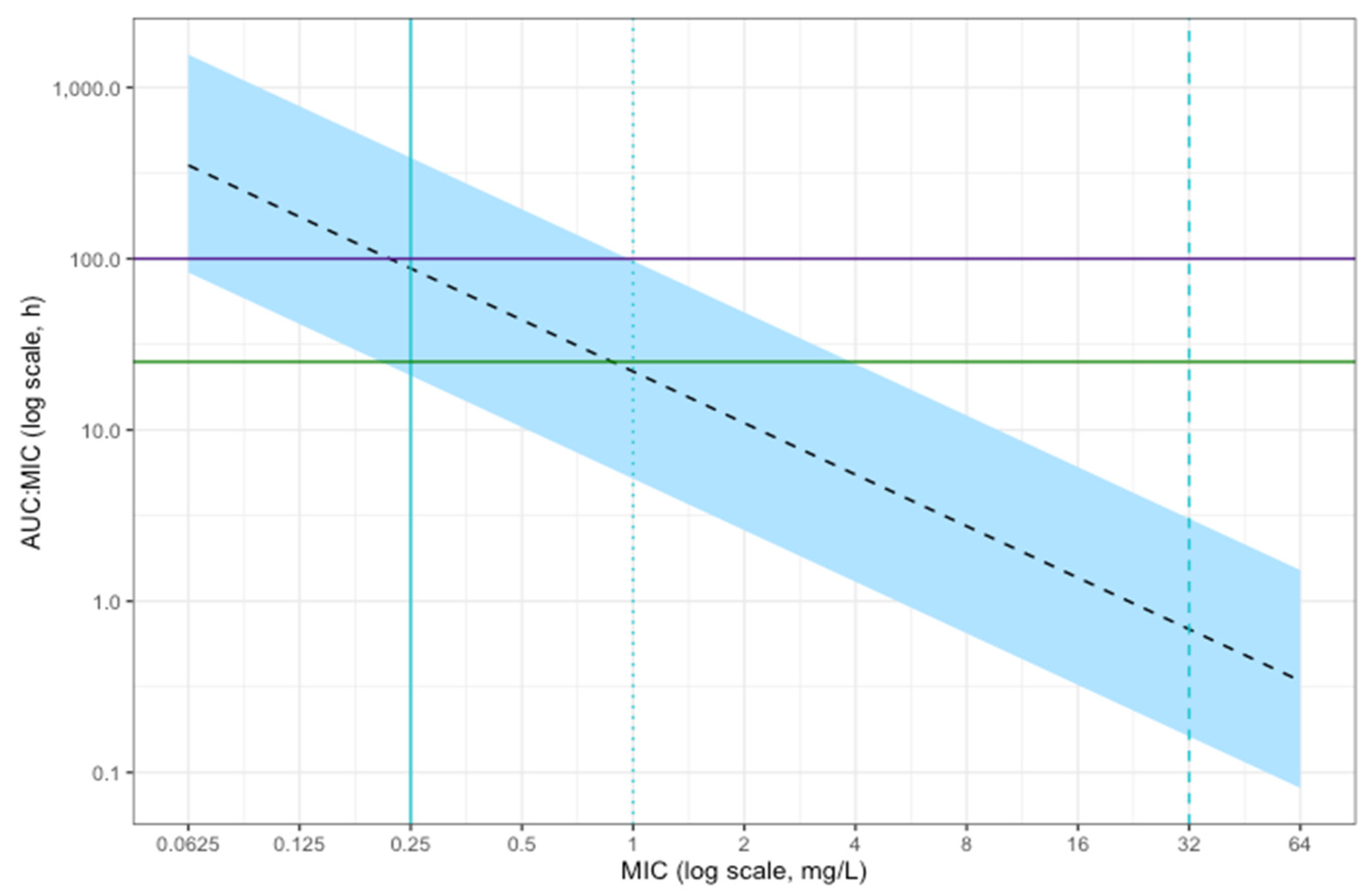
2.4. Simulations
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Peters, D.H.; Clissold, S.P. Clarithromycin. A review of its antimicrobial activity, pharmacokinetic properties and therapeutic potential. Drugs 1992, 44, 117–164. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.A.; Lipman, J. Pharmacokinetic issues for antibiotics in the critically ill patient. Crit. Care Med. 2009, 37, 840–851, quiz 859. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.A.; Paul, S.K.; Akova, M.; Bassetti, M.; De Waele, J.J.; Dimopoulos, G.; Kaukonen, K.M.; Koulenti, D.; Martin, C.; Montravers, P.; et al. DALI: Defining antibiotic levels in intensive care unit patients: Are current β-lactam antibiotic doses sufficient for critically ill patients? Clin. Infect. Dis. 2014, 58, 1072–1083. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Aziz, M.H.; Hammond, N.E.; Brett, S.J.; Cotta, M.O.; De Waele, J.J.; Devaux, A.; Di Tanna, G.L.; Dulhunty, J.M.; Elkady, H.; Eriksson, L.; et al. Prolonged vs Intermittent Infusions of β-Lactam Antibiotics in Adults With Sepsis or Septic Shock: A Systematic Review and Meta-Analysis. JAMA 2024, 332, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Davey, P.G. The pharmacokinetics of clarithromycin and its 14-OH metabolite. J. Hosp. Infect. 1991, 19 (Suppl. A), 29–37. [Google Scholar] [CrossRef]
- Traunmüller, F.; Zeitlinger, M.; Zeleny, P.; Müller, M.; Joukhadar, C. Pharmacokinetics of single- and multiple-dose oral clarithromycin in soft tissues determined by microdialysis. Antimicrob. Agents Chemother. 2007, 51, 3185–3189. [Google Scholar] [CrossRef]
- Abduljalil, K.; Kinzig, M.; Bulitta, J.; Horkovics-Kovats, S.; Sörgel, F.; Rodamer, M.; Fuhr, U. Modeling the autoinhibition of clarithromycin metabolism during repeated oral administration. Antimicrob. Agents Chemother. 2009, 53, 2892–2901. [Google Scholar] [CrossRef]
- Womble, A.Y.; GiguËre, S.; Lee, E.A.; Vickroy, T.W. Pharmacokinetics of clarithromycin and concentrations in body fluids and bronchoalveolar cells of foals. Am. J. Vet. Res. 2006, 67, 1681–1686. [Google Scholar] [CrossRef]
- Fish, D.N.; Abraham, E. Pharmacokinetics of a clarithromycin suspension administered via nasogastric tube to seriously ill patients. Antimicrob. Agents Chemother. 1999, 43, 1277–1280. [Google Scholar] [CrossRef]
- Kays, M.B.; Denys, G.A. In vitro activity and pharmacodynamics of azithromycin and clarithromycin against Streptococcus pneumoniae based on serum and intrapulmonary pharmacokinetics. Clin. Ther. 2001, 23, 413–424. [Google Scholar] [CrossRef]
- Novelli, A.; Fallani, S.; Cassetta, M.I.; Arrigucci, S.; Mazzei, T. In vivo pharmacodynamic evaluation of clarithromycin in comparison to erythromycin. J. Chemother. 2002, 14, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Tessier, P.R.; Kim, M.K.; Zhou, W.; Xuan, D.; Li, C.; Ye, M.; Nightingale, C.H.; Nicolau, D.P. Pharmacodynamic assessment of clarithromycin in a murine model of pneumococcal pneumonia. Antimicrob. Agents Chemother. 2002, 46, 1425–1434. [Google Scholar] [CrossRef] [PubMed]
- Electronic Medicines Compendium. Summary of Product Characteristics: Clarithromycin 125 mg/5ml Suspension. Available online: https://www.medicines.org.uk/emc/product/515/smpc (accessed on 4 June 2024).
- Anon, J.B.; Jacobs, M.R.; Poole, M.D.; Ambrose, P.G.; Benninger, M.S.; Hadley, J.A.; Craig, W.A.; Partnership, S.A.A.H. Antimicrobial treatment guidelines for acute bacterial rhinosinusitis. Otolaryngol. Head Neck Surg. 2004, 130, 1–45. [Google Scholar] [CrossRef]
- Noreddin, A.M.; El-Khatib, W.F.; Aolie, J.; Salem, A.H.; Zhanel, G.G. Pharmacodynamic target attainment potential of azithromycin, clarithromycin, and telithromycin in serum and epithelial lining fluid of community-acquired pneumonia patients with penicillin-susceptible, intermediate, and resistant Streptococcus pneumoniae. Int. J. Infect. Dis. 2009, 13, 483–487. [Google Scholar] [CrossRef] [PubMed]
- European Committee on Antimicrobial Susceptibility Testing. Breakpoint Tables for Interpretation of MICs and Zone Diameters. Version 14.0. 2024. Available online: https://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Breakpoint_tables/v_14.0_Breakpoint_Tables.pdf (accessed on 19 March 2025).
- Waites, K.B.; Bade, D.J.; Bébéar, C.; Brown, S.D.; Davidson, M.K.; Duffy, L.B.; Kenny, G.; Matlow, A.; Shortridge, D.; Talkington, D.; et al. Methods for Antimicrobial Susceptibility Testing for Human Mycoplasmas: Approved Guideline; CLSI Document; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2011. [Google Scholar]
- Wang, G.; Wu, P.; Tang, R.; Zhang, W. Global prevalence of resistance to macrolides in Mycoplasma pneumoniae: A systematic review and meta-analysis. J. Antimicrob. Chemother. 2022, 77, 2353–2363. [Google Scholar] [CrossRef]
- Giamarellos-Bourboulis, E.J.; Siampanos, A.; Bolanou, A.; Doulou, S.; Kakavoulis, N.; Tsiakos, K.; Katopodis, S.; Schinas, G.; Skorda, L.; Alexiou, Z.; et al. Clarithromycin for early anti-inflammatory responses in community-acquired pneumonia in Greece (ACCESS): A randomised, double-blind, placebo-controlled trial. Lancet Respir. Med. 2024, 12, 294–304. [Google Scholar] [CrossRef]
- British National Formulary. Clarithromycin Interactions Checker. 2025. Available online: https://bnf.nice.org.uk/interactions/clarithromycin/ (accessed on 19 March 2025).
- Chu, S.; Wilson, D.S.; Deaton, R.L.; Mackenthun, A.V.; Eason, C.N.; Cavanaugh, J.H. Single- and multiple-dose pharmacokinetics of clarithromycin, a new macrolide antimicrobial. J. Clin. Pharmacol. 1993, 33, 719–726. [Google Scholar] [CrossRef]
- Lonsdale, D.O.; Kipper, K.; Baker, E.H.; Barker, C.I.S.; Oldfield, I.; Philips, B.J.; Johnston, A.; Rhodes, A.; Sharland, M.; Standing, J.F. β-Lactam antimicrobial pharmacokinetics and target attainment in critically ill patients aged 1 day to 90 years: The ABDose study. J. Antimicrob. Chemother. 2020, 75, 3625–3634. [Google Scholar] [CrossRef]
- Shah, R.V.; Kipper, K.; Baker, E.H.; Barker, C.I.S.; Oldfield, I.; Philips, B.J.; Johnston, A.; Lipman, J.; Rhodes, A.; Basarab, M.; et al. Population Pharmacokinetic Study of Benzylpenicillin in Critically Unwell Adults. Antibiotics 2023, 12, 643. [Google Scholar] [CrossRef]
- Kipper, K.; Barker, C.I.S.; Standing, J.F.; Sharland, M.; Johnston, A. Development of a Novel Multipenicillin Assay and Assessment of the Impact of Analyte Degradation: Lessons for Scavenged Sampling in Antimicrobial Pharmacokinetic Study Design. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef]
- Beal, S.; Sheine, L.B.; Boeckmann, A.; Bauer, R.J. NONMEM User’s Guides (1989–2011); Icon Development Solutions: Ellicott City, MD, USA, 2011. [Google Scholar]
- Lonsdale, D.O.; Baker, E.H.; Kipper, K.; Barker, C.; Philips, B.; Rhodes, A.; Sharland, M.; Standing, J.F. Scaling beta-lactam antimicrobial pharmacokinetics from early life to old age. Br. J. Clin. Pharmacol. 2019, 85, 316–346. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, E.N.; Karlsson, M.O. Xpose—An S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput. Methods Programs Biomed. 1999, 58, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Keizer, R.J.; Karlsson, M.O.; Hooker, A. Modeling and Simulation Workbench for NONMEM: Tutorial on Pirana, PsN, and Xpose. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, e50. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. The WHO AWaRe (Access, Watch, Reserve) Antibiotic Book; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- Metlay, J.P.; Waterer, G.W.; Long, A.C.; Anzueto, A.; Brozek, J.; Crothers, K.; Cooley, L.A.; Dean, N.C.; Fine, M.J.; Flanders, S.A.; et al. Diagnosis and Treatment of Adults with Community-acquired Pneumonia. An Official Clinical Practice Guideline of the American Thoracic Society and Infectious Diseases Society of America. Am. J. Respir. Crit. Care Med. 2019, 200, e45–e67. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Median or Total | Range (Interquartile Range) |
|---|---|---|
| Age (years) | 66 | 25–85.8 (56.7–72.7) |
| Weight (kg) | 80 | 53–120 (65–95) |
| Height (cm) * | 173 | 150–192 (166–178) |
| BMI (kg/m2) * | 26.9 | 18.3–35.2 (22.9–31.0) |
| Creatinine (µmol/L) | 89 | 40–276 (71–121) |
| Albumin (g/L) | 25 | 12–38 (21–29) |
| APACHE II score | 20 | 0–28 (16–23) |
| CRP (mg/L) | 70 | 3.5–478 (50.3–169.8) |
| ALT (U/L) | 34 | 9–166 (24–48) |
| Male:Female | 12:7 | |
| Ethnicity | ||
| White | 12 | |
| Black British | 1 | |
| Asian | 1 | |
| Not stated | 5 | |
| Infection source ** | ||
| Chest | 14 | |
| Skin | 1 | |
| Central nervous system | 1 | |
| Ear nose and throat | 2 | |
| Gastrointestinal | 1 | |
| Unknown | 1 | |
| Vasopressors (no. of patients) | 12 | |
| Ventilation status ** | ||
| Self-ventilating | 14 | |
| Non-invasive ventilation | 4 | |
| Invasive ventilation | 12 | |
| Clinical outcome (90 days) | ||
| Alive | 13 | |
| Deceased (infection-attributable) | 3 | |
| Deceased (not attributable to infection) | 3 |
| Mean Parameter Estimates | Relative Standard Error (%) | Range of Individual Estimates | Bootstrap (n = 500) Median (95% CI) | |
|---|---|---|---|---|
| Fixed effects | ||||
| θCL (L/h/70 kg) | 8.17 | 17 | 2.1–55.7 | 7.8 (5.6–10.7) |
| θV1 (L/70 kg) | 25.7 | 29 | 3.1–766.4 | 25.3 (5.4–45.0) |
| θQ (L/h/70 kg) | 62.0 | 18 | 62.4 (43.4–112.5) | |
| θV2 (L/h/70 kg) | 60.6 | 16 | 45.9–112.5 | 62.0 (44.7–108.0) |
| OMEGA | ||||
| η2 CL | 0.53 | 32 | 0.51 (0.21–0.87) | |
| η2 V1 | 1.55 | 70 | 1.58 (0.15–7.95) | |
| SIGMA | ||||
| σ2Proportional | 0.034 | 30 | 0.0317 (0.0161–0.0553) |
| Dosing Interval 1 | Dosing Interval 2 | |
|---|---|---|
| Hours after dose | 0.5/1, 2, 6, 11.75 | 0.5/1, 8, 10, 11.75 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shah, R.V.; Kipper, K.; Baker, E.H.; Barker, C.I.S.; Oldfield, I.; Davidson, H.C.; Swire, C.C.; Philips, B.J.; Johnston, A.; Rhodes, A.; et al. Intravenous Clarithromycin in Critically Ill Adults: A Population Pharmacokinetic Study. Antibiotics 2025, 14, 559. https://doi.org/10.3390/antibiotics14060559
Shah RV, Kipper K, Baker EH, Barker CIS, Oldfield I, Davidson HC, Swire CC, Philips BJ, Johnston A, Rhodes A, et al. Intravenous Clarithromycin in Critically Ill Adults: A Population Pharmacokinetic Study. Antibiotics. 2025; 14(6):559. https://doi.org/10.3390/antibiotics14060559
Chicago/Turabian StyleShah, Reya V., Karin Kipper, Emma H. Baker, Charlotte I. S. Barker, Isobel Oldfield, Harriet C. Davidson, Cleodie C. Swire, Barbara J. Philips, Atholl Johnston, Andrew Rhodes, and et al. 2025. "Intravenous Clarithromycin in Critically Ill Adults: A Population Pharmacokinetic Study" Antibiotics 14, no. 6: 559. https://doi.org/10.3390/antibiotics14060559
APA StyleShah, R. V., Kipper, K., Baker, E. H., Barker, C. I. S., Oldfield, I., Davidson, H. C., Swire, C. C., Philips, B. J., Johnston, A., Rhodes, A., Sharland, M., Standing, J. F., & Lonsdale, D. O. (2025). Intravenous Clarithromycin in Critically Ill Adults: A Population Pharmacokinetic Study. Antibiotics, 14(6), 559. https://doi.org/10.3390/antibiotics14060559