
Anti-Staphylococcus aureus Activity and Structural Characterization of Rationally Designed Peptides
 , ,
, ,  , ,
, ,  , ,
, ,  , , and
, , and 
Abstract
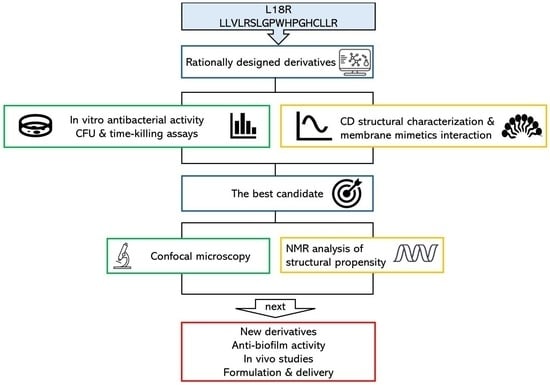
1. Introduction
2. Results
2.1. Peptide Design
2.2. Cytotoxicity
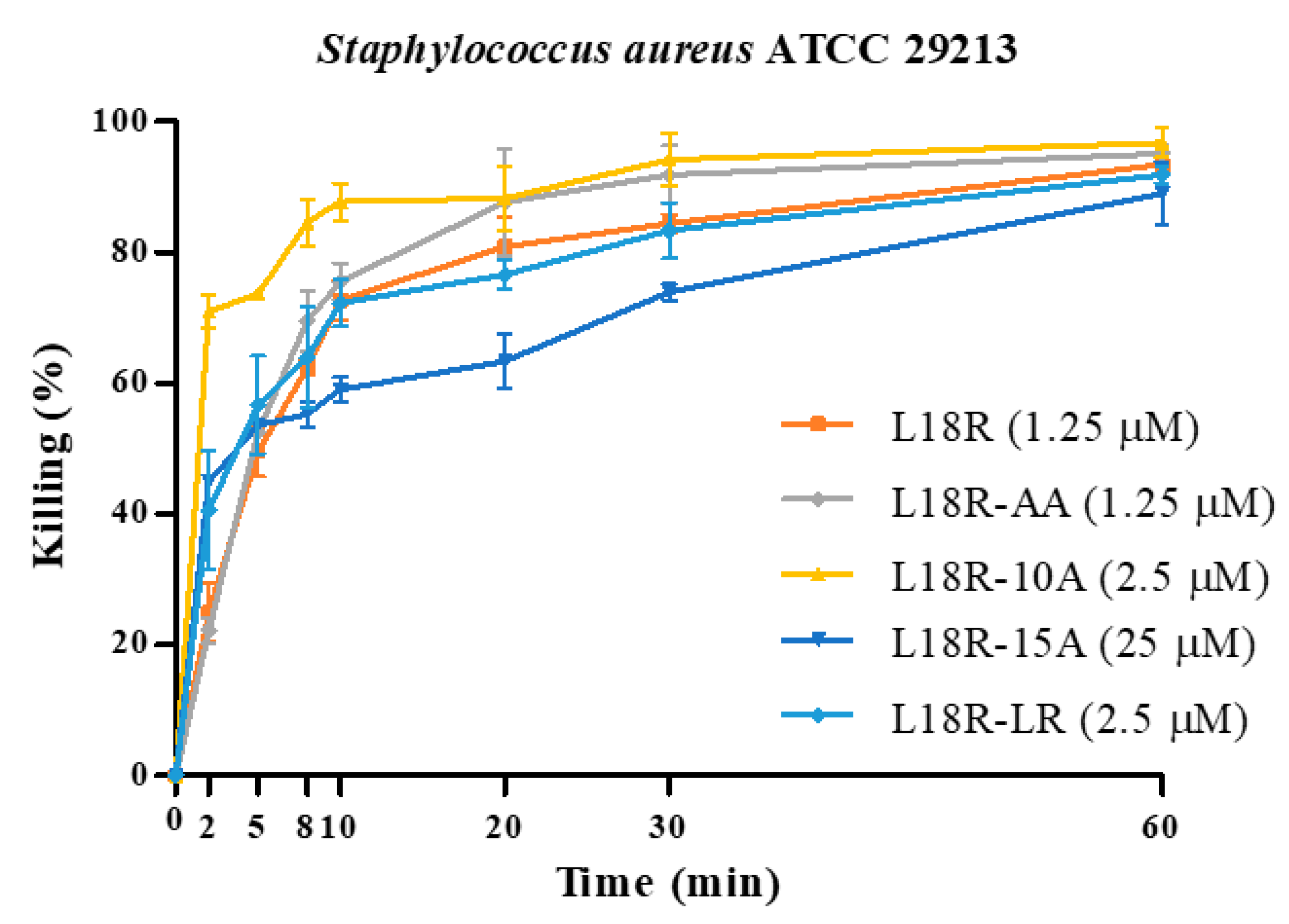
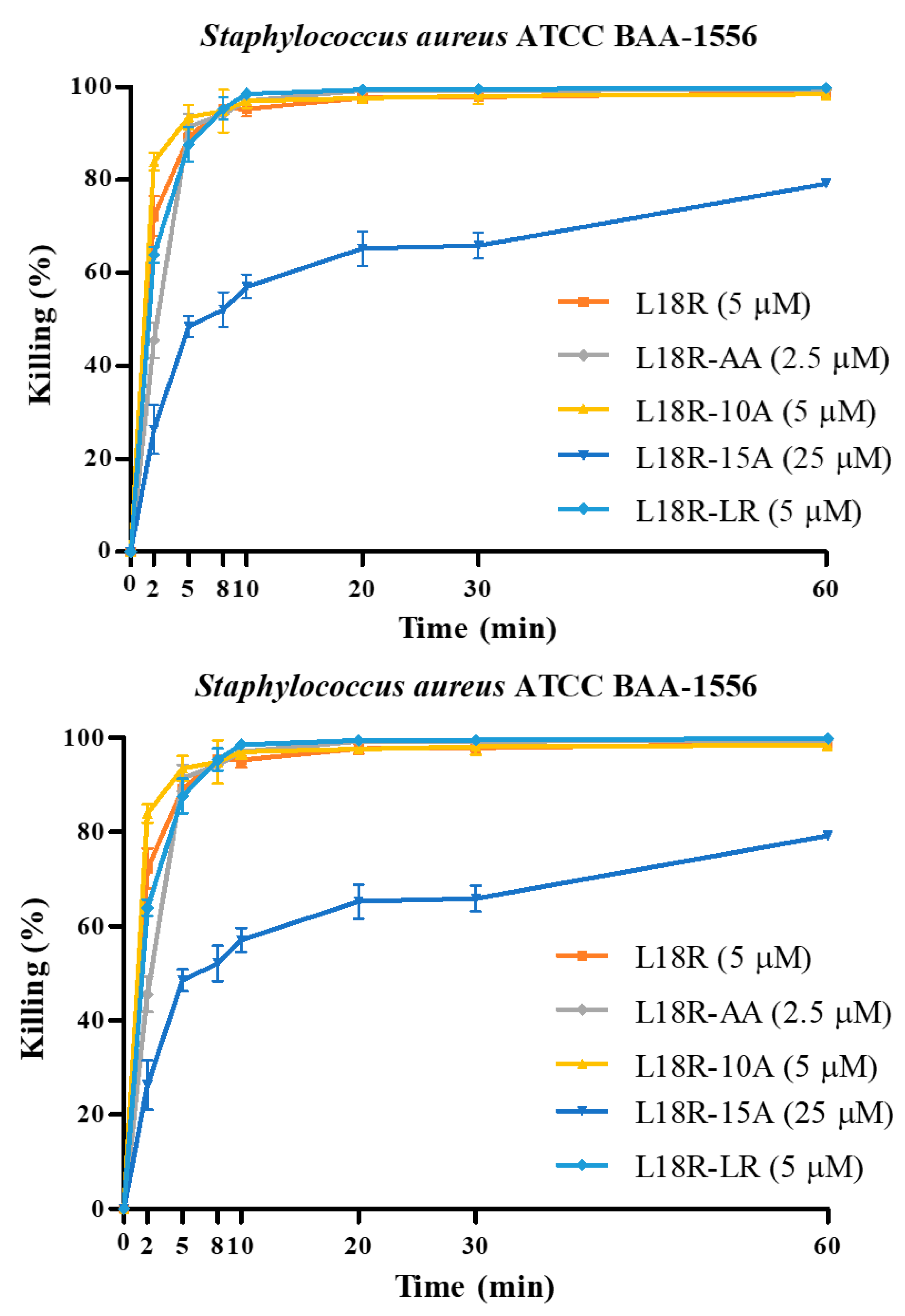
2.3. In Vitro Antibacterial Activity
2.4. Confocal Laser Scanning Microscopy (CLSM) Study
2.5. Structural Characterization of Investigated Peptides
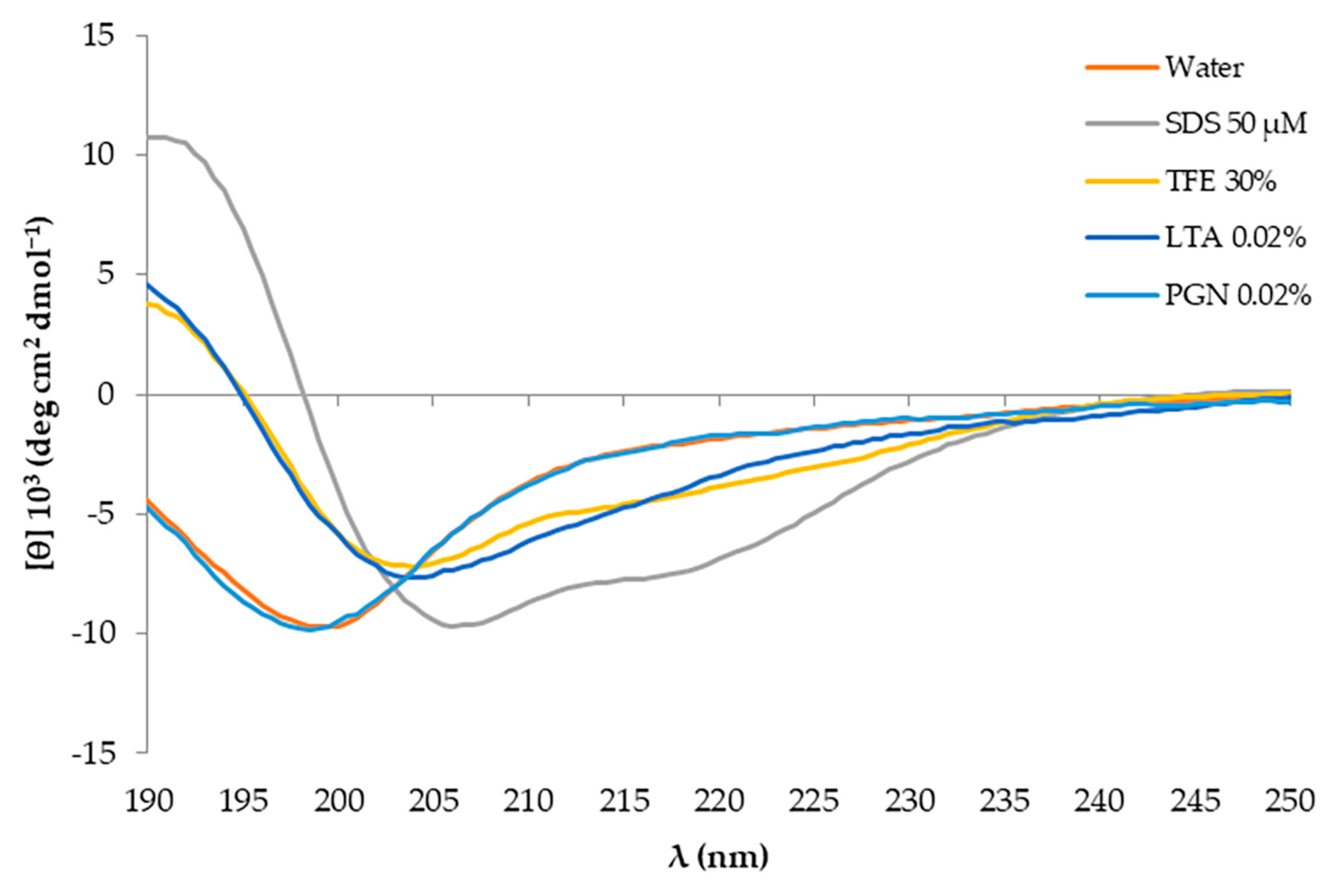
2.5.1. Circular Dichroism Analysis and Interaction with System Mimetics
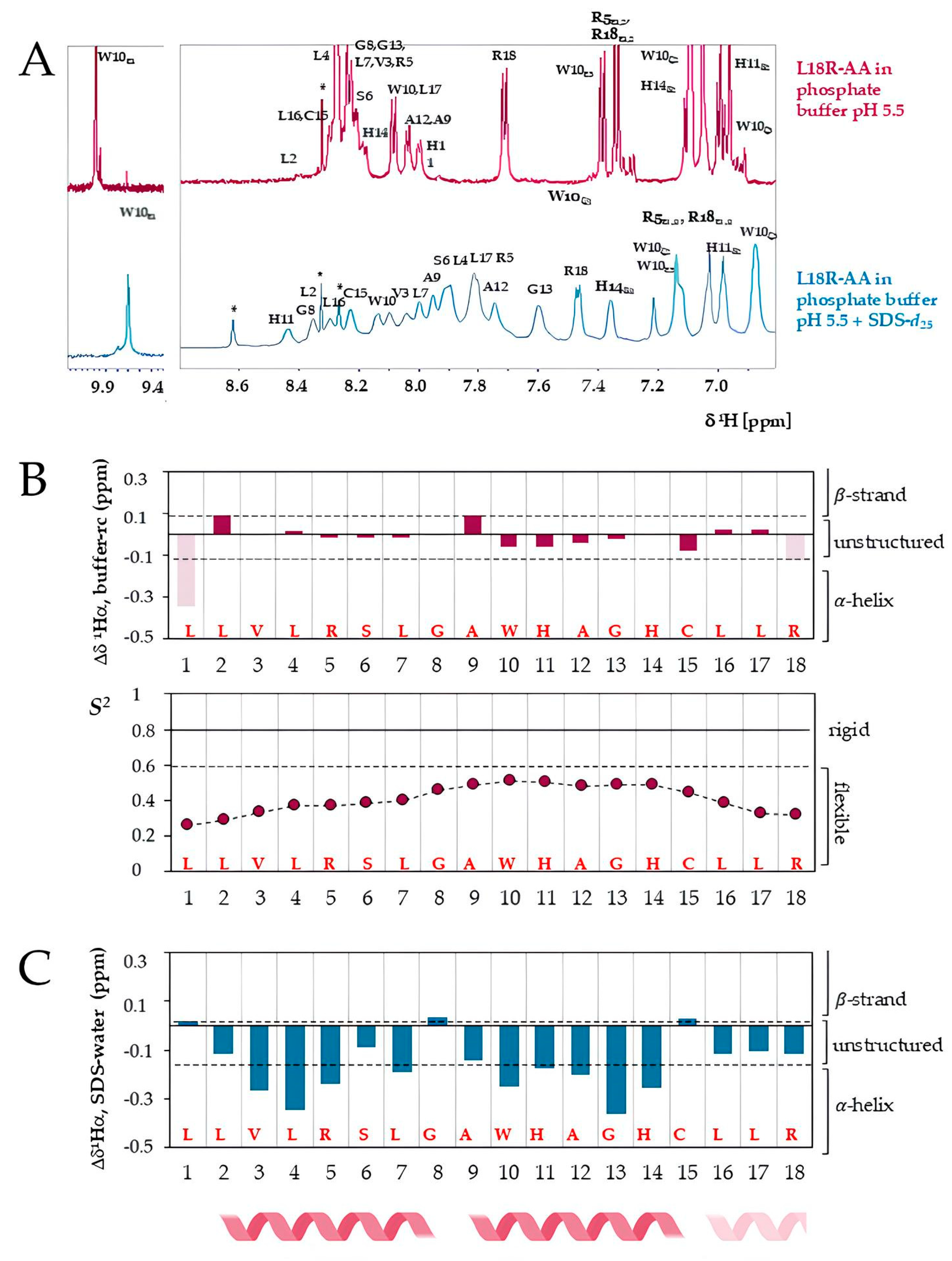
2.5.2. Structural Characterization of L18R-AA by Nuclear Magnetic Resonance
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains
4.2. In Silico Analysis of Peptides
4.3. Peptide Synthesis
4.4. Peptide Cytotoxicity
4.5. Evaluation of In Vitro Antibacterial Activity of Peptides
4.6. CLSM Study
4.7. CD Studies
4.8. NMR Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AI | Aliphatic index |
| AMPs | Antimicrobial peptides |
| AMR | Antimicrobial resistance |
| CD | Circular dichroism |
| CFU | Colony-forming unit |
| CLSM | Confocal laser scanning microscopy |
| CS | Chemical shift |
| Da | Dalton |
| DMSO | Dimethyl sulfoxide |
| DMSO-d6 | Deuterated DMSO |
| EC50 | Half-maximal effective concentration |
| Fmoc | Fluoren-9-ylmethoxycarbonyl |
| GRAVY | Grand average of hydropathy |
| HIV | Human immunodeficiency virus |
| IGHJ2 | Immunoglobulin heavy chain J gene |
| LTA | Lipoteichoic acid |
| MHA | Mueller–Hinton agar |
| MM | Molecular mass |
| MRSA | Methicillin-resistant Staphylococcus aureus |
| NMR | Nuclear magnetic resonance spectroscopy |
| PGN | Peptidoglycan |
| pI | Isoelectric point |
| PI | Propidium iodide |
| SDS | Sodium dodecyl sulphate |
| SDS-d25 | Deuterated SDS |
| TFE | 2,2,2-Trifluoroethanol |
| VRSA | Vancomycin-resistant Staphylococcus aureus |
| WHO | World Health Organization |
References
- Mestrovic, T.; Robles Aguilar, G.; Swetschinski, L.R.; Ikuta, K.S.; Gray, A.P.; Davis Weaver, N.; Han, C.; Wool, E.E.; Gershberg Hayoon, A.; Hay, S.I.; et al. The burden of bacterial antimicrobial resistance in the WHO European region in 2019: A cross-country systematic analysis. Lancet Public Health 2022, 7, e897–e913. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.S.; Wong, C.T.H.; Aung, T.T.; Lakshminarayanan, R.; Mehta, J.S.; Rauz, S.; McNally, A.; Kintses, B.; Peacock, S.J.; de la Fuente-Nunez, C.; et al. Antimicrobial resistance: A concise update. Lancet Microbe 2025, 6, 100947. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Antimicrobial Resistance. Available online: https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance (accessed on 18 March 2025).
- Naghavi, M.; Vollset, S.E.; Ikuta, K.S.; Swetschinski, L.R.; Gray, A.P.; Wool, E.E.; Robles Aguilar, G.; Mestrovic, T.; Smith, G.; Han, C.; et al. Global burden of bacterial antimicrobial resistance 1990–2021: A systematic analysis with forecasts to 2050. Lancet 2024, 404, 1199–1226. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.J.L.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Tong, S.Y.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G., Jr. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef]
- Khairullah, A.R.; Widodo, A.; Riwu, K.H.P.; Yanestria, S.M.; Moses, I.B.; Effendi, M.H.; Fauzia, K.A.; Fauziah, I.; Hasib, A.; Kusala, M.K.J.; et al. Spread of livestock-associated methicillin-resistant Staphylococcus aureus in poultry and its risks to public health: A comprehensive review. Open Vet. J. 2024, 14, 2116–2128. [Google Scholar] [CrossRef]
- Piewngam, P.; Otto, M. Staphylococcus aureus colonisation and strategies for decolonisation. Lancet Microbe 2024, 5, e606–e618. [Google Scholar] [CrossRef]
- Bashabsheh, R.H.F.; Al-Fawares, O.; Natsheh, I.; Bdeir, R.; Al-Khreshieh, R.O.; Bashabsheh, H.H.F. Staphylococcus aureus epidemiology, pathophysiology, clinical manifestations and application of nano-therapeutics as a promising approach to combat methicillin resistant Staphylococcus aureus. Pathog. Glob. Health 2024, 118, 209–231. [Google Scholar] [CrossRef]
- Lam, J.C.; Stokes, W. The Golden Grapes of Wrath—Staphylococcus aureus Bacteremia: A Clinical Review. Am. J. Med. 2023, 136, 19–26. [Google Scholar] [CrossRef]
- Munro, C.; Zilberberg, M.D.; Shorr, A.F. Bloodstream Infection in the Intensive Care Unit: Evolving Epidemiology and Microbiology. Antibiotcs 2024, 13, 123. [Google Scholar] [CrossRef]
- Cheung, G.Y.C.; Bae, J.S.; Otto, M. Pathogenicity and virulence of Staphylococcus aureus. Virulence 2021, 12, 547–569. [Google Scholar] [CrossRef] [PubMed]
- Borah, A.; Srivastava, A. Impact of extracellular enzymes on Staphylococcus aureus host tissue adaptation and infection. Acta Pathol. Microbiol. Immunol. Scand. 2025, 133, e13502. [Google Scholar] [CrossRef] [PubMed]
- Silva-Santana, G. Staphylococcus aureus: Dynamics of pathogenicity and antimicrobial-resistance in hospital and community environments—Comprehensive overview. Res. Microbiol. 2025, 176, 104267. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wang, S.; Liu, X.; Li, M.; Wang, X.; Chen, H.; Qu, C.; Liu, Y.; Liu, J. Strategies for Survival of Staphylococcus aureus in Host Cells. Int. J. Mol. Sci. 2025, 26, 720. [Google Scholar] [CrossRef]
- World Health Organization. WHO Bacterial Priority Pathogens List, 2024: Bacterial Pathogens of Public Health Importance to guide Research, Development and Strategies to Prevent and Control Antimicrobial Resistance; World Health Organization: Geneva, Switzerland, 2024; Available online: https://iris.who.int/bitstream/handle/10665/376776/9789240093461-eng.pdf?sequence=1 (accessed on 18 March 2025).
- Bellis, K.L.; Dissanayake, O.M.; Harrison, E.M.; Aggarwal, D. Community methicillin-resistant Staphylococcus aureus outbreaks in areas of low prevalence. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2025, 31, 182–189. [Google Scholar] [CrossRef]
- Hiramatsu, K.; Hanaki, H.; Ino, T.; Yabuta, K.; Oguri, T.; Tenover, F.C. Methicillin-resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. J. Antimicrob. Chemother. 1997, 40, 135–136. [Google Scholar] [CrossRef]
- Sievert, D.M.; Rudrik, J.T.; Patel, J.B.; McDonald, L.C.; Wilkins, M.J.; Hageman, J.C. Vancomycin-resistant Staphylococcus aureus in the United States, 2002–2006. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2008, 46, 668–674. [Google Scholar] [CrossRef]
- Yang, W.; Chen, T.; Zhou, Q.; Xu, J. Resistance to linezolid in Staphylococcus aureus by mutation, modification, and acquisition of genes. J. Antibiot. 2025, 78, 4–13. [Google Scholar] [CrossRef]
- Nazli, A.; Tao, W.; You, H.; He, X.; He, Y. Treatment of MRSA Infection: Where are We? Curr. Med. Chem. 2024, 31, 4425–4460. [Google Scholar] [CrossRef]
- Subbarayudu, S.; Namasivayam, S.K.R.; Arockiaraj, J. Immunomodulation in Non-traditional Therapies for Methicillin-resistant Staphylococcus aureus (MRSA) Management. Curr. Microbiol. 2024, 81, 346. [Google Scholar] [CrossRef]
- Golban, M.; Charostad, J.; Kazemian, H.; Heidari, H. Phage-Derived Endolysins Against Resistant Staphylococcus spp.: A Review of Features, Antibacterial Activities, and Recent Applications. Infect. Dis. Ther. 2025, 14, 13–57. [Google Scholar] [CrossRef] [PubMed]
- Westgeest, A.C.; Hanssen, J.L.J.; de Boer, M.G.J.; Schippers, E.F.; Lambregts, M.M.C. Eradication of community-onset Methicillin-resistant Staphylococcus aureus carriage: A narrative review. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2025, 31, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Bucataru, C.; Ciobanasu, C. Antimicrobial peptides: Opportunities and challenges in overcoming resistance. Microbiol. Res. 2024, 286, 127822. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yang, J.; Qu, C.; Zhang, Q.; Sun, S.; Liu, L. Anti-Staphylococcus aureus effects of natural antimicrobial peptides and the underlying mechanisms. Future Microbiol. 2024, 19, 355–372. [Google Scholar] [CrossRef]
- Gani, Z.; Kumar, A.; Raje, M.; Raje, C.I. Antimicrobial peptides: An alternative strategy to combat antimicrobial resistance. Drug Discov. Today 2025, 30, 104305. [Google Scholar] [CrossRef]
- Michalik, M.; Podbielska-Kubera, A.; Dmowska-Koroblewska, A. Antibiotic Resistance of Staphylococcus aureus Strains-Searching for New Antimicrobial Agents-Review. Pharmaceuticals 2025, 18, 81. [Google Scholar] [CrossRef]
- Luo, Y.; Song, Y. Mechanism of Antimicrobial Peptides: Antimicrobial, Anti-Inflammatory and Antibiofilm Activities. Int. J. Mol. Sci. 2021, 22, 11401. [Google Scholar] [CrossRef]
- Neghabi Hajigha, M.; Hajikhani, B.; Vaezjalali, M.; Samadi Kafil, H.; Kazemzadeh Anari, R.; Goudarzi, M. Antiviral and antibacterial peptides: Mechanisms of action. Heliyon 2024, 10, e40121. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, J.; Feng, Z.; Ma, Y. The Role and Mechanisms of Antimicrobial Peptides in Overcoming Multidrug-Resistant Bacteria. Molecules 2024, 30, 128. [Google Scholar] [CrossRef]
- Zhang, T.; Jin, Q.; Ji, J. Antimicrobial Peptides and Their Mimetics: Promising Candidates of Next-Generation Therapeutic Agents Combating Multidrug-Resistant Bacteria. Adv. Biol. 2025, 9, 2400461. [Google Scholar] [CrossRef]
- Polonelli, L.; Ciociola, T.; Sperinde, M.; Giovati, L.; D’Adda, T.; Galati, S.; Travassos, L.R.; Magliani, W.; Conti, S. Fungicidal activity of peptides encoded by immunoglobulin genes. Sci. Rep. 2017, 7, 10896. [Google Scholar] [CrossRef] [PubMed]
- Di Fermo, P.; Ciociola, T.; Di Lodovico, S.; D’Ercole, S.; Petrini, M.; Giovati, L.; Conti, S.; Di Giulio, M.; Cellini, L. Antimicrobial Peptide L18R Displays a Modulating Action against Inter-Kingdom Biofilms in the Lubbock Chronic Wound Biofilm Model. Microorganisms 2021, 9, 1779. [Google Scholar] [CrossRef] [PubMed]
- Mergoni, G.; Manfredi, M.; Bertani, P.; Ciociola, T.; Conti, S.; Giovati, L. Activity of Two Antimicrobial Peptides against Enterococcus faecalis in a Model of Biofilm-Mediated Endodontic Infection. Antibiotics 2021, 10, 1220. [Google Scholar] [CrossRef] [PubMed]
- Gagat, P.; Ostrówka, M.; Duda-Madej, A.; Mackiewicz, P. Enhancing Antimicrobial Peptide Activity through Modifications of Charge, Hydrophobicity, and Structure. Int. J. Mol. Sci. 2024, 25, 10821. [Google Scholar] [CrossRef]
- Ciociola, T.; Pertinhez, T.A.; Giovati, L.; Sperindè, M.; Magliani, W.; Ferrari, E.; Gatti, R.; D’Adda, T.; Spisni, A.; Conti, S.; et al. Dissecting the Structure-Function Relationship of a Fungicidal Peptide Derived from the Constant Region of Human Immunoglobulins. Antimicrob. Agents Chemother. 2016, 60, 2435–2442. [Google Scholar] [CrossRef]
- Martin, O.A.; Villegas, M.E.; Vila, J.A.; Scheraga, H.A. Analysis of 13Calpha and 13Cbeta chemical shifts of cysteine and cystine residues in proteins: A quantum chemical approach. J. Biomol. NMR 2010, 46, 217–225. [Google Scholar] [CrossRef]
- Berjanskii, M.V.; Wishart, D.S. Unraveling the meaning of chemical shifts in protein NMR. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 1564–1576. [Google Scholar] [CrossRef]
- Chen, C.; Shi, J.; Wang, D.; Kong, P.; Wang, Z.; Liu, Y. Antimicrobial peptides as promising antibiotic adjuvants to combat drug-resistant pathogens. Crit. Rev. Microbiol. 2024, 50, 267–284. [Google Scholar] [CrossRef]
- Datta, M.; Rajeev, A.; Chattopadhyay, I. Application of antimicrobial peptides as next-generation therapeutics in the biomedical world. Biotechnol. Genet. Eng. Rev. 2024, 40, 2458–2496. [Google Scholar] [CrossRef]
- Gonçalves, R.; Monges, B.E.D.; Oshiro, K.G.N.; Cândido, E.S.; Pimentel, J.P.F.; Franco, O.L.; Cardoso, M.H. Advantages and Challenges of Using Antimicrobial Peptides in Synergism with Antibiotics for Treating Multidrug-Resistant Bacteria. ACS Infect. Dis. 2025, 11, 323–334. [Google Scholar] [CrossRef]
- Roque-Borda, C.A.; Primo, L.M.D.G.; Medina-Alarcón, K.P.; Campos, I.C.; Nascimento, C.F.; Saraiva, M.M.S.; Berchieri Junior, A.; Fusco-Almeida, A.M.; Mendes-Giannini, M.J.S.; Perdigão, J.; et al. Antimicrobial Peptides: A Promising Alternative to Conventional Antimicrobials for Combating Polymicrobial Biofilms. Adv. Sci. 2025, 12, e2410893. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Tu, Y.; Li, B.; Qu, G.; Li, A.; Peng, X.; Li, S.; Shao, C. Antimicrobial peptide biological activity, delivery systems and clinical translation status and challenges. J. Transl. Med. 2025, 23, 292. [Google Scholar] [CrossRef] [PubMed]
- Lv, S.; Wang, J.; You, R.; Liu, S.; Ding, Y.; Hadianamrei, R.; Tomeh, M.A.; Pan, F.; Cai, Z.; Zhao, X. Highly selective performance of rationally designed antimicrobial peptides based on ponericin-W1. Biomater. Sci. 2022, 10, 4848–4865. [Google Scholar] [CrossRef] [PubMed]
- Ageitos, L.; Boaro, A.; Cesaro, A.; Torres, M.D.T.; Broset, E.; de la Fuente-Nunez, C. Frog-derived synthetic peptides display anti-infective activity against Gram-negative pathogens. Trends Biotechnol. 2025, 24, S0167–S7799. [Google Scholar] [CrossRef]
- Zhang, J.; Luan, L.; Xu, Y.; Jiang, S.; Zhang, W.; Tian, L.; Ye, W.; Han, J.; Zhang, C.; Wang, T.; et al. Development of novel broad-spectrum amphipathic antimicrobial peptides against multidrug-resistant bacteria through a rational combination strategy. J. Adv. Res. 2025, in press. [Google Scholar] [CrossRef]
- Islam, M.Z.; Hossain, F.; Ali, M.H.; Yamazaki, M. Relationship between antimicrobial peptides-induced cell membrane damage and bactericidal activity. Biophys. J. 2023, 122, 4645–4655. [Google Scholar] [CrossRef]
- Schmidtchen, A.; Pasupuleti, M.; Malmsten, M. Effect of hydrophobic modifications in antimicrobial peptides. Adv. Colloid Interface Sci. 2014, 205, 265–274. [Google Scholar] [CrossRef]
- He, S.; Stone, T.A.; Deber, C.M. Uncoupling Amphipathicity and Hydrophobicity: Role of Charge Clustering in Membrane Interactions of Cationic Antimicrobial Peptides. Biochemistry 2021, 60, 2586–2592. [Google Scholar] [CrossRef]
- Takechi-Haraya, Y.; Ohgita, T.; Kotani, M.; Kono, H.; Saito, C.; Tamagaki-Asahina, H.; Nishitsuji, K.; Uchimura, K.; Sato, T.; Kawano, R.; et al. Effect of hydrophobic moment on membrane interaction and cell penetration of apolipoprotein E-derived arginine-rich amphipathic α-helical peptides. Sci. Rep. 2022, 12, 4959. [Google Scholar] [CrossRef]
- Meier, S.; Ridgway, Z.M.; Picciano, A.L.; Caputo, G.A. Impacts of Hydrophobic Mismatch on Antimicrobial Peptide Efficacy and Bilayer Permeabilization. Antibiotics 2023, 12, 1624. [Google Scholar] [CrossRef]
- Choi, Y.; Choe, H.W.; Kook, M.; Choo, S.; Park, T.W.; Bae, S.; Kim, H.; Yang, J.; Jeong, W.S.; Yu, J.; et al. Proline-Hinged α-Helical Peptides Sensitize Gram-Positive Antibiotics, Expanding Their Physicochemical Properties to Be Used as Gram-Negative Antibiotics. J. Med. Chem. 2024, 67, 1825–1842. [Google Scholar] [CrossRef] [PubMed]
- Rončević, T.; Gerdol, M.; Pacor, S.; Cvitanović, A.; Begić, A.; Weber, I.; Krce, L.; Caporale, A.; Mardirossian, M.; Tossi, A.; et al. Antimicrobial Peptide with a Bent Helix Motif Identified in Parasitic Flatworm Mesocestoides corti. Int. J. Mol. Sci. 2024, 25, 11690. [Google Scholar] [CrossRef] [PubMed]
- Qu, P.; Gao, W.; Chen, H.; Li, D.; Yang, N.; Zhu, J.; Feng, X.; Liu, C.; Li, Z. The Central Hinge Link Truncation of the Antimicrobial Peptide Fowlicidin-3 Enhances Its Cell Selectivity without Antibacterial Activity Loss. Antimicrob. Agents Chemother. 2016, 60, 2798–2806. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Bae, T.; Schneewind, O.; Takeuchi, F.; Hiramatsu, K. Genome Sequence of Staphylococcus aureus Strain Newman and Comparative Analysis of Staphylococcal Genomes: Polymorphism and Evolution of Two Major Pathogenicity Islands. J. Bacteriol. 2008, 190, 300–310. [Google Scholar] [CrossRef]
- Kuroda, M.; Ohta, T.; Uchiyama, I.; Baba, T.; Yuzawa, H.; Kobayashi, I.; Cui, L.; Oguchi, A.; Aoki, K.; Nagai, Y.; et al. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet 2001, 357, 1225–1240. [Google Scholar] [CrossRef]
- Ciociola, T.; Giovati, L.; Giovannelli, A.; Conti, S.; Castagnola, M.; Vitali, A. The activity of a mammalian proline-rich peptide against Gram-negative bacteria, including drug-resistant strains, relies on a nonmembranolytic mode of action. Infect. Drug Resist. 2018, 11, 969–979. [Google Scholar] [CrossRef]
- Skinner, S.P.; Fogh, R.H.; Boucher, W.; Ragan, T.J.; Mureddu, L.G.; Vuister, G.W. CcpNmr AnalysisAssign: A flexible platform for integrated NMR analysis. J. Biomol. NMR 2016, 66, 111–124. [Google Scholar] [CrossRef]
- Bischetti, M.; Alaimo, N.; Nardelli, F.; Punzi, P.; Amariei, C.; Ingenito, R.; Musco, G.; Gallo, M.; Cicero, D.O. Structural insights on the selective interaction of the histidine-rich piscidin antimicrobial peptide Of-Pis1 with membranes. Biochim. Biophys. Acta Biomembr. 2023, 1865, 184080. [Google Scholar] [CrossRef]
- Hendus-Altenburger, R.; Fernandes, C.B.; Bugge, K.; Kunze, M.B.A.; Boomsma, W.; Kragelund, B.B. Random coil chemical shifts for serine, threonine and tyrosine phosphorylation over a broad pH range. J. Biomol. NMR 2019, 73, 713–725. [Google Scholar] [CrossRef]
- Shen, Y.; Bax, A. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 2013, 56, 227–241. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence | MM (Da) | pI | Charge | AI | GRAVY |
|---|---|---|---|---|---|---|
| L18R | LLVLRSLGPWHPGHCLLR | 2067.53 | 10.35 | 2+ | 146.11 | 0.467 |
| L18R-AA | LLVLRSLGAWHAGHCLLR | 2015.45 | 10.35 | 2+ | 157.22 | 0.844 |
| L18R-10A | LLVLRSLGPAHPGHCLLR | 1952.39 | 10.35 | 2+ | 151.67 | 0.617 |
| L18R-15A | LLVLRSLGPWHPGHALLR | 2035.47 | 12.00 | 2+ | 151.67 | 0.428 |
| L18R-LR | LLVLLSRGPWHPGHCLLR | 2067.53 | 10.35 | 2+ | 146.11 | 0.467 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Artesani, L.; Gallo, M.; Giovati, L.; Bisignano, F.M.; Ferrari, E.; Castronovo, L.M.; Conti, S.; Santoro, F.; Pertinhez, T.A.; Ciociola, T. Anti-Staphylococcus aureus Activity and Structural Characterization of Rationally Designed Peptides. Antibiotics 2025, 14, 437. https://doi.org/10.3390/antibiotics14050437
Artesani L, Gallo M, Giovati L, Bisignano FM, Ferrari E, Castronovo LM, Conti S, Santoro F, Pertinhez TA, Ciociola T. Anti-Staphylococcus aureus Activity and Structural Characterization of Rationally Designed Peptides. Antibiotics. 2025; 14(5):437. https://doi.org/10.3390/antibiotics14050437
Chicago/Turabian StyleArtesani, Lorenza, Mariana Gallo, Laura Giovati, Francesca Maria Bisignano, Elena Ferrari, Lara M. Castronovo, Stefania Conti, Francesco Santoro, Thelma A. Pertinhez, and Tecla Ciociola. 2025. "Anti-Staphylococcus aureus Activity and Structural Characterization of Rationally Designed Peptides" Antibiotics 14, no. 5: 437. https://doi.org/10.3390/antibiotics14050437
APA StyleArtesani, L., Gallo, M., Giovati, L., Bisignano, F. M., Ferrari, E., Castronovo, L. M., Conti, S., Santoro, F., Pertinhez, T. A., & Ciociola, T. (2025). Anti-Staphylococcus aureus Activity and Structural Characterization of Rationally Designed Peptides. Antibiotics, 14(5), 437. https://doi.org/10.3390/antibiotics14050437