

Combatting Pseudomonas aeruginosa with β-Lactam Antibiotics: A Revived Weapon?


Abstract

1. Introduction
2. β-Lactam Antibiotics and Mechanisms of Resistance
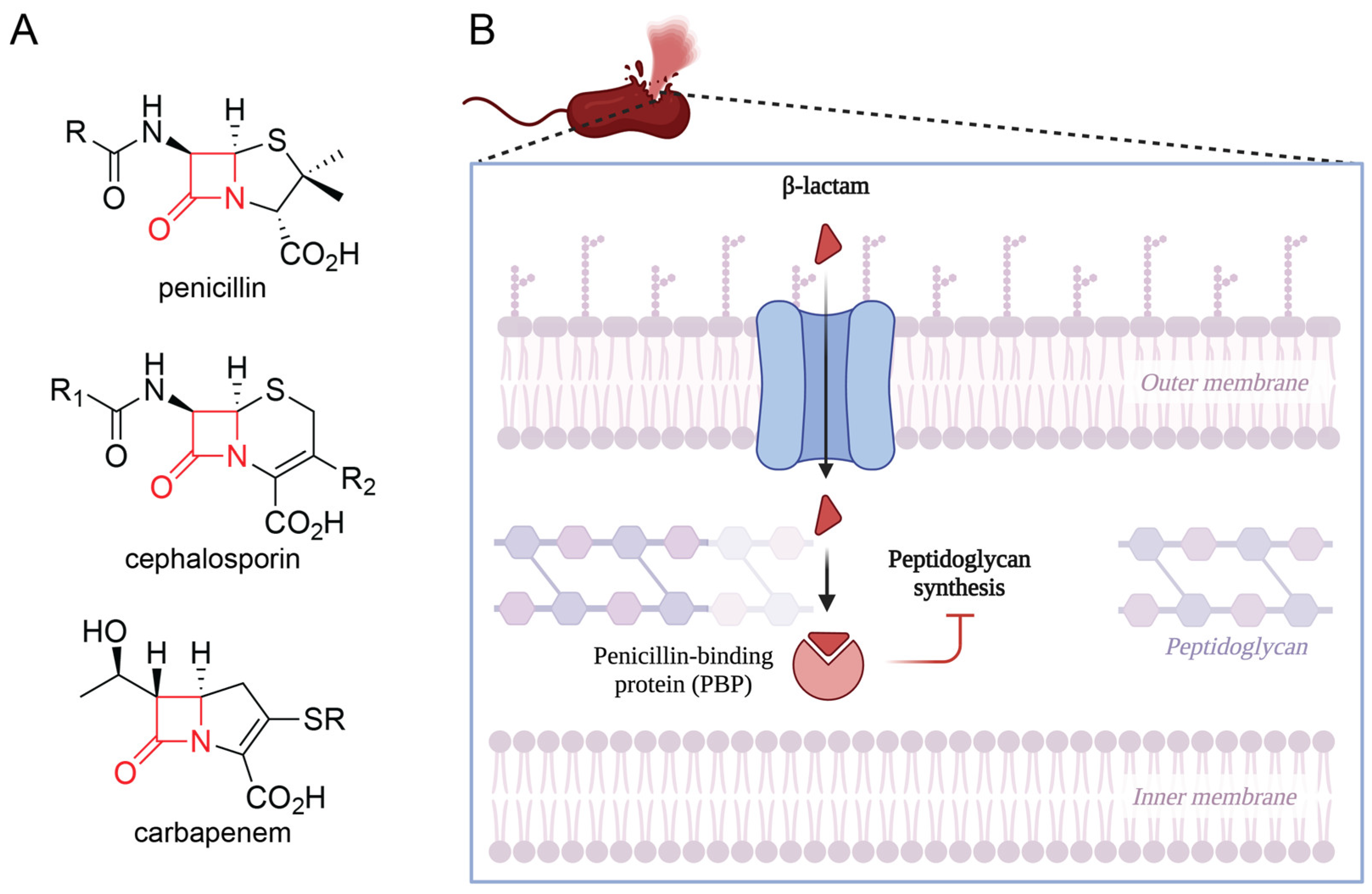
2.1. Mechanism and Classification of β-Lactams
2.2. β-Lactamase-Mediated Resistance
2.3. Target Modification
2.4. Decreased Membrane Permeability: Role of Porins
2.5. Efflux Pumps
3. Adjuvant Agents: Mechanisms and Use
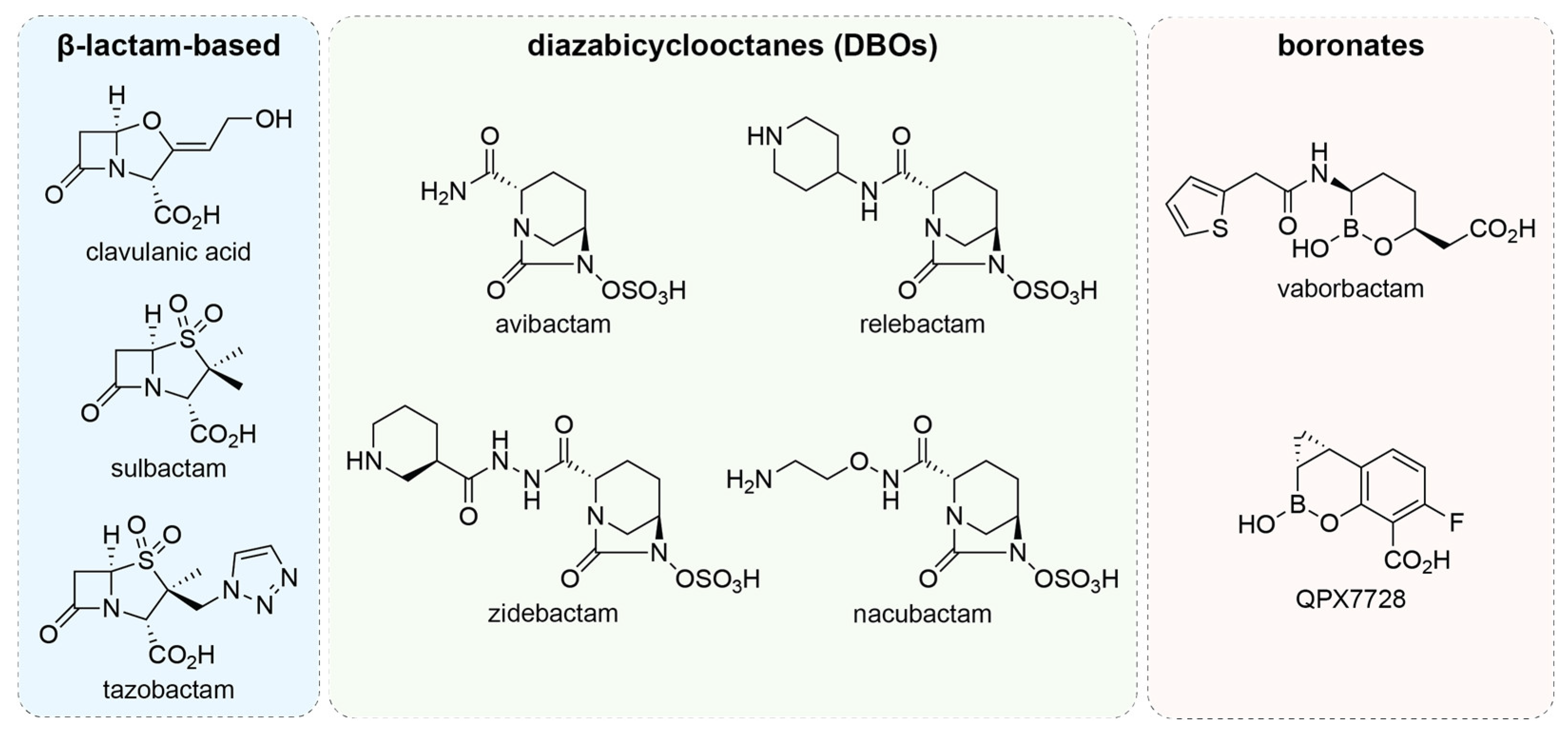
3.1. β-Lactamase Inhibitors
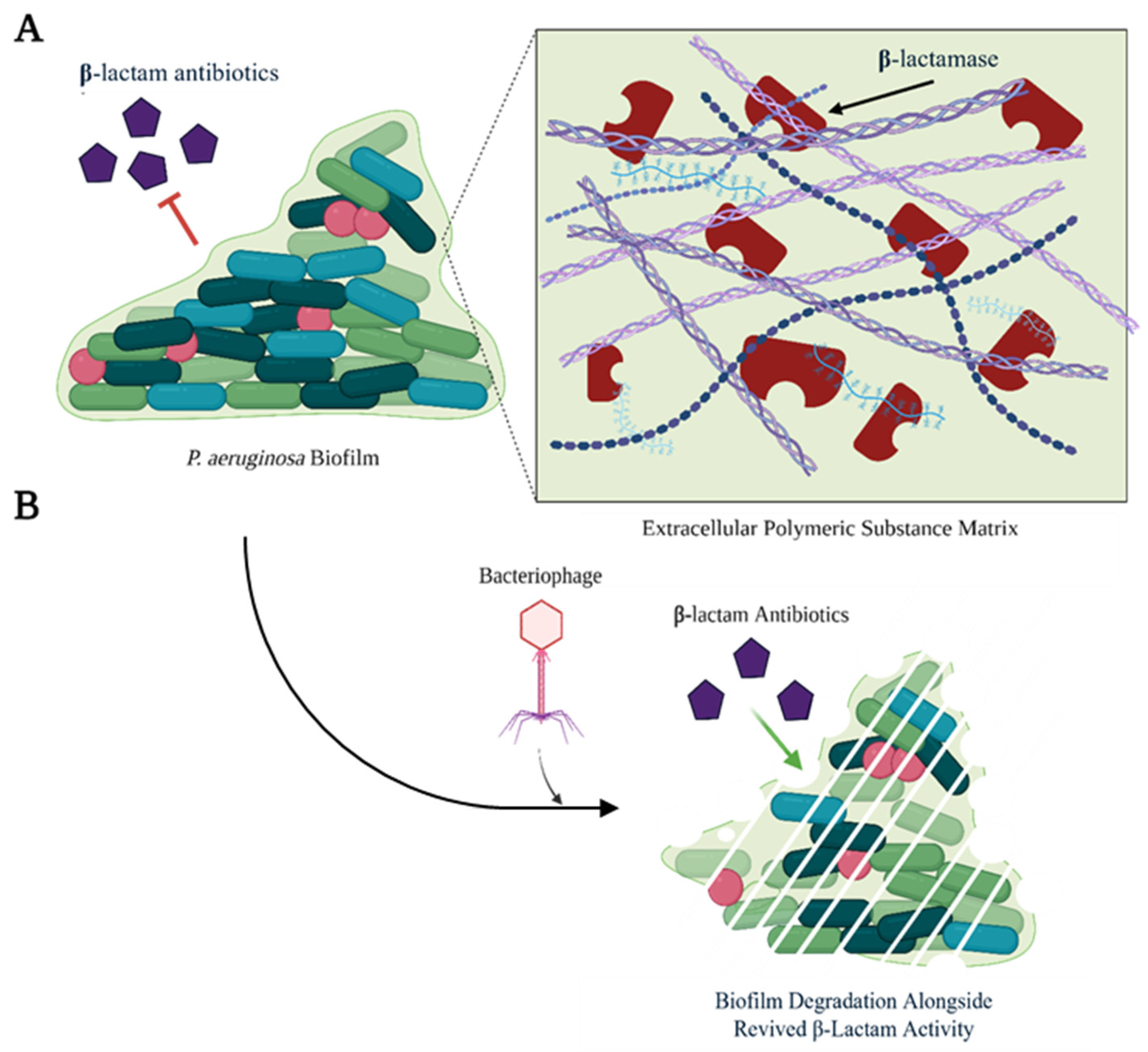
3.2. Bacteriophages
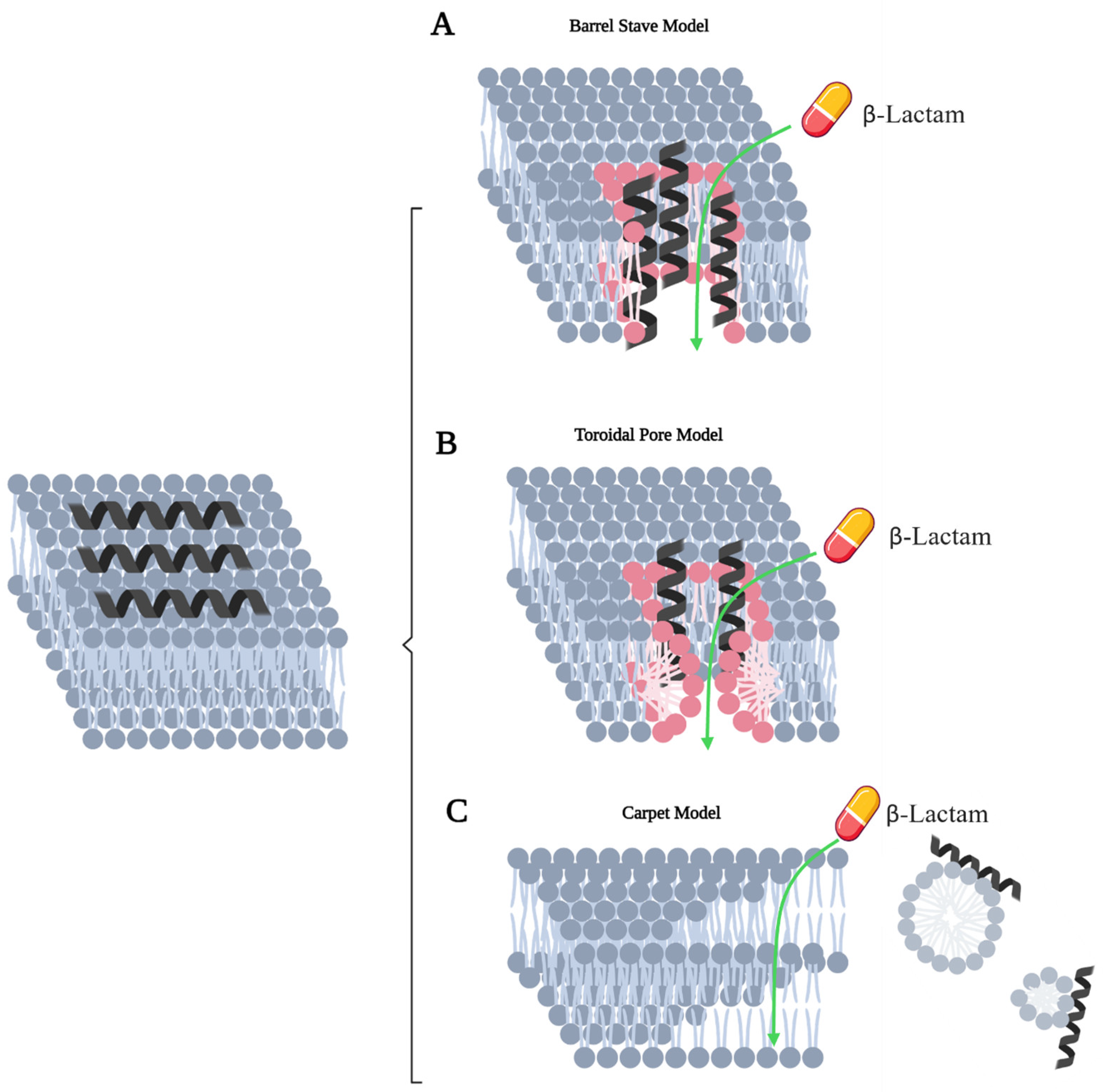
3.3. Antimicrobial Peptides
4. Future Directions for Identification of β-Lactam Adjuvant Agents
4.1. AI-Based Identification of Novel Synergistic Agents
4.2. Challenges Moving Forward
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| MDR | Multidrug resistant |
| PBP | Penicillin-binding protein |
| SBL | Serine β-lactamase |
| MBL | Metallo-β-lactamase |
| ESBL | Extended-spectrum β-lactamase |
| RND | Resistance-nodulation-division |
| DBO | Diazabicyclooctane |
| MIC | Minimum inhibitory concentration |
| EPS | Extracellular polymeric substances |
| AMP | Antimicrobial peptide |
| FIC | Fractional inhibitory concentration |
| Trp | Tryptophan |
References
- Magill, S.S.; Edwards, J.R.; Bamberg, W.; Beldavs, Z.G.; Dumyati, G.; Kainer, M.A.; Lynfield, R.; Maloney, M.; McAllister-Hollod, L.; Nadle, J. Multistate point-prevalence survey of health care–associated infections. N. Engl. J. Med. 2014, 370, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, D.; Kollef, M. The Epidemiology and Pathogenesis and Treatment of Pseudomonas aeruginosa Infections: An Update. Drugs 2021, 81, 2117–2131. [Google Scholar] [CrossRef] [PubMed]
- Weiner, L.M.; Webb, A.K.; Limbago, B.; Dudeck, M.A.; Patel, J.; Kallen, A.J.; Edwards, J.R.; Sievert, D.M. Antimicrobial-resistant pathogens associated with healthcare-associated infections: Summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2011–2014. Infect. Control Hosp. Epidemiol. 2016, 37, 1288–1301. [Google Scholar] [CrossRef]
- Ikuta, K.S.; Swetschinski, L.R.; Aguilar, G.R.; Sharara, F.; Mestrovic, T.; Gray, A.P.; Weaver, N.D.; Wool, E.E.; Han, C.; Hayoon, A.G. Global mortality associated with 33 bacterial pathogens in 2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2022, 400, 2221–2248. [Google Scholar] [CrossRef]
- López-Calleja, A.I.; Morales, E.M.; Medina, R.N.; Esgueva, M.F.; Pareja, J.S.; Moya, J.M.G.-L.; Cerón, I.F.; Bayon, J.V.; López, A.R. Antimicrobial activity of ceftolozane-tazobactam against multidrug-resistant and extensively drug-resistant Pseudomonas aeruginosa clinical isolates from a Spanish hospital. Rev. Esp. Quimioter. 2019, 32, 68. [Google Scholar]
- Zarb, P.; Coignard, B.; Griskeviciene, J.; Muller, A.; Vankerckhoven, V.; Weist, K.; Goossens, M.M.; Vaerenberg, S.; Hopkins, S.; Catry, B. The European Centre for Disease Prevention and Control (ECDC) pilot point prevalence survey of healthcare-associated infections and antimicrobial use. Eurosurveillance 2012, 17, 20316. [Google Scholar] [CrossRef]
- Cooke, A.C.; Nello, A.V.; Ernst, R.K.; Schertzer, J.W. Analysis of Pseudomonas aeruginosa biofilm membrane vesicles supports multiple mechanisms of biogenesis. PLoS ONE 2019, 14, e0212275. [Google Scholar] [CrossRef]
- Metruccio, M.M.; Evans, D.J.; Gabriel, M.M.; Kadurugamuwa, J.L.; Fleiszig, S.M. Pseudomonas aeruginosa outer membrane vesicles triggered by human mucosal fluid and lysozyme can prime host tissue surfaces for bacterial adhesion. Front. Microbiol. 2016, 7, 871. [Google Scholar] [CrossRef]
- Kadurugamuwa, J.L.; Beveridge, T.J. Bacteriolytic effect of membrane vesicles from Pseudomonas aeruginosa on other bacteria including pathogens: Conceptually new antibiotics. J. Bacteriol. 1996, 178, 2767–2774. [Google Scholar] [CrossRef]
- Koeppen, K.; Barnaby, R.; Jackson, A.A.; Gerber, S.A.; Hogan, D.A.; Stanton, B.A. Tobramycin reduces key virulence determinants in the proteome of Pseudomonas aeruginosa outer membrane vesicles. PLoS ONE 2019, 14, e0211290. [Google Scholar] [CrossRef]
- He, C.; Zhou, Y.; Liu, F.; Liu, H.; Tan, H.; Jin, S.; Wu, W.; Ge, B. Bacterial nucleotidyl cyclase inhibits the host innate immune response by suppressing TAK1 activation. Infect. Immun. 2017, 85, e00239-17. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.K.; Dhasmana, N.; Dubey, N.; Kumar, N.; Gangwal, A.; Gupta, M.; Singh, Y. Bacterial virulence factors: Secreted for survival. Indian J. Microbiol. 2017, 57, 1–10. [Google Scholar] [CrossRef]
- Rasko, D.A.; Sperandio, V. Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. Drug Discov. 2010, 9, 117–128. [Google Scholar] [CrossRef]
- Hauser, A.R. The type III secretion system of Pseudomonas aeruginosa: Infection by injection. Nat. Rev. Microbiol. 2009, 7, 654–665. [Google Scholar] [CrossRef]
- Bagayoko, S.; Leon-Icaza, S.A.; Pinilla, M.; Hessel, A.; Santoni, K.; Péricat, D.; Bordignon, P.-J.; Moreau, F.; Eren, E.; Boyancé, A. Host phospholipid peroxidation fuels ExoU-dependent cell necrosis and supports Pseudomonas aeruginosa-driven pathology. PLoS Path. 2021, 17, e1009927. [Google Scholar] [CrossRef]
- Finck-Barbançon, V.; Goranson, J.; Zhu, L.; Sawa, T.; Wiener-Kronish, J.P.; Fleiszig, S.M.; Wu, C.; Mende-Mueller, L.; Frank, D.W. ExoU expression by Pseudomonas aeruginosa correlates with acute cytotoxicity and epithelial injury. Mol. Microbiol. 1997, 25, 547–557. [Google Scholar] [CrossRef]
- Zhao, F.; Wang, Q.; Zhang, Y.; Lei, L. Anaerobic biosynthesis of rhamnolipids by Pseudomonas aeruginosa: Performance, mechanism and its application potential for enhanced oil recovery. Microb. Cell Factories 2021, 20, 103. [Google Scholar] [CrossRef]
- Eder, K.; Vizler, C.; Kusz, E.; Karcagi, I.; Glavinas, H.; Balogh, G.E.; Vigh, L.; Duda, E.; Gyorfy, Z. The role of lipopolysaccharide moieties in macrophage response to Escherichia coli. Biochem. Biophys. Res. Commun. 2009, 389, 46–51. [Google Scholar] [CrossRef]
- Gellatly, S.L.; Hancock, R.E. Pseudomonas aeruginosa: New insights into pathogenesis and host defenses. Pathog. Dis. 2013, 67, 159–173. [Google Scholar] [CrossRef]
- Banerjee, D.; Stableforth, D. The treatment of respiratory pseudomonas infection in cystic fibrosis: What drug and which way? Drugs 2000, 60, 1053–1064. [Google Scholar] [CrossRef]
- Bush, K.; Bradford, P.A. β-Lactams and β-lactamase inhibitors: An overview. Cold Spring Harb. Perspect. Med. 2016, 6, a025247. [Google Scholar] [CrossRef] [PubMed]
- Thakuria, B.; Lahon, K. The Beta Lactam Antibiotics as an Empirical Therapy in a Developing Country: An Update on Their Current Status and Recommendations to Counter the Resistance against Them. J. Clin. Diagn. Res. 2013, 7, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.Y.; Van Boeckel, T.P.; Martinez, E.M.; Pant, S.; Gandra, S.; Levin, S.A.; Goossens, H.; Laxminarayan, R. Global increase and geographic convergence in antibiotic consumption between 2000 and 2015. Proc. Natl. Acad. Sci. USA 2018, 115, E3463–E3470. [Google Scholar] [CrossRef] [PubMed]
- Bankar, N.J.; Ugemuge, S.; Ambad, R.S.; Hawale, D.V.; Timilsina, D.R. Implementation of Antimicrobial Stewardship in the Healthcare Setting. Cureus 2022, 14, e26664. [Google Scholar] [CrossRef]
- Poole, K. Pseudomonas aeruginosa: Resistance to the max. Front. Microbiol. 2011, 2, 65. [Google Scholar] [CrossRef]
- Breidenstein, E.B.; de la Fuente-Núñez, C.; Hancock, R.E. Pseudomonas aeruginosa: All roads lead to resistance. Trends Microbiol. 2011, 19, 419–426. [Google Scholar] [CrossRef]
- Cholley, P.; Thouverez, M.; Hocquet, D.; van der Mee-Marquet, N.; Talon, D.; Bertrand, X. Most multidrug-resistant Pseudomonas aeruginosa isolates from hospitals in eastern France belong to a few clonal types. J. Clin. Microbiol. 2011, 49, 2578–2583. [Google Scholar] [CrossRef]
- Treepong, P.; Kos, V.N.; Guyeux, C.; Blanc, D.S.; Bertrand, X.; Valot, B.; Hocquet, D. Global emergence of the widespread Pseudomonas aeruginosa ST235 clone. Clin. Microbiol. Infect. 2018, 24, 258–266. [Google Scholar] [CrossRef]
- Feretzakis, G.; Loupelis, E.; Sakagianni, A.; Skarmoutsou, N.; Michelidou, S.; Velentza, A.; Martsoukou, M.; Valakis, K.; Petropoulou, S.; Koutalas, E. A 2-year single-centre audit on antibiotic resistance of Pseudomonas aeruginosa, Acinetobacter baumannii and Klebsiella pneumoniae strains from an intensive care unit and other wards in a general public hospital in Greece. Antibiotics 2019, 8, 62. [Google Scholar] [CrossRef]
- Matos, E.C.O.d.; Andriolo, R.B.; Rodrigues, Y.C.; Lima, P.D.L.d.; Carneiro, I.C.d.R.S.; Lima, K.V.B. Mortality in patients with multidrug-resistant Pseudomonas aeruginosa infections: A meta-analysis. Rev. Soc. Bras. Med. Trop. 2018, 51, 415–420. [Google Scholar] [CrossRef]
- Nathwani, D.; Raman, G.; Sulham, K.; Gavaghan, M.; Menon, V. Clinical and economic consequences of hospital-acquired resistant and multidrug-resistant Pseudomonas aeruginosa infections: A systematic review and meta-analysis. Antimicrob. Resist. Infect. Control. 2014, 3, 32. [Google Scholar] [CrossRef] [PubMed]
- Tängdén, T. Combination antibiotic therapy for multidrug-resistant Gram-negative bacteria. Ups. J. Med. Sci. 2014, 119, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Worthington, R.J.; Melander, C. Combination approaches to combat multidrug-resistant bacteria. Trends Biotechnol. 2013, 31, 177–184. [Google Scholar] [CrossRef]
- Ahmed, A.; Azim, A.; Gurjar, M.; Baronia, A.K. Current concepts in combination antibiotic therapy for critically ill patients. Indian J. Crit. Care Med. 2014, 18, 310–314. [Google Scholar] [CrossRef]
- Pletz, M.W.; Hagel, S.; Forstner, C. Who benefits from antimicrobial combination therapy? Lancet Infect. Dis. 2017, 17, 677–678. [Google Scholar] [CrossRef]
- Wang, N.; Luo, J.; Deng, F.; Huang, Y.; Zhou, H. Antibiotic combination therapy: A strategy to overcome bacterial resistance to aminoglycoside antibiotics. Front. Pharmacol. 2022, 13, 839808. [Google Scholar] [CrossRef]
- Kim, D.; Kim, S.; Kwon, Y.; Kim, Y.; Park, H.; Kwak, K.; Lee, H.; Lee, J.H.; Jang, K.M.; Kim, D.; et al. Structural Insights for β-Lactam Antibiotics. Biomol. Ther. 2023, 31, 141–147. [Google Scholar] [CrossRef]
- Dalhoff, A.; Janjic, N.; Echols, R. Redefining penems. Biochem. Pharmacol. 2006, 71, 1085–1095. [Google Scholar] [CrossRef]
- Papp-Wallace, K.M.; Endimiani, A.; Taracila, M.A.; Bonomo, R.A. Carbapenems: Past, present, and future. Antimicrob. Agents Chemother. 2011, 55, 4943–4960. [Google Scholar] [CrossRef]
- Flores-Kim, J.; Dobihal, G.S.; Fenton, A.; Rudner, D.Z.; Bernhardt, T.G. A switch in surface polymer biogenesis triggers growth-phase-dependent and antibiotic-induced bacteriolysis. Elife 2019, 8, e44912. [Google Scholar] [CrossRef]
- Mora-Ochomogo, M.; Lohans, C.T. β-Lactam antibiotic targets and resistance mechanisms: From covalent inhibitors to substrates. RSC Med. Chem. 2021, 12, 1623–1639. [Google Scholar] [CrossRef] [PubMed]
- Ambler, R.P. The structure of β-lactamases. Philos. Trans. R. Soc. London. B Biol. Sci. 1980, 289, 321–331. [Google Scholar]
- Zhao, W.-H.; Hu, Z.-Q. β-lactamases identified in clinical isolates of Pseudomonas aeruginosa. Crit. Rev. Microbiol. 2010, 36, 245–258. [Google Scholar] [CrossRef]
- Tooke, C.L.; Hinchliffe, P.; Bragginton, E.C.; Colenso, C.K.; Hirvonen, V.H.A.; Takebayashi, Y.; Spencer, J. β-Lactamases and β-Lactamase Inhibitors in the 21st Century. J. Mol. Biol. 2019, 431, 3472–3500. [Google Scholar] [CrossRef]
- Bush, K. Bench-to-bedside review: The role of β-lactamases in antibiotic-resistant Gram-negative infections. Crit. Care 2010, 14, 224. [Google Scholar] [CrossRef]
- Jacoby George, A. AmpC β-Lactamases. Clin. Microbiol. Rev. 2009, 22, 161–182. [Google Scholar] [CrossRef]
- Evans Benjamin, A.; Amyes Sebastian, G.B. OXA β-Lactamases. Clin. Microbiol. Rev. 2014, 27, 241–263. [Google Scholar] [CrossRef]
- Naas, T.; Oueslati, S.; Bonnin, R.A.; Dabos, M.L.; Zavala, A.; Dortet, L.; Retailleau, P.; Iorga, B.I. Beta-lactamase database (BLDB)–structure and function. J. Enzyme Inhib. Med. Chem. 2017, 32, 917–919. [Google Scholar] [CrossRef]
- Bonomo, R.A. β-Lactamases: A Focus on Current Challenges. Cold Spring Harb. Perspect. Med. 2017, 7, a025239. [Google Scholar] [CrossRef]
- Bush, K.; Bradford, P.A. Epidemiology of β-lactamase-producing pathogens. Clin. Microbiol. Rev. 2020, 33, e00047-19. [Google Scholar] [CrossRef]
- Bush, K.; Bradford, P.A. Interplay between β-lactamases and new β-lactamase inhibitors. Nat. Rev. Microbiol. 2019, 17, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Castanheira, M.; Simner, P.J.; Bradford, P.A. Extended-spectrum β-lactamases: An update on their characteristics, epidemiology and detection. JAC-Antimicrob. Resist. 2021, 3, dlab092. [Google Scholar] [CrossRef]
- Walther-Rasmussen, J.; Høiby, N. OXA-type carbapenemases. J. Antimicrob. Chemother. 2006, 57, 373–383. [Google Scholar] [CrossRef]
- Medeiros, A.A. Evolution and dissemination of β-lactamases accelerated by generations of β-lactam antibiotics. Clin. Infect. Dis. 1997, 24, S19–S45. [Google Scholar] [CrossRef]
- Walther-Rasmussen, J.; Høiby, N. Class A carbapenemases. J. Antimicrob. Chemother. 2007, 60, 470–482. [Google Scholar] [CrossRef]
- Poirel, L.; Potron, A.; Nordmann, P. OXA-48-like carbapenemases: The phantom menace. J. Antimicrob. Chemother. 2012, 67, 1597–1606. [Google Scholar] [CrossRef]
- Boyd, S.E.; Holmes, A.; Peck, R.; Livermore, D.M.; Hope, W. OXA-48-like β-lactamases: Global epidemiology, treatment options, and development pipeline. Antimicrob. Agents Chemother. 2022, 66, e00216–e00222. [Google Scholar] [CrossRef]
- Halat, D.H.; Moubareck, C.A. The Intriguing Carbapenemases of Pseudomonas aeruginosa: Current Status, Genetic Profile, and Global Epidemiology. Yale J. Biol. Med. 2022, 95, 507. [Google Scholar]
- Potron, A.; Poirel, L.; Nordmann, P. Emerging broad-spectrum resistance in Pseudomonas aeruginosa and Acinetobacter baumannii: Mechanisms and epidemiology. Int. J. Antimicrob. Agents 2015, 45, 568–585. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.R.; Toleman, M.A.; Poirel, L.; Nordmann, P. Metallo-β-lactamases: The quiet before the storm? Clin. Microbiol. Rev. 2005, 18, 306–325. [Google Scholar] [CrossRef]
- Ju, L.C.; Cheng, Z.; Fast, W.; Bonomo, R.A.; Crowder, M.W. The Continuing Challenge of Metallo-β-Lactamase Inhibition: Mechanism Matters. Trends Pharmacol. Sci. 2018, 39, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wu, W.; Wang, N.; Feng, H.; Wang, J.; Wang, F.; Zhang, Y.; Chen, H.; Yang, Q.; Jiang, Y. Pseudomonas aeruginosa High-Risk Sequence Type 463 Co-Producing KPC-2 and AFM-1 Carbapenemases, China, 2020–2022. Emerging Infect. Dis. 2023, 29, 2136. [Google Scholar] [CrossRef] [PubMed]
- Peña, C.; Suarez, C.; Gozalo, M.; Murillas, J.; Almirante, B.; Pomar, V.; Aguilar, M.; Granados, A.; Calbo, E.; Rodríguez-Baño, J. Prospective multicenter study of the impact of carbapenem resistance on mortality in Pseudomonas aeruginosa bloodstream infections. Antimicrob. Agents Chemother. 2012, 56, 1265–1272. [Google Scholar] [CrossRef]
- Reyes, J.; Komarow, L.; Chen, L.; Ge, L.; Hanson, B.M.; Cober, E.; Herc, E.; Alenazi, T.; Kaye, K.S.; Garcia-Diaz, J. Global epidemiology and clinical outcomes of carbapenem-resistant Pseudomonas aeruginosa and associated carbapenemases (POP): A prospective cohort study. Lancet Microbe 2023, 4, e159–e170. [Google Scholar] [CrossRef]
- Thaden, J.T.; Park, L.P.; Maskarinec, S.A.; Ruffin, F.; Fowler, V.G., Jr.; Van Duin, D. Results from a 13-year prospective cohort study show increased mortality associated with bloodstream infections caused by Pseudomonas aeruginosa compared to other bacteria. Antimicrob. Agents Chemother. 2017, 61, e02671-16. [Google Scholar] [CrossRef]
- Yuan, Q.; Guo, L.; Li, B.; Zhang, S.; Feng, H.; Zhang, Y.; Yu, M.; Hu, H.; Chen, H.; Yang, Q. Risk factors and outcomes of inpatients with carbapenem-resistant Pseudomonas aeruginosa bloodstream infections in China: A 9-year trend and multicenter cohort study. Front. Microbiol. 2023, 14, 1137811. [Google Scholar] [CrossRef]
- Liu, Q.; Li, X.; Li, W.; Du, X.; He, J.-Q.; Tao, C.; Feng, Y. Influence of carbapenem resistance on mortality of patients with Pseudomonas aeruginosa infection: A meta-analysis. Sci. Rep. 2015, 5, 11715. [Google Scholar] [CrossRef]
- Kang, C.-I.; Kim, S.-H.; Kim, H.-B.; Park, S.-W.; Choe, Y.-J.; Oh, M.-d.; Kim, E.-C.; Choe, K.-W. Pseudomonas aeruginosa bacteremia: Risk factors for mortality and influence of delayed receipt of effective antimicrobial therapy on clinical outcome. Clin. Infect. Dis. 2003, 37, 745–751. [Google Scholar] [CrossRef]
- Kang, C.-I.; Kim, S.-H.; Park, W.B.; Lee, K.-D.; Kim, H.-B.; Kim, E.-C.; Oh, M.-d.; Choe, K.-W. Bloodstream infections caused by antibiotic-resistant gram-negative bacilli: Risk factors for mortality and impact of inappropriate initial antimicrobial therapy on outcome. Antimicrob. Agents Chemother. 2005, 49, 760–766. [Google Scholar] [CrossRef]
- Cosgrove, S.E. The relationship between antimicrobial resistance and patient outcomes: Mortality, length of hospital stay, and health care costs. Clin. Infect. Dis. 2006, 42, S82–S89. [Google Scholar] [CrossRef]
- Kadri, S.S.; Lai, Y.L.; Warner, S.; Strich, J.R.; Babiker, A.; Ricotta, E.E.; Demirkale, C.Y.; Dekker, J.P.; Palmore, T.N.; Rhee, C. Inappropriate empirical antibiotic therapy for bloodstream infections based on discordant in-vitro susceptibilities: A retrospective cohort analysis of prevalence, predictors, and mortality risk in US hospitals. Lancet Infect. Dis. 2021, 21, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Chumbita, M.; Puerta-Alcalde, P.; Yáñez, L.; Angeles Cuesta, M.; Chinea, A.; Español-Morales, I.; Fernandez-Abellán, P.; Gudiol, C.; González-Sierra, P.; Rojas, R. High rate of inappropriate antibiotics in patients with hematologic malignancies and Pseudomonas aeruginosa bacteremia following international guideline recommendations. Microbiol. Spectr. 2023, 11, e00674-23. [Google Scholar] [CrossRef] [PubMed]
- Suarez, C.; Pena, C.; Gavalda, L.; Tubau, F.; Manzur, A.; Dominguez, M.A.; Pujol, M.; Gudiol, F.; Ariza, J. Influence of carbapenem resistance on mortality and the dynamics of mortality in Pseudomonas aeruginosa bloodstream infection. Int. J. Infect. Dis. 2010, 14, e73–e78. [Google Scholar] [CrossRef]
- Sid Ahmed, M.A.; Khan, F.A.; Sultan, A.A.; Söderquist, B.; Ibrahim, E.B.; Jass, J.; Omrani, A.S. β-lactamase-mediated resistance in MDR-Pseudomonas aeruginosa from Qatar. Antimicrob. Resist. Infect. Control. 2020, 9, 170. [Google Scholar] [CrossRef]
- Costa-Júnior, S.D.; da Silva, A.M.C.M.; Niedja da Paz Pereira, J.; da Costa Lima, J.L.; Cavalcanti, I.M.F.; Maciel, M.A.V. Emergence of rmtD1 gene in clinical isolates of Pseudomonas aeruginosa carrying bla KPC and/or bla VIM-2 genes in Brazil. Braz. J. Microbiol. 2021, 52, 1959–1965. [Google Scholar] [CrossRef]
- Murray, C.J.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Aguilar, G.R.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, Y.-M.; Davies, C. Penicillin-binding protein 3 is essential for growth of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2017, 61, e01651-16. [Google Scholar] [CrossRef]
- Falagas, M.E.; Mavroudis, A.D.; Vardakas, K.Z. The antibiotic pipeline for multi-drug resistant gram negative bacteria: What can we expect? Expert Rev. Anti Infect. Ther. 2016, 14, 747–763. [Google Scholar] [CrossRef]
- Theuretzbacher, U.; Gottwalt, S.; Beyer, P.; Butler, M.; Czaplewski, L.; Lienhardt, C.; Moja, L.; Paul, M.; Paulin, S.; Rex, J.H. Analysis of the clinical antibacterial and antituberculosis pipeline. Lancet Infect. Dis. 2019, 19, e40–e50. [Google Scholar] [CrossRef]
- Diaz Caballero, J.; Clark, S.T.; Coburn, B.; Zhang, Y.; Wang, P.W.; Donaldson, S.L.; Tullis, D.E.; Yau, Y.C.; Waters, V.J.; Hwang, D.M. Selective sweeps and parallel pathoadaptation drive Pseudomonas aeruginosa evolution in the cystic fibrosis lung. mBio 2015, 6, e00981-15. [Google Scholar] [CrossRef]
- Clark, S.T.; Sinha, U.; Zhang, Y.; Wang, P.W.; Donaldson, S.L.; Coburn, B.; Waters, V.J.; Yau, Y.C.; Tullis, D.E.; Guttman, D.S. Penicillin-binding protein 3 is a common adaptive target among Pseudomonas aeruginosa isolates from adult cystic fibrosis patients treated with β-lactams. Int. J. Antimicrob. Agents 2019, 53, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [Google Scholar] [CrossRef] [PubMed]
- Pagès, J.-M.; James, C.E.; Winterhalter, M. The porin and the permeating antibiotic: A selective diffusion barrier in Gram-negative bacteria. Nat. Rev. Microbiol. 2008, 6, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, S.; Bouffartigues, E.; Bodilis, J.; Maillot, O.; Lesouhaitier, O.; Feuilloley, M.G.; Orange, N.; Dufour, A.; Cornelis, P. Structure, function and regulation of Pseudomonas aeruginosa porins. FEMS Microbiol. Rev. 2017, 41, 698–722. [Google Scholar] [CrossRef]
- Llanes, C.; Pourcel, C.; Richardot, C.; Plésiat, P.; Fichant, G.; Cavallo, J.-D.; Mérens, A.; Group, G.S.; Vu-Thien, H.; Leclercq, R. Diversity of β-lactam resistance mechanisms in cystic fibrosis isolates of Pseudomonas aeruginosa: A French multicentre study. J. Antimicrob. Chemother. 2013, 68, 1763–1771. [Google Scholar] [CrossRef]
- Giske, C.G.; Buarø, L.; Sundsfjord, A.; Wretlind, B. Alterations of porin, pumps, and penicillin-binding proteins in carbapenem resistant clinical isolates of Pseudomonas aeruginosa. Microb. Drug Resist. 2008, 14, 23–30. [Google Scholar] [CrossRef]
- Li, H.; Luo, Y.-F.; Williams, B.J.; Blackwell, T.S.; Xie, C.-M. Structure and function of OprD protein in Pseudomonas aeruginosa: From antibiotic resistance to novel therapies. Int. J. Med. Microbiol. 2012, 302, 63–68. [Google Scholar] [CrossRef]
- Ochs, M.M.; McCusker, M.P.; Bains, M.; Hancock, R.E. Negative regulation of the Pseudomonas aeruginosa outer membrane porin OprD selective for imipenem and basic amino acids. Antimicrob. Agents Chemother. 1999, 43, 1085–1090. [Google Scholar] [CrossRef]
- Iyer, R.; Sylvester, M.A.; Velez-Vega, C.; Tommasi, R.; Durand-Reville, T.F.; Miller, A.A. Whole-cell-based assay to evaluate structure permeation relationships for carbapenem passage through the Pseudomonas aeruginosa porin OprD. ACS Infect. Dis. 2017, 3, 310–319. [Google Scholar] [CrossRef]
- Ude, J.; Tripathi, V.; Buyck, J.M.; Söderholm, S.; Cunrath, O.; Fanous, J.; Claudi, B.; Egli, A.; Schleberger, C.; Hiller, S. Outer membrane permeability: Antimicrobials and diverse nutrients bypass porins in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2021, 118, e2107644118. [Google Scholar] [CrossRef]
- Isabella, V.M.; Campbell, A.J.; Manchester, J.; Sylvester, M.; Nayar, A.S.; Ferguson, K.E.; Tommasi, R.; Miller, A.A. Toward the rational design of carbapenem uptake in Pseudomonas aeruginosa. Chem. Biol. 2015, 22, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Tamber, S.; Maier, E.; Benz, R.; Hancock, R.E. Characterization of OpdH, a Pseudomonas aeruginosa porin involved in the uptake of tricarboxylates. J. Bacteriol. 2007, 189, 929–939. [Google Scholar] [CrossRef]
- Kabra, R.; Chauhan, N.; Kumar, A.; Ingale, P.; Singh, S. Efflux pumps and antimicrobial resistance: Paradoxical components in systems genomics. Prog. Biophys. Mol. Biol. 2019, 141, 15–24. [Google Scholar] [CrossRef]
- Nikaido, H.; Takatsuka, Y. Mechanisms of RND multidrug efflux pumps. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2009, 1794, 769–781. [Google Scholar] [CrossRef]
- Fernando, D.M.; Kumar, A. Resistance-nodulation-division multidrug efflux pumps in gram-negative bacteria: Role in virulence. Antibiotics 2013, 2, 163–181. [Google Scholar] [CrossRef]
- Alav, I.; Kobylka, J.; Kuth, M.S.; Pos, K.M.; Picard, M.; Blair, J.M.; Bavro, V.N. Structure, assembly, and function of tripartite efflux and type 1 secretion systems in gram-negative bacteria. Chem. Rev. 2021, 121, 5479–5596. [Google Scholar] [CrossRef]
- del Barrio-Tofiño, E.; López-Causapé, C.; Cabot, G.; Rivera, A.; Benito, N.; Segura, C.; Montero, M.M.; Sorlí, L.; Tubau, F.; Gómez-Zorrilla, S. Genomics and susceptibility profiles of extensively drug-resistant Pseudomonas aeruginosa isolates from Spain. Antimicrob. Agents Chemother. 2017, 61, e01589-17. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Ko, K.S. OprD mutations and inactivation, expression of efflux pumps and AmpC, and metallo-β-lactamases in carbapenem-resistant Pseudomonas aeruginosa isolates from South Korea. Int. J. Antimicrob. Agents 2012, 40, 168–172. [Google Scholar] [CrossRef]
- Moubareck, C.A.; Halat, D.H.; Akkawi, C.; Nabi, A.; AlSharhan, M.A.; AlDeesi, Z.O.; Peters, C.C.; Celiloglu, H.; Sarkis, D.K. Role of outer membrane permeability, efflux mechanism, and carbapenemases in carbapenem-nonsusceptible Pseudomonas aeruginosa from Dubai hospitals: Results of the first cross-sectional survey. Int. J. Infect. Dis. 2019, 84, 143–150. [Google Scholar] [CrossRef]
- Köhler, T.; Michéa-Hamzehpour, M.; Henze, U.; Gotoh, N.; Kocjancic Curty, L.; Pechère, J.C. Characterization of MexE–MexF–OprN, a positively regulated multidrug efflux system of Pseudomonas aeruginosa. Mol. Microbiol. 1997, 23, 345–354. [Google Scholar] [CrossRef]
- Moyá, B.; Beceiro, A.; Cabot, G.; Juan, C.; Zamorano, L.; Alberti, S.; Oliver, A. Pan-β-lactam resistance development in Pseudomonas aeruginosa clinical strains: Molecular mechanisms, penicillin-binding protein profiles, and binding affinities. Antimicrob. Agents Chemother. 2012, 56, 4771–4778. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.-p.; Xu, Y.-h.; Wang, Z.-x.; Fang, Y.-p.; Shen, J.-l. Overexpression of MexAB-OprM efflux pump in carbapenem-resistant Pseudomonas aeruginosa. Arch. Microbiol. 2016, 198, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Chalhoub, H.; Pletzer, D.; Weingart, H.; Braun, Y.; Tunney, M.M.; Elborn, J.S.; Rodriguez-Villalobos, H.; Plésiat, P.; Kahl, B.C.; Denis, O. Mechanisms of intrinsic resistance and acquired susceptibility of Pseudomonas aeruginosa isolated from cystic fibrosis patients to temocillin, a revived antibiotic. Sci. Rep. 2017, 7, 40208. [Google Scholar] [CrossRef]
- Masuda, N.; Sakagawa, E.; Ohya, S.; Gotoh, N.; Tsujimoto, H.; Nishino, T. Substrate specificities of MexAB-OprM, MexCD-OprJ, and MexXY-oprM efflux pumps in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2000, 44, 3322–3327. [Google Scholar] [CrossRef]
- Frimodt-Møller, J.; Rossi, E.; Haagensen, J.A.J.; Falcone, M.; Molin, S.; Johansen, H.K. Mutations causing low level antibiotic resistance ensure bacterial survival in antibiotic-treated hosts. Sci. Rep. 2018, 8, 12512. [Google Scholar] [CrossRef]
- Masuda, N.; Sakagawa, E.; Ohya, S.; Gotoh, N.; Tsujimoto, H.; Nishino, T. Contribution of the MexX-MexY-OprM efflux system to intrinsic resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2000, 44, 2242–2246. [Google Scholar] [CrossRef]
- Gomis-Font, M.A.; Pitart, C.; del Barrio-Tofiño, E.; Zboromyrska, Y.; Cortes-Lara, S.; Mulet, X.; Marco, F.; Vila, J.; López-Causapé, C.; Oliver, A. Emergence of Resistance to Novel Cephalosporin–β-lactamase Inhibitor Combinations through the Modification of the Pseudomonas aeruginosa MexCD-OprJ Efflux Pump. Antimicrob. Agents Chemother. 2021, 65, e00089-21. [Google Scholar] [CrossRef]
- Evans, K.; Adewoye, L.; Poole, K. MexR repressor of the mexAB-oprM multidrug efflux operon of Pseudomonas aeruginosa: Identification of MexR binding sites in the mexA-mexR intergenic region. J. Bacteriol. 2001, 183, 807–812. [Google Scholar] [CrossRef]
- Saito, K.; Eda, S.; Maseda, H.; Nakae, T. Molecular mechanism of MexR-mediated regulation of MexAB–OprM efflux pump expression in Pseudomonas aeruginosa. FEMS Microbiol. Lett. 2001, 195, 23–28. [Google Scholar] [CrossRef]
- Poole, K.; Tetro, K.; Zhao, Q.; Neshat, S.; Heinrichs, D.E.; Bianco, N. Expression of the multidrug resistance operon mexA-mexB-oprM in Pseudomonas aeruginosa: mexR encodes a regulator of operon expression. Antimicrob. Agents Chemother. 1996, 40, 2021–2028. [Google Scholar] [CrossRef]
- Sobel, M.L.; Hocquet, D.; Cao, L.; Plesiat, P.; Poole, K. Mutations in PA3574 (nalD) lead to increased MexAB-OprM expression and multidrug resistance in laboratory and clinical isolates of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2005, 49, 1782–1786. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Yoneyama, H.; Nakae, T. nalB-type mutations causing the overexpression of the MexAB-OprM efflux pump are located in the mexR gene of the Pseudomonas aeruginosa chromosome. FEMS Microbiol. Lett. 1999, 179, 67–72. [Google Scholar] [CrossRef]
- Morita, Y.; Cao, L.; Gould, V.C.; Avison, M.B.; Poole, K. nalD encodes a second repressor of the mexAB-oprM multidrug efflux operon of Pseudomonas aeruginosa. J. Bacteriol. 2006, 188, 8649–8654. [Google Scholar] [CrossRef]
- Köhler, T.; Michea-Hamzehpour, M.; Epp, S.F.; Pechere, J.-C. Carbapenem activities against Pseudomonas aeruginosa: Respective contributions of OprD and efflux systems. Antimicrob. Agents Chemother. 1999, 43, 424–427. [Google Scholar] [CrossRef]
- Sobel, M.L.; Neshat, S.; Poole, K. Mutations in PA2491 (mexS) promote MexT-dependent mexEF-oprN expression and multidrug resistance in a clinical strain of Pseudomonas aeruginosa. J. Bacteriol. 2005, 187, 1246–1253. [Google Scholar] [CrossRef]
- Köhler, T.; Epp, S.F.; Curty, L.K.; Pechère, J.-C. Characterization of MexT, the regulator of the MexE-MexF-OprN multidrug efflux system of Pseudomonas aeruginosa. J. Bacteriol. 1999, 181, 6300–6305. [Google Scholar] [CrossRef]
- Reading, C.; Cole, M. Clavulanic acid: A beta-lactamase-inhibiting beta-lactam from Streptomyces clavuligerus. Antimicrob. Agents Chemother. 1977, 11, 852–857. [Google Scholar] [CrossRef]
- Charnas, R.L.; Fisher, J.; Knowles, J.R. Chemical studies on the inactivation of Escherichia coli RTEM β-lactamase by clavulanic acid. Biochemistry 1978, 17, 2185–2189. [Google Scholar] [CrossRef]
- Charnas, R.L.; Knowles, J.R. Inactivation of radiolabeled RTEM. beta.-lactamase from Escherichia coli by clavulanic acid and 9-deoxyclavulanic acid. Biochemistry 1981, 20, 3214–3219. [Google Scholar] [CrossRef]
- Bush, K. A resurgence of β-lactamase inhibitor combinations effective against multidrug-resistant Gram-negative pathogens. Int. J. Antimicrob. Agents 2015, 46, 483–493. [Google Scholar] [CrossRef]
- Yigit, H.; Queenan, A.M.; Rasheed, J.K.; Biddle, J.W.; Domenech-Sanchez, A.; Alberti, S.; Bush, K.; Tenover, F.C. Carbapenem-resistant strain of Klebsiella oxytoca harboring carbapenem-hydrolyzingβ-lactamase KPC-2. Antimicrob. Agents Chemother. 2003, 47, 3881–3889. [Google Scholar] [CrossRef] [PubMed]
- Woodford, N.; Tierno, P.M., Jr.; Young, K.; Tysall, L.; Palepou, M.-F.I.; Ward, E.; Painter, R.E.; Suber, D.F.; Shungu, D.; Silver, L.L. Outbreak of Klebsiella pneumoniae producing a new carbapenem-hydrolyzing class A β-lactamase, KPC-3, in a New York medical center. Antimicrob. Agents Chemother. 2004, 48, 4793–4799. [Google Scholar] [CrossRef]
- Moland, E.S.; Hong, S.G.; Thomson, K.S.; Larone, D.H.; Hanson, N.D. Klebsiella pneumoniae isolate producing at least eight different β-lactamases, including AmpC and KPC β-lactamases. Antimicrob. Agents Chemother. 2007, 51, 800. [Google Scholar] [CrossRef]
- Yahav, D.; Giske, C.G.; Grāmatniece, A.; Abodakpi, H.; Tam, V.H.; Leibovici, L. New β-lactam–β-lactamase inhibitor combinations. Clin. Microbiol. Rev. 2020, 34. [Google Scholar] [CrossRef]
- Coleman, K. Diazabicyclooctanes (DBOs): A potent new class of non-β-lactam β-lactamase inhibitors. Curr. Opin. Microbiol. 2011, 14, 550–555. [Google Scholar] [CrossRef]
- Rubino, C.M.; Bhavnani, S.M.; Loutit, J.S.; Morgan, E.E.; White, D.; Dudley, M.N.; Griffith, D.C. Phase 1 study of the safety, tolerability, and pharmacokinetics of vaborbactam and meropenem alone and in combination following single and multiple doses in healthy adult subjects. Antimicrob. Agents Chemother. 2018, 62, e02228-17. [Google Scholar] [CrossRef]
- Hecker, S.J.; Reddy, K.R.; Totrov, M.; Hirst, G.C.; Lomovskaya, O.; Griffith, D.C.; King, P.; Tsivkovski, R.; Sun, D.; Sabet, M. Discovery of a cyclic boronic acid β-lactamase inhibitor (RPX7009) with utility vs. class A serine carbapenemases. J. Med. Chem. 2015, 58, 3682–3692. [Google Scholar] [CrossRef]
- Lee, S.Y.; Gill, C.M.; Nicolau, D.P. Activity of novel β-lactam/β-lactamase inhibitor combinations against serine carbapenemase-producing carbapenem-resistant Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2023, 78, 2795–2800. [Google Scholar] [CrossRef]
- Ruiz, V.H.; Gill, C.M.; Nicolau, D.P. Assessing the in vivo impact of novel β-lactamase inhibitors on the efficacy of their partner β-lactams against serine carbapenemase-producing Pseudomonas aeruginosa using human-simulated exposures. J. Antimicrob. Chemother. 2024, 79, 546–551. [Google Scholar] [CrossRef]
- González-Pinto, L.; Alonso-García, I.; Blanco-Martín, T.; Camacho-Zamora, P.; Fraile-Ribot, P.A.; Outeda-García, M.; Lasarte-Monterrubio, C.; Guijarro-Sánchez, P.; Maceiras, R.; Moya, B.; et al. Impact of chromosomally encoded resistance mechanisms and transferable β-lactamases on the activity of cefiderocol and innovative β-lactam/β-lactamase inhibitor combinations against Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2024, 79, 2591–2597. [Google Scholar] [CrossRef]
- Carvalhaes, C.G.; Shortridge, D.; Sader, H.S.; Castanheira, M. Activity of Meropenem-Vaborbactam against Bacterial Isolates Causing Pneumonia in Patients in U.S. Hospitals during 2014 to 2018. Antimicrob. Agents Chemother. 2020, 64, e02177-19. [Google Scholar] [CrossRef] [PubMed]
- Mushtaq, S.; Meunier, D.; Vickers, A.; Woodford, N.; Livermore, D.M. Activity of imipenem/relebactam against Pseudomonas aeruginosa producing ESBLs and carbapenemases. J. Antimicrob. Chemother. 2021, 76, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Livermore, D.M.; Mushtaq, S.; Warner, M.; Woodford, N. Activity of OP0595/β-lactam combinations against Gram-negative bacteria with extended-spectrum, AmpC and carbapenem-hydrolysing β-lactamases. J. Antimicrob. Chemother. 2015, 70, 3032–3041. [Google Scholar] [CrossRef] [PubMed]
- Livermore, D.M.; Warner, M.; Mushtaq, S. Activity of MK-7655 combined with imipenem against Enterobacteriaceae and Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2013, 68, 2286–2290. [Google Scholar] [CrossRef]
- Le Terrier, C.; Nordmann, P.; Poirel, L. In vitro activity of aztreonam in combination with newly developed β-lactamase inhibitors against MDR Enterobacterales and Pseudomonas aeruginosa producing metallo-β-lactamases. J. Antimicrob. Chemother. 2023, 78, 101–107. [Google Scholar] [CrossRef]
- Thomson, K.S.; AbdelGhani, S.; Snyder, J.W.; Thomson, G.K. Activity of cefepime-zidebactam against multidrug-resistant (MDR) Gram-negative pathogens. Antibiotics 2019, 8, 32. [Google Scholar] [CrossRef]
- Lomovskaya, O.; Rubio-Aparicio, D.; Nelson, K.; Sun, D.; Tsivkovski, R.; Castanheira, M.; Lindley, J.; Loutit, J.; Dudley, M. In Vitro Activity of the Ultrabroad-Spectrum Beta-Lactamase Inhibitor QPX7728 in Combination with Multiple Beta-Lactam Antibiotics against Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2021, 65, e00210-21. [Google Scholar] [CrossRef]
- Slater, C.L.; Winogrodzki, J.; Fraile-Ribot, P.A.; Oliver, A.; Khajehpour, M.; Mark, B.L. Adding Insult to Injury: Mechanistic Basis for How AmpC Mutations Allow Pseudomonas aeruginosa To Accelerate Cephalosporin Hydrolysis and Evade Avibactam. Antimicrob. Agents Chemother. 2020, 64, e00894-20. [Google Scholar] [CrossRef]
- Alonso-García, I.; Vázquez-Ucha, J.C.; Lasarte-Monterrubio, C.; González-Mayo, E.; Lada-Salvador, P.; Vela-Fernández, R.; Aja-Macaya, P.; Guijarro-Sánchez, P.; Rumbo-Feal, S.; Muíño-Andrade, M.; et al. Simultaneous and divergent evolution of resistance to cephalosporin/β-lactamase inhibitor combinations and imipenem/relebactam following ceftazidime/avibactam treatment of MDR Pseudomonas aeruginosa infections. J. Antimicrob. Chemother. 2023, 78, 1195–1200. [Google Scholar] [CrossRef]
- Ruedas-López, A.; Alonso-García, I.; Lasarte-Monterrubio, C.; Guijarro-Sánchez, P.; Gato, E.; Vázquez-Ucha, J.C.; Vallejo, J.A.; Fraile-Ribot, P.A.; Fernández-Pérez, B.; Velasco, D.; et al. Selection of AmpC β-Lactamase Variants and Metallo-β-Lactamases Leading to Ceftolozane/Tazobactam and Ceftazidime/Avibactam Resistance during Treatment of MDR/XDR Pseudomonas aeruginosa Infections. Antimicrob. Agents Chemother. 2022, 66, e0206721. [Google Scholar] [CrossRef]
- Hujer, A.M.; Bethel, C.R.; Taracila, M.A.; Marshall, S.H.; Rojas, L.J.; Winkler, M.L.; Painter, R.E.; Domitrovic, T.N.; Watkins, R.R.; Abdelhamed, A.M.; et al. Imipenem/Relebactam Resistance in Clinical Isolates of Extensively Drug Resistant Pseudomonas aeruginosa: Inhibitor-Resistant β-Lactamases and Their Increasing Importance. Antimicrob. Agents Chemother. 2022, 66, e0179021. [Google Scholar] [CrossRef] [PubMed]
- Bruynoghe, R.; Maisin, J. Essais de thérapeutique au moyen du bacteriophage. CR Soc. Biol. 1921, 85, 1120–1121. [Google Scholar]
- Stone, R. Stalin’s forgotten cure. Science 2002, 298, 728–731. [Google Scholar] [CrossRef]
- Kutateladze, Á.; Adamia, R. Phage therapy experience at the Eliava Institute. Med. Mal. Infect. 2008, 38, 426–430. [Google Scholar] [CrossRef]
- Abedon, S.T.; Kuhl, S.J.; Blasdel, B.G.; Kutter, E.M. Phage treatment of human infections. Bacteriophage 2011, 1, 66–85. [Google Scholar] [CrossRef]
- Schooley, R.T.; Biswas, B.; Gill, J.J.; Hernandez-Morales, A.; Lancaster, J.; Lessor, L.; Barr, J.J.; Reed, S.L.; Rohwer, F.; Benler, S. Development and use of personalized bacteriophage-based therapeutic cocktails to treat a patient with a disseminated resistant Acinetobacter baumannii infection. Antimicrob. Agents Chemother. 2017, 61, e00954-17. [Google Scholar] [CrossRef]
- El Haddad, L.; Harb, C.P.; Gebara, M.A.; Stibich, M.A.; Chemaly, R.F. A systematic and critical review of bacteriophage therapy against multidrug-resistant ESKAPE organisms in humans. Clin. Infect. Dis. 2019, 69, 167–178. [Google Scholar] [CrossRef]
- Dedrick, R.M.; Guerrero-Bustamante, C.A.; Garlena, R.A.; Russell, D.A.; Ford, K.; Harris, K.; Gilmour, K.C.; Soothill, J.; Jacobs-Sera, D.; Schooley, R.T. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 2019, 25, 730–733. [Google Scholar] [CrossRef]
- Thi, M.T.T.; Wibowo, D.; Rehm, B.H.A. Pseudomonas aeruginosa Biofilms. Int. J. Mol. Sci. 2020, 21, 8671. [Google Scholar] [CrossRef]
- Dibdin, G.H.; Assinder, S.J.; Nichols, W.W.; Lambert, P.A. Mathematical model of β-lactam penetration into a biofilm of Pseudomonas aeruginosa while undergoing simultaneous inactivation by released β-lactamases. J. Antimicrob. Chemother. 1996, 38, 757–769. [Google Scholar] [CrossRef]
- Wang, H.; Ciofu, O.; Yang, L.; Wu, H.; Song, Z.; Oliver, A.; Høiby, N. High β-lactamase levels change the pharmacodynamics of β-lactam antibiotics in Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 2013, 57, 196–204. [Google Scholar]
- Knecht, L.E.; Veljkovic, M.; Fieseler, L. Diversity and function of phage encoded depolymerases. Front. Microbiol. 2020, 10, 2949. [Google Scholar] [CrossRef]
- Danis-Wlodarczyk, K.; Olszak, T.; Arabski, M.; Wasik, S.; Majkowska-Skrobek, G.; Augustyniak, D.; Gula, G.; Briers, Y.; Jang, H.B.; Vandenheuvel, D. Characterization of the newly isolated lytic bacteriophages KTN6 and KT28 and their efficacy against Pseudomonas aeruginosa biofilm. PLoS ONE 2015, 10, e0127603. [Google Scholar]
- Hanlon, G.W.; Denyer, S.P.; Olliff, C.J.; Ibrahim, L.J. Reduction in exopolysaccharide viscosity as an aid to bacteriophage penetration through Pseudomonas aeruginosa biofilms. Appl. Environ. Microbiol. 2001, 67, 2746–2753. [Google Scholar] [CrossRef]
- Henriksen, K.; Rørbo, N.; Rybtke, M.L.; Martinet, M.G.; Tolker-Nielsen, T.; Høiby, N.; Middelboe, M.; Ciofu, O.P. aeruginosa flow-cell biofilms are enhanced by repeated phage treatments but can be eradicated by phage–ciprofloxacin combination: —Monitoring the phage–P. aeruginosa biofilms interactions. Pathog. Dis. 2019, 77, ftz011. [Google Scholar] [CrossRef]
- Franklin, M.J.; Nivens, D.E.; Weadge, J.T.; Howell, P.L. Biosynthesis of the Pseudomonas aeruginosa extracellular polysaccharides, alginate, Pel, and Psl. Front. Microbiol. 2011, 2, 167. [Google Scholar] [CrossRef]
- Glonti, T.; Chanishvili, N.; Taylor, P. Bacteriophage-derived enzyme that depolymerizes the alginic acid capsule associated with cystic fibrosis isolates of Pseudomonas aeruginosa. J. Appl. Microbiol. 2010, 108, 695–702. [Google Scholar] [CrossRef]
- Ertesvåg, H. Alginate-modifying enzymes: Biological roles and biotechnological uses. Front. Microbiol. 2015, 6, 523. [Google Scholar] [CrossRef]
- Yan, J.; Mao, J.; Xie, J. Bacteriophage polysaccharide depolymerases and biomedical applications. Biodrugs 2014, 28, 265–274. [Google Scholar] [CrossRef]
- Wong, T.Y.; Preston, L.A.; Schiller, N.L. Alginate lyase: Review of major sources and enzyme characteristics, structure-function analysis, biological roles, and applications. Annu. Rev. Microbiol. 2000, 54, 289–340. [Google Scholar] [CrossRef]
- Osawa, T.; Matsubara, Y.; Muramatsu, T.; Kimura, M.; Kakuta, Y. Crystal structure of the alginate (poly α-L-guluronate) lyase from Corynebacterium sp. at 1.2 Å resolution. J. Mol. Biol. 2005, 345, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Adnan, M.; Shah, M.R.A.; Jamal, M.; Jalil, F.; Andleeb, S.; Nawaz, M.A.; Pervez, S.; Hussain, T.; Shah, I.; Imran, M. Isolation and characterization of bacteriophage to control multidrug-resistant Pseudomonas aeruginosa planktonic cells and biofilm. Biologicals 2020, 63, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Pei, R.; Lamas-Samanamud, G.R. Inhibition of biofilm formation by T7 bacteriophages producing quorum-quenching enzymes. Appl. Environ. Microbiol. 2014, 80, 5340–5348. [Google Scholar] [CrossRef] [PubMed]
- Latz, S.; Krüttgen, A.; Häfner, H.; Buhl, E.M.; Ritter, K.; Horz, H.-P. Differential effect of newly isolated phages belonging to PB1-like, phiKZ-like and LUZ24-like viruses against multi-drug resistant Pseudomonas aeruginosa under varying growth conditions. Viruses 2017, 9, 315. [Google Scholar] [CrossRef]
- Alves, D.R.; Perez-Esteban, P.; Kot, W.; Bean, J.; Arnot, T.; Hansen, L.; Enright, M.C.; Jenkins, A.T.A. A novel bacteriophage cocktail reduces and disperses P seudomonas aeruginosa biofilms under static and flow conditions. Microb. Biotechnol. 2016, 9, 61–74. [Google Scholar] [CrossRef]
- Shafique, M.; Alvi, I.A.; Abbas, Z.; ur Rehman, S. Assessment of biofilm removal capacity of a broad host range bacteriophage JHP against Pseudomonas aeruginosa. APMIS 2017, 125, 579–584. [Google Scholar] [CrossRef]
- Lehman, S.M.; Donlan, R.M. Bacteriophage-mediated control of a two-species biofilm formed by microorganisms causing catheter-associated urinary tract infections in an in vitro urinary catheter model. Antimicrob. Agents Chemother. 2015, 59, 1127–1137. [Google Scholar] [CrossRef]
- Pires, D.; Sillankorva, S.; Faustino, A.; Azeredo, J. Use of newly isolated phages for control of Pseudomonas aeruginosa PAO1 and ATCC 10145 biofilms. Res. Microbiol. 2011, 162, 798–806. [Google Scholar] [CrossRef]
- Knezevic, P.; Obreht, D.; Curcin, S.; Petrusic, M.; Aleksic, V.; Kostanjsek, R.; Petrovic, O. Phages of Pseudomonas aeruginosa: Response to environmental factors and in vitro ability to inhibit bacterial growth and biofilm formation. J. Appl. Microbiol. 2011, 111, 245–254. [Google Scholar] [CrossRef]
- Fu, W.; Forster, T.; Mayer, O.; Curtin, J.J.; Lehman, S.M.; Donlan, R.M. Bacteriophage cocktail for the prevention of biofilm formation by Pseudomonas aeruginosa on catheters in an in vitro model system. Antimicrob. Agents Chemother. 2010, 54, 397–404. [Google Scholar] [CrossRef]
- Jamal, M.; Andleeb, S.; Jalil, F.; Imran, M.; Nawaz, M.A.; Hussain, T.; Ali, M.; Das, C.R. Isolation and characterization of a bacteriophage and its utilization against multi-drug resistant Pseudomonas aeruginosa-2995. Life Sci. 2017, 190, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Kwiatek, M.; Parasion, S.; Rutyna, P.; Mizak, L.; Gryko, R.; Niemcewicz, M.; Olender, A.; Łobocka, M. Isolation of bacteriophages and their application to control Pseudomonas aeruginosa in planktonic and biofilm models. Res. Microbiol. 2017, 168, 194–207. [Google Scholar] [CrossRef]
- Fong, S.A.; Drilling, A.; Morales, S.; Cornet, M.E.; Woodworth, B.A.; Fokkens, W.J.; Psaltis, A.J.; Vreugde, S.; Wormald, P.-J. Activity of bacteriophages in removing biofilms of Pseudomonas aeruginosa isolates from chronic rhinosinusitis patients. Front. Cell. Infect. Microbiol. 2017, 7, 418. [Google Scholar] [CrossRef]
- Jeon, J.; Yong, D. Two novel bacteriophages improve survival in Galleria mellonella infection and mouse acute pneumonia models infected with extensively drug-resistant Pseudomonas aeruginosa. Appl. Environ. Microbiol. 2019, 85, e02900–e02918. [Google Scholar] [CrossRef]
- Holguín, A.V.; Rangel, G.; Clavijo, V.; Prada, C.; Mantilla, M.; Gomez, M.C.; Kutter, E.; Taylor, C.; Fineran, P.C.; Barrios, A.F.G. Phage ΦPan70, a putative temperate phage, controls Pseudomonas aeruginosa in planktonic, biofilm and burn mouse model assays. Viruses 2015, 7, 4602–4623. [Google Scholar] [CrossRef]
- Alemayehu, D.; Casey, P.; McAuliffe, O.; Guinane, C.; Martin, J.; Shanahan, F.; Coffey, A.; Ross, R.; Hill, C. Bacteriophages MR299-2 and NH-4 can eliminate Pseudomonas aeruginosa in the murine lung and on cystic fibrosis lung airway cells. mBio 2012, 3, e00029-12. [Google Scholar] [CrossRef]
- Forti, F.; Roach, D.R.; Cafora, M.; Pasini, M.E.; Horner, D.S.; Fiscarelli, E.V.; Rossitto, M.; Cariani, L.; Briani, F.; Debarbieux, L. Design of a broad-range bacteriophage cocktail that reduces Pseudomonas aeruginosa biofilms and treats acute infections in two animal models. Antimicrob. Agents Chemother. 2018, 62, e02573-17. [Google Scholar] [CrossRef]
- Basu, S.; Agarwal, M.; Nath, G. An In vivo Wound Model Utilizing Bacteriophage Therapy of Pseudomonas aeruginosa Biofilms. Ostomy Wound Manag. 2015, 61, 16–23. [Google Scholar]
- Ageitos, J.M.; Sánchez-Pérez, A.; Calo-Mata, P.; Villa, T.G. Antimicrobial peptides (AMPs): Ancient compounds that represent novel weapons in the fight against bacteria. Biochem. Pharmacol. 2017, 133, 117–138. [Google Scholar] [CrossRef]
- Savini, F.; Loffredo, M.; Troiano, C.; Bobone, S.; Malanovic, N.; Eichmann, T.; Caprio, L.; Canale, V.; Park, Y.; Mangoni, M. Binding of an antimicrobial peptide to bacterial cells: Interaction with different species, strains and cellular components. Biochim. Biophys. Acta (BBA)-Biomembr. 2020, 1862, 183291. [Google Scholar] [CrossRef]
- Bahar, A.A.; Ren, D. Antimicrobial peptides. Pharmaceuticals 2013, 6, 1543–1575. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Liu, Q.; Wang, D.; Li, J. Characterization and antimicrobial mechanism of CF-14, a new antimicrobial peptide from the epidermal mucus of catfish. Fish Shellfish Immunol. 2019, 92, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Song, Y. Mechanism of Antimicrobial Peptides: Antimicrobial, Anti-Inflammatory and Antibiofilm Activities. Int. J. Mol. Sci. 2021, 22, 11401. [Google Scholar] [CrossRef]
- Bobone, S.; Stella, L. Selectivity of Antimicrobial Peptides: A Complex Interplay of Multiple Equilibria. Adv. Exp. Med. Biol. 2019, 1117, 175–214. [Google Scholar] [CrossRef]
- Harder, J.; Schröder, J.-M. Antimicrobial Peptides: Role in Human Health and Disease; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Pushpanathan, M.; Gunasekaran, P.; Rajendhran, J. Antimicrobial peptides: Versatile biological properties. Int. J. Pept. 2013, 2013, 675391. [Google Scholar] [CrossRef]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Murase, O.; Fujii, N.; Miyajima, K. Translocation of a channel-forming antimicrobial peptide, magainin 2, across lipid bilayers by forming a pore. Biochemistry 1995, 34, 6521–6526. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Murase, O.; Fujii, N.; Miyajima, K. An antimicrobial peptide, magainin 2, induced rapid flip-flop of phospholipids coupled with pore formation and peptide translocation. Biochemistry 1996, 35, 11361–11368. [Google Scholar] [CrossRef]
- Lee, T.-H.; N Hall, K.; Aguilar, M.-I. Antimicrobial peptide structure and mechanism of action: A focus on the role of membrane structure. Curr. Top. Med. Chem. 2016, 16, 25–39. [Google Scholar] [CrossRef]
- Kumar, P.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial peptides: Diversity, mechanism of action and strategies to improve the activity and biocompatibility in vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef]
- Shai, Y. Mode of action of membrane active antimicrobial peptides. Pept. Sci. Orig. Res. Biomol. 2002, 66, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef]
- Lohner, K. Membrane-active antimicrobial peptides as template structures for novel antibiotic agents. Curr. Top. Med. Chem. 2017, 17, 508–519. [Google Scholar] [CrossRef]
- Clement, N.R.; Gould, J.M. Pyranine (8-hydroxy-1, 3, 6-pyrenetrisulfonate) as a probe of internal aqueous hydrogen ion concentration in phospholipid vesicles. Biochemistry 1981, 20, 1534–1538. [Google Scholar] [CrossRef]
- Jahangiri, A.; Neshani, A.; Mirhosseini, S.A.; Ghazvini, K.; Zare, H.; Sedighian, H. Synergistic effect of two antimicrobial peptides, Nisin and P10 with conventional antibiotics against extensively drug-resistant Acinetobacter baumannii and colistin-resistant Pseudomonas aeruginosa isolates. Microb. Pathog. 2021, 150, 104700. [Google Scholar] [CrossRef]
- Field, D.; Seisling, N.; Cotter, P.D.; Ross, R.P.; Hill, C. Synergistic nisin-polymyxin combinations for the control of Pseudomonas biofilm formation. Front. Microbiol. 2016, 7, 1713. [Google Scholar] [CrossRef]
- Thomas, V.M.; Brown, R.M.; Ashcraft, D.S.; Pankey, G.A. Synergistic effect between nisin and polymyxin B against pandrug-resistant and extensively drug-resistant Acinetobacter baumannii. Int. J. Antimicrob. Agents 2019, 53, 663–668. [Google Scholar] [CrossRef]
- Naghmouchi, K.; Baah, J.; Hober, D.; Jouy, E.; Rubrecht, C.; Sané, F.; Drider, D. Synergistic effect between colistin and bacteriocins in controlling Gram-negative pathogens and their potential to reduce antibiotic toxicity in mammalian epithelial cells. Antimicrob. Agents Chemother. 2013, 57, 2719–2725. [Google Scholar] [CrossRef]
- de Leeuw, E.; Li, C.; Zeng, P.; Li, C.; Diepeveen-de Buin, M.; Lu, W.-Y.; Breukink, E.; Lu, W. Functional interaction of human neutrophil peptide-1 with the cell wall precursor lipid II. FEBS Lett. 2010, 584, 1543–1548. [Google Scholar] [CrossRef]
- Münch, D.; Sahl, H.-G. Structural variations of the cell wall precursor lipid II in Gram-positive bacteria—Impact on binding and efficacy of antimicrobial peptides. Biochim. Biophys. Acta (BBA)-Biomembr. 2015, 1848, 3062–3071. [Google Scholar] [CrossRef]
- Xhindoli, D.; Pacor, S.; Benincasa, M.; Scocchi, M.; Gennaro, R.; Tossi, A. The human cathelicidin LL-37—A pore-forming antibacterial peptide and host-cell modulator. Biochim. Biophys. Acta (BBA)-Biomembr. 2016, 1858, 546–566. [Google Scholar] [CrossRef] [PubMed]
- Ridyard, K.E.; Overhage, J. The potential of human peptide LL-37 as an antimicrobial and anti-biofilm agent. Antibiotics 2021, 10, 650. [Google Scholar] [CrossRef] [PubMed]
- Haisma, E.M.; de Breij, A.; Chan, H.; van Dissel, J.T.; Drijfhout, J.W.; Hiemstra, P.S.; El Ghalbzouri, A.; Nibbering, P.H. LL-37-derived peptides eradicate multidrug-resistant Staphylococcus aureus from thermally wounded human skin equivalents. Antimicrob. Agents Chemother. 2014, 58, 4411–4419. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.; Gonçalves, S.; Felício, M.R.; Maturana, P.; Santos, N.C.; Semorile, L.; Hollmann, A.; Maffía, P.C. Synergistic and antibiofilm activity of the antimicrobial peptide P5 against carbapenem-resistant Pseudomonas aeruginosa. Biochim. Biophys. Acta (BBA)-Biomembr. 2019, 1861, 1329–1337. [Google Scholar] [CrossRef]
- Hollmann, A.; Martínez, M.; Noguera, M.E.; Augusto, M.T.; Disalvo, A.; Santos, N.C.; Semorile, L.; Maffía, P.C. Role of amphipathicity and hydrophobicity in the balance between hemolysis and peptide–membrane interactions of three related antimicrobial peptides. Colloids Surf. B. Biointerfaces 2016, 141, 528–536. [Google Scholar] [CrossRef]
- Faccone, D.; Veliz, O.; Corso, A.; Noguera, M.; Martínez, M.; Payes, C.; Semorile, L.; Maffía, P.C. Antimicrobial activity of de novo designed cationic peptides against multi-resistant clinical isolates. Eur. J. Med. Chem. 2014, 71, 31–35. [Google Scholar] [CrossRef]
- Rudilla, H.; Fusté, E.; Cajal, Y.; Rabanal, F.; Vinuesa, T.; Viñas, M. Synergistic antipseudomonal effects of synthetic peptide AMP38 and carbapenems. Molecules 2016, 21, 1223. [Google Scholar] [CrossRef]
- Wu, M.; Maier, E.; Benz, R.; Hancock, R.E. Mechanism of interaction of different classes of cationic antimicrobial peptides with planar bilayers and with the cytoplasmic membrane of Escherichia coli. Biochemistry 1999, 38, 7235–7242. [Google Scholar] [CrossRef]
- Shang, D.; Han, X.; Du, W.; Kou, Z.; Jiang, F. Trp-containing antibacterial peptides impair quorum sensing and biofilm development in multidrug-resistant Pseudomonas aeruginosa and exhibit synergistic effects with antibiotics. Front. Microbiol. 2021, 12, 611009. [Google Scholar] [CrossRef]
- Gusmão, K.A.; Santos, D.M.d.; Santos, V.M.; Cortés, M.E.; Reis, P.V.; Santos, V.L.; Piló-Veloso, D.; Verly, R.M.; Lima, M.E.d.; Resende, J.M. Ocellatin peptides from the skin secretion of the South American frog Leptodactylus labyrinthicus (Leptodactylidae): Characterization, antimicrobial activities and membrane interactions. J. Venom. Anim. Tox. incl. Trop. Dis. 2017, 23, 4. [Google Scholar] [CrossRef]
- Bessa, L.J.; Eaton, P.; Dematei, A.; Placido, A.; Vale, N.; Gomes, P.; Delerue-Matos, C.; SA Leite, J.R.; Gameiro, P. Synergistic and antibiofilm properties of ocellatin peptides against multidrug-resistant Pseudomonas aeruginosa. Future Microbiol. 2018, 13, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Pandidan, S.; Mechler, A. Nano-viscosimetry analysis of the membrane disrupting action of the bee venom peptide melittin. Sci. Rep. 2019, 9, 10841. [Google Scholar] [CrossRef] [PubMed]
- Akbari, R.; Hakemi-Vala, M.; Pashaie, F.; Bevalian, P.; Hashemi, A.; Pooshang Bagheri, K. Highly synergistic effects of melittin with conventional antibiotics against multidrug-resistant isolates of Acinetobacter baumannii and Pseudomonas aeruginosa. Microb. Drug Resist. 2019, 25, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Stoner, I. Biopharma has abandoned antibiotic development. Here’s why we did too. Endpoints News, 13 December 2019. [Google Scholar]
- Geneva Pharmaceuticals v. GlaxoSmithKline; Court of Appeals, Federal Circuit: Washington, DC, USA, 2003; p. 1373.
- Mullard, A. Achaogen bankruptcy highlights antibacterial development woes. Nat. Rev. Drug Discov. 2019, 18, 411–412. [Google Scholar] [CrossRef]
- de la Fuente-Nunez, C. Antibiotic discovery with machine learning. Nat. Biotechnol. 2022, 40, 833–834. [Google Scholar] [CrossRef]
- Stokes, J.M.; Yang, K.; Swanson, K.; Jin, W.; Cubillos-Ruiz, A.; Donghia, N.M.; MacNair, C.R.; French, S.; Carfrae, L.A.; Bloom-Ackermann, Z. A deep learning approach to antibiotic discovery. Cell 2020, 180, 688–702.e613. [Google Scholar] [CrossRef]
- Jukič, M.; Bren, U. Machine learning in antibacterial drug design. Front. Pharmacol. 2022, 13, 864412. [Google Scholar] [CrossRef]
- Liu, G.; Stokes, J.M. A brief guide to machine learning for antibiotic discovery. Curr. Opin. Microbiol. 2022, 69, 102190. [Google Scholar] [CrossRef]
- David, L.; Brata, A.M.; Mogosan, C.; Pop, C.; Czako, Z.; Muresan, L.; Ismaiel, A.; Dumitrascu, D.I.; Leucuta, D.C.; Stanculete, M.F.; et al. Artificial Intelligence and Antibiotic Discovery. Antibiotics 2021, 10, 1376. [Google Scholar] [CrossRef]
- Liu, G.; Catacutan, D.B.; Rathod, K.; Swanson, K.; Jin, W.; Mohammed, J.C.; Chiappino-Pepe, A.; Syed, S.A.; Fragis, M.; Rachwalski, K. Deep learning-guided discovery of an antibiotic targeting Acinetobacter baumannii. Nat. Chem. Biol. 2023, 19, 1342–1350. [Google Scholar] [CrossRef]
- Parvaiz, N.; Ahmad, F.; Yu, W.; MacKerell, A.D., Jr.; Azam, S.S. Discovery of beta-lactamase CMY-10 inhibitors for combination therapy against multi-drug resistant Enterobacteriaceae. PLoS ONE 2021, 16, e0244967. [Google Scholar] [CrossRef] [PubMed]
- McNair, K.; Aziz, R.K.; Pusch, G.D.; Overbeek, R.; Dutilh, B.E.; Edwards, R. Phage genome annotation using the RAST pipeline. Bacteriophages Methods Protoc. 2018, 3, 231–238. [Google Scholar]
- McNair, K.; Zhou, C.; Dinsdale, E.A.; Souza, B.; Edwards, R.A. PHANOTATE: A novel approach to gene identification in phage genomes. Bioinformatics 2019, 35, 4537–4542. [Google Scholar] [CrossRef]
- Thung, T.Y.; White, M.E.; Dai, W.; Wilksch, J.J.; Bamert, R.S.; Rocker, A.; Stubenrauch, C.J.; Williams, D.; Huang, C.; Schittelhelm, R. Component parts of bacteriophage virions accurately defined by a machine-learning approach built on evolutionary features. Msystems 2021, 6, e00242-21. [Google Scholar] [CrossRef]
- Wang, J.; Dai, W.; Li, J.; Xie, R.; Dunstan, R.A.; Stubenrauch, C.; Zhang, Y.; Lithgow, T. PaCRISPR: A server for predicting and visualizing anti-CRISPR proteins. Nucleic Acids Res. 2020, 48, W348–W357. [Google Scholar] [CrossRef]
- Bhadra, P.; Yan, J.; Li, J.; Fong, S.; Siu, S.W. AmPEP: Sequence-based prediction of antimicrobial peptides using distribution patterns of amino acid properties and random forest. Sci. Rep. 2018, 8, 1697. [Google Scholar] [CrossRef]
- Karp, N.A. Reproducible preclinical research-Is embracing variability the answer? PLoS Biol. 2018, 16, e2005413. [Google Scholar] [CrossRef]
- Dirnagl, U.; Duda, G.N.; Grainger, D.W.; Reinke, P.; Roubenoff, R. Reproducibility, relevance and reliability as barriers to efficient and credible biomedical technology translation. Adv. Drug Deliv. Rev. 2022, 182, 114118. [Google Scholar] [CrossRef]
- Samuel, S.; König-Ries, B. Understanding experiments and research practices for reproducibility: An exploratory study. PeerJ 2021, 9, e11140. [Google Scholar] [CrossRef]
- Hillary, F.G.; Medaglia, J.D. What the replication crisis means for intervention science. Int. J. Psychophysiol. 2020, 154, 3–5. [Google Scholar] [CrossRef]
- Papp-Wallace, K.M.; Bethel, C.R.; Caillon, J.; Barnes, M.D.; Potel, G.; Bajaksouzian, S.; Rutter, J.D.; Reghal, A.; Shapiro, S.; Taracila, M.A. Beyond piperacillin-tazobactam: Cefepime and AAI101 as a potent β-lactam− β-lactamase inhibitor combination. Antimicrob. Agents Chemother. 2019, 63, e00105-19. [Google Scholar] [CrossRef] [PubMed]
- Dalhoff, A. Are antibacterial effects of non-antibiotic drugs random or purposeful because of a common evolutionary origin of bacterial and mammalian targets? Infection 2021, 49, 569–589. [Google Scholar] [CrossRef]
- Casteels, R.; Login, I. Reserpine has a direct action as a calcium antagonist on mammalian smooth muscle cells. J. Physiol. 1983, 340, 403–414. [Google Scholar] [CrossRef]
- Campoli-Richards, D.M.; Brogden, R.N. Sulbactam/ampicillin: A review of its antibacterial activity, pharmacokinetic properties, and therapeutic use. Drugs 1987, 33, 577–609. [Google Scholar] [CrossRef]
- Cady Kyle, C.; Bondy-Denomy, J.; Heussler Gary, E.; Davidson Alan, R.; O’Toole George, A. The CRISPR/Cas Adaptive Immune System of Pseudomonas aeruginosa Mediates Resistance to Naturally Occurring and Engineered Phages. J. Bacteriol. 2012, 194, 5728–5738. [Google Scholar] [CrossRef]
- Gooderham, W.J.; Gellatly, S.L.; Sanschagrin, F.; McPhee, J.B.; Bains, M.; Cosseau, C.; Levesque, R.C.; Hancock, R.E. The sensor kinase PhoQ mediates virulence in Pseudomonas aeruginosa. Microbiology 2009, 155, 699–711. [Google Scholar] [CrossRef]
- McPhee, J.B.; Lewenza, S.; Hancock, R.E. Cationic antimicrobial peptides activate a two-component regulatory system, PmrA-PmrB, that regulates resistance to polymyxin B and cationic antimicrobial peptides in Pseudomonas aeruginosa. Mol. Microbiol. 2003, 50, 205–217. [Google Scholar] [CrossRef]
- Llobet, E.; Tomas, J.M.; Bengoechea, J.A. Capsule polysaccharide is a bacterial decoy for antimicrobial peptides. Microbiology 2008, 154, 3877–3886. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ambler Class | Representative Members Found in P. aeruginosa | Substrate Scope and Relevance |
|---|---|---|
| A (SBLs) | KPC-2, GES-2 | Certain members of this class can degrade all β-lactam subclasses; includes ESBLs and carbapenemases [44] |
| B (MBLs) | NDM-1, IMP-1, VIM-2 | Most noted for carbapenemase activity; some members degrade penicillins and cephalosporins; do not protect against monobactams [45] |
| C (SBLs) | AmpC, PDC-1 | Generally effective against penicillins and cephalosporins; some have ESBL activity; limited ability to degrade carbapenems [46] |
| D (SBLs) | OXA-10, OXA-48 | Degrade certain penicillins and cephalosporins; some members have ESBL or carbapenemase activity [47] |
| Efflux System | β-Lactam Substrates |
|---|---|
| MexAB-OprM | All tested β-lactams, * excluding imipenem [104,114] |
| MexXY-OprM | All tested β-lactams, * excluding carbenicillin, sulbenicillin, cefsulodin, ceftazidime, moxalactam, flomoxef, aztreonam, and imipenem [104] |
| MexCD-OprJ | All tested β-lactams, * excluding carbenicillin, sulbenicillin, cefsulodin, ceftazidime, moxalactam, aztreonam, and imipenem [104] |
| MexEF-OprN | Imipenem [115,116] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, D.W.; Lohans, C.T. Combatting Pseudomonas aeruginosa with β-Lactam Antibiotics: A Revived Weapon? Antibiotics 2025, 14, 526. https://doi.org/10.3390/antibiotics14050526
Zhao DW, Lohans CT. Combatting Pseudomonas aeruginosa with β-Lactam Antibiotics: A Revived Weapon? Antibiotics. 2025; 14(5):526. https://doi.org/10.3390/antibiotics14050526
Chicago/Turabian StyleZhao, Dylan W., and Christopher T. Lohans. 2025. "Combatting Pseudomonas aeruginosa with β-Lactam Antibiotics: A Revived Weapon?" Antibiotics 14, no. 5: 526. https://doi.org/10.3390/antibiotics14050526
APA StyleZhao, D. W., & Lohans, C. T. (2025). Combatting Pseudomonas aeruginosa with β-Lactam Antibiotics: A Revived Weapon? Antibiotics, 14(5), 526. https://doi.org/10.3390/antibiotics14050526