
The Role of PI3k-Gamma Modulation in Bacterial Infection: A Review of the Literature and Selected Experimental Observations

 , , , , , and
, , , , , and
Abstract
1. Introduction
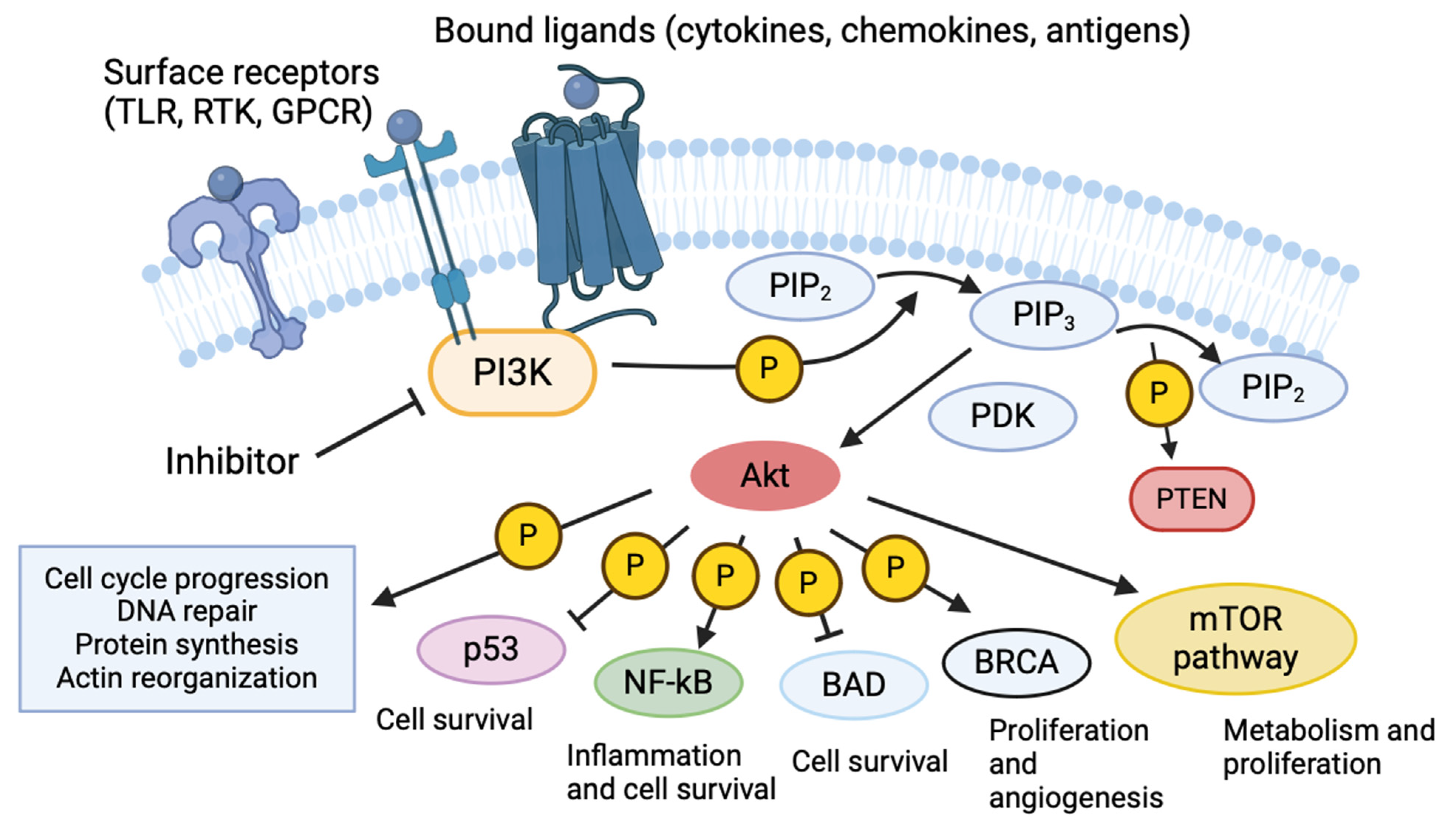
2. Phosphoinositide 3-Kinases
3. PI3k in Infectious Disease
4. PI3k Regulation of Macrophage Responses to Infection

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Stimulant | Effect | Reference |
|---|---|---|---|
| A549 (Epithelial) | Bacillus anthracis | Blocks actin activity and attenuates spore internalization | [47] |
| HeLa (Epithelial) | Chlamydia trachomatis | Restores pro-apoptotic functionality | [48] |
| Group B Streptococcus | Reduces bacterial internalization | [49] | |
| MDCK (Epithelial) | Pseudomonas aeruginosa | Reduces bacterial internalization | [51] |
| GES-1 (Epithelial) | Helicobacter pylori | Inhibits bacteria-induced PI3k overactivation and excessive reactive oxygen species production | [52] |
| BEC (Endothelial) | Staphylococcus aureus | Reduces bacterial internalization | [50] |
| Human Neutrophils | Francisella tularensis | Restores homeostatic apoptotic functions during infection | [56] |
| Chlamydia pneumoniae,C. psittaci | Reverses infection-induced delay of apoptosis | [57,58] | |
| Murine Neutrophils | LPS | Increases apoptosis | [55] |
| Campylobacter jejuni | Reduces migration and infiltration | [59] | |
| Zebrafish Neutrophils | Pseudomonas aeruginosa | Reduces motility and infiltration to site of infection | [60] |
| Various Murine and Hamster Cells | SARS-CoV-2 | Downregulates inflammatory cytokine expression, improves survival, and reduces immune cell recruitment | [67] |
| Human Monocytes | LPS | Reduces production of IFN-γ | [78] |
| THP-1 (Human Monocytic Cell Line) | LPS | Enhances TNF and TF expression | [73] |
| Group B Streptococcus | Reduces actin projections, phagocytic uptake, and NF-κB localization | [87] | |
| Murine Macrophages | LPS | Increases nitric oxide and TNF production | [72] |
| LPS | Increases release of NF-κB from inhibitory complex | [42] | |
| LPS | Enhances TNF, IL-6, and TF expression | [74] | |
| Helicobacter pylori | Blocks internalization of bacteria | [84] | |
| Streptococcus pneumoniae | Reduces macrophage recruitment, lung bacterial clearance, and survival | [89] | |
| RAW264.7 (Murine Macrophage Cell Line) | LPS | Attenuates NF-κB binding to DNA | [77] |
| LPS | Reduces LPS-induced nitric oxide, PGE2, TNF, IL-6, and IL-1β production | [82] | |
| Staphylococcus aureus | Reduces autophagy and phagocytosis while increasing NF-κB-mediated cytokine production | [90] | |
| J774A.1 (Murine Macrophage Cell Line) | Legionella pneumophila | Prevents intracellular replication by reducing bacterial invasion | [86] |
| MH-S (Murine Macrophage Cell Line) | Pseudomonas aeruginosa | Blocks phagocytosis | [88] |
| Chick Microglial Cells | LPS | Inhibited nitric oxide production | [80] |
| BV2 (Microglial Cell Line) | LPS | Attenuates NF-κB binding to DNA | [76] |
| LPS | Reduces LPS-induced NF-κB activity, nitric oxide, PGE2 IL-1β, and TNF production | [81] |
5. Selected Experimentation
5.1. Bacterial Strains, Cell Lines, and Reagents Used
5.2. Statistical Analysis and Programs
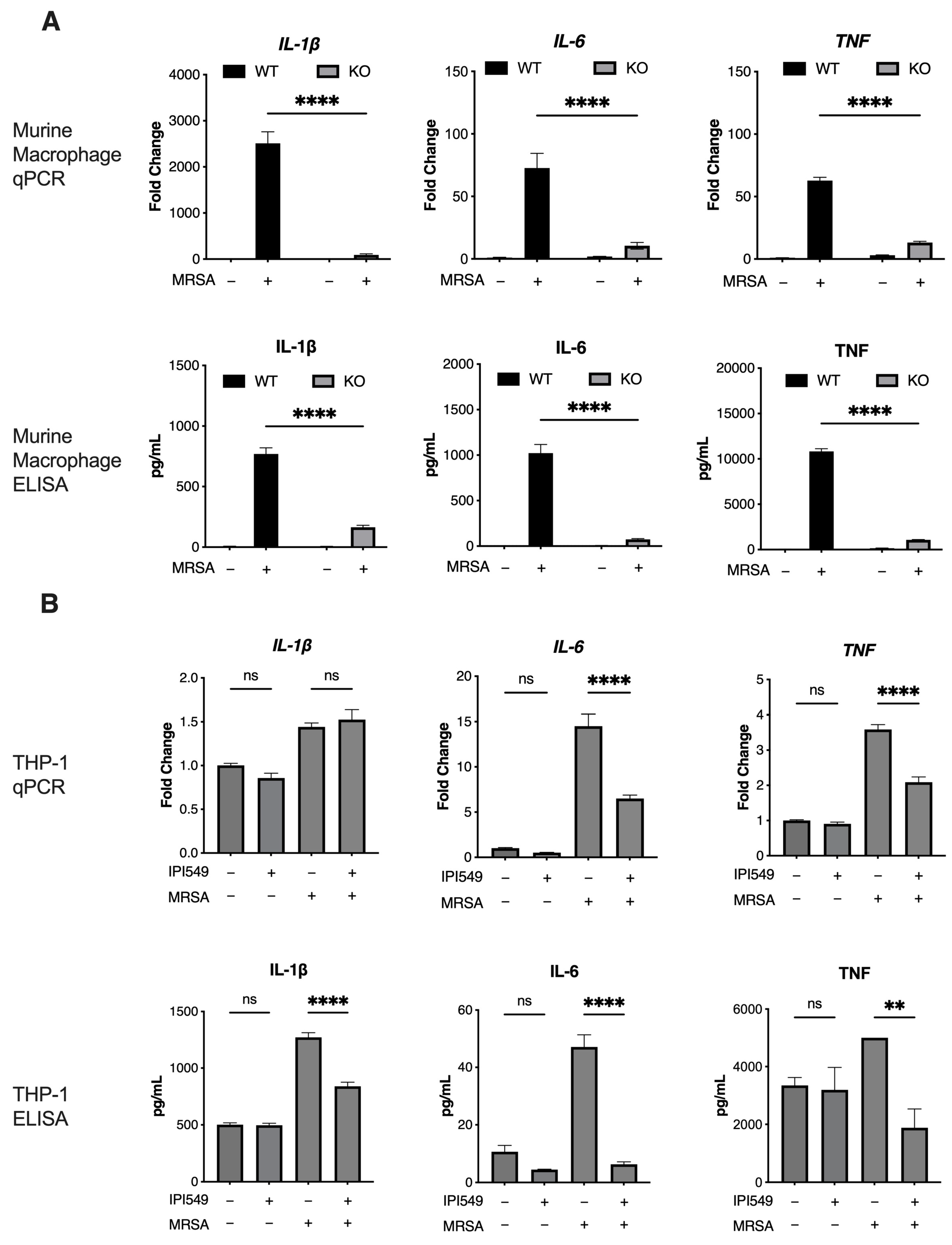
5.3. PI3k Knockout and Inhibition Reduces Inflammation in Human/Murine Macrophages
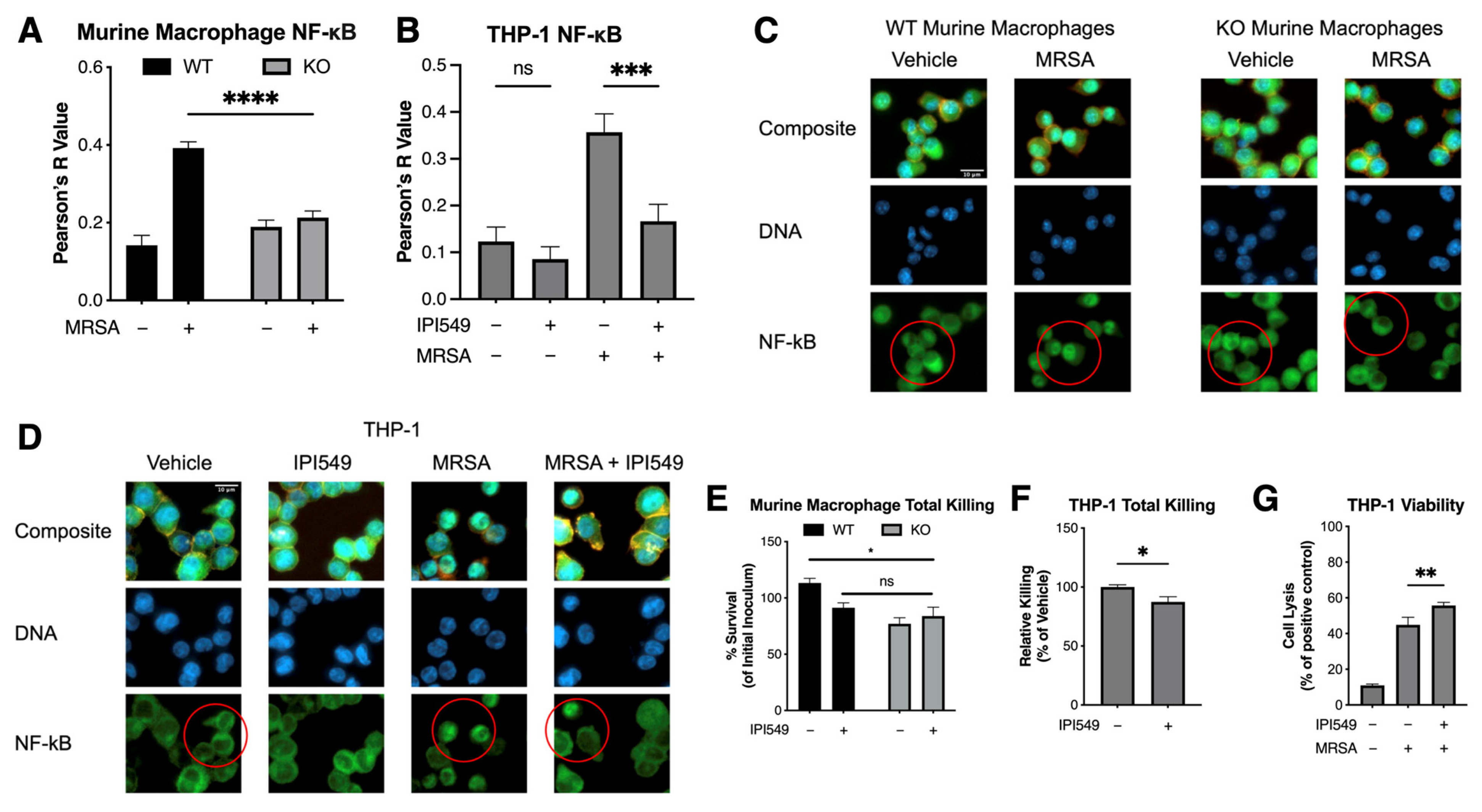
5.4. PI3k Knockout and Inhibition Reduces NF-κB Colocalization
5.5. Pharmacological and Genetic Inhibition of PI3kγ Does Not Compromise Bacterial Killing
6. Discussion and Areas of Future Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McIntosh, S.A.; Alam, F.; Adams, L.; Boon, I.S.; Callaghan, J.; Conti, I.; Copson, E.; Carson, V.; Davidson, M.; Fitzgerald, H.; et al. Global Funding for Cancer Research between 2016 and 2020: A Content Analysis of Public and Philanthropic Investments. Lancet Oncol. 2023, 24, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in Cancer: Mechanisms and Advances in Clinical Trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef]
- Antimicrobial Resistance Collaborators. Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.J.; Maher, T.J.; Zhang, Y.; Lanahan, S.M.; Bucklin, M.L.; Compton, S.R.; Tyler, P.M.; Comrie, W.A.; Matsuda, M.; Olivier, K.N.; et al. Human PI3Kγ Deficiency and Its Microbiota-Dependent Mouse Model Reveal Immunodeficiency and Tissue Immunopathology. Nat. Commun. 2019, 10, 4364. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Martin-Conte, E.L.; Hirsch, E. Phosphoinositide 3-Kinase p110γ in Immunity. IUBMB Life 2011, 63, 707–713. [Google Scholar] [CrossRef]
- Sasaki, T.; Irie-Sasaki, J.; Jones, R.G.; Oliveira-dos-Santos, A.J.; Stanford, W.L.; Bolon, B.; Wakeham, A.; Itie, A.; Bouchard, D.; Kozieradzki, I.; et al. Function of PI3Kgamma in Thymocyte Development, T Cell Activation, and Neutrophil Migration. Science 2000, 287, 1040–1046. [Google Scholar] [CrossRef]
- Sadhu, C.; Masinovsky, B.; Dick, K.; Sowell, C.G.; Staunton, D.E. Essential Role of Phosphoinositide 3-Kinase Delta in Neutrophil Directional Movement. J. Immunol. 2003, 170, 2647–2654. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Rascio, F.; Spadaccino, F.; Rocchetti, M.T.; Castellano, G.; Stallone, G.; Netti, G.S.; Ranieri, E. The Pathogenic Role of PI3K/AKT Pathway in Cancer Onset and Drug Resistance: An Updated Review. Cancers 2021, 13, 3949. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and Saturation Analysis of Cancer Genes across 21 Tumour Types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef]
- Bilanges, B.; Posor, Y.; Vanhaesebroeck, B. PI3K Isoforms in Cell Signalling and Vesicle Trafficking. Nat. Rev. Mol. Cell Biol. 2019, 20, 515–534. [Google Scholar] [CrossRef] [PubMed]
- Rathinaswamy, M.K.; Burke, J.E. Class I Phosphoinositide 3-Kinase (PI3K) Regulatory Subunits and Their Roles in Signaling and Disease. Adv. Biol. Regul. 2020, 75, 100657. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [PubMed]
- Fox, M.; Mott, H.R.; Owen, D. Class IA PI3K Regulatory Subunits: p110-Independent Roles and Structures. Biochem. Soc. Trans. 2020, 48, 1397–1417. [Google Scholar] [CrossRef]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the Phosphoinositide 3-Kinase Pathway in Cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef]
- Hennessy, B.T.; Smith, D.L.; Ram, P.T.; Lu, Y.; Mills, G.B. Exploiting the PI3K/AKT Pathway for Cancer Drug Discovery. Nat. Rev. Drug Discov. 2005, 4, 988–1004. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT Network at the Interface of Oncogenic Signalling and Cancer Metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Yu, M.; Chen, J.; Xu, Z.; Yang, B.; He, Q.; Luo, P.; Yan, H.; Yang, X. Development and Safety of PI3K Inhibitors in Cancer. Arch. Toxicol. 2023, 97, 635–650. [Google Scholar] [CrossRef]
- Mishra, R.; Patel, H.; Alanazi, S.; Kilroy, M.K.; Garrett, J.T. PI3K Inhibitors in Cancer: Clinical Implications and Adverse Effects. Int. J. Mol. Sci. 2021, 22, 3464. [Google Scholar] [CrossRef]
- Walker, E.H.; Pacold, M.E.; Perisic, O.; Stephens, L.; Hawkins, P.T.; Wymann, M.P.; Williams, R.L. Structural Determinants of Phosphoinositide 3-Kinase Inhibition by Wortmannin, LY294002, Quercetin, Myricetin, and Staurosporine. Mol. Cell 2000, 6, 909–919. [Google Scholar] [CrossRef]
- Vlahos, C.J.; Matter, W.F.; Hui, K.Y.; Brown, R.F. A Specific Inhibitor of Phosphatidylinositol 3-Kinase, 2-(4-Morpholinyl)-8-Phenyl-4H-1-Benzopyran-4-One (LY294002). J. Biol. Chem. 1994, 269, 5241–5248. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.; Silveira, G.G.; Soave, D.F.; Costa, J.P.O.; Silva, A.R. The Role of the LY294002—A Non-Selective Inhibitor of Phosphatidylinositol 3-Kinase (PI3K) Pathway- in Cell Survival and Proliferation in Cell Line SCC-25. Asian Pac. J. Cancer Prev. 2019, 20, 3377–3383. [Google Scholar] [CrossRef]
- Lee, W.-H.; Loo, C.-Y.; Ghadiri, M.; Leong, C.-R.; Young, P.M.; Traini, D. The Potential to Treat Lung Cancer via Inhalation of Repurposed Drugs. Adv. Drug Deliv. Rev. 2018, 133, 107–130. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Postow, M.; Chmielowski, B.; Sullivan, R.; Patnaik, A.; Cohen, E.E.W.; Shapiro, G.; Steuer, C.; Gutierrez, M.; Yeckes-Rodin, H.; et al. Eganelisib, a First-in-Class PI3Kγ Inhibitor, in Patients with Advanced Solid Tumors: Results of the Phase 1/1b MARIO-1 Trial. Clin. Cancer Res. 2023, 29, 2210–2219. [Google Scholar] [CrossRef]
- Trigueiros, B.A.F.D.S.; Santos, I.J.S.; Pimenta, F.P.; Ávila, A.R. A Long Way to Go: A Scenario for Clinical Trials of PI3K Inhibitors in Treating Cancer. Cancer Control 2024, 31, 10732748241238047. [Google Scholar] [CrossRef]
- Fritsch, C.; Huang, A.; Chatenay-Rivauday, C.; Schnell, C.; Reddy, A.; Liu, M.; Kauffmann, A.; Guthy, D.; Erdmann, D.; De Pover, A.; et al. Characterization of the Novel and Specific PI3Kα Inhibitor NVP-BYL719 and Development of the Patient Stratification Strategy for Clinical Trials. Mol. Cancer Ther. 2014, 13, 1117–1129. [Google Scholar] [CrossRef] [PubMed]
- Mensah, F.A.; Blaize, J.-P.; Bryan, L.J. Spotlight on Copanlisib and Its Potential in the Treatment of Relapsed/refractory Follicular Lymphoma: Evidence to Date. Onco Targets Ther. 2018, 11, 4817–4827. [Google Scholar] [CrossRef]
- Winkler, D.G.; Faia, K.L.; DiNitto, J.P.; Ali, J.A.; White, K.F.; Brophy, E.E.; Pink, M.M.; Proctor, J.L.; Lussier, J.; Martin, C.M.; et al. PI3K-δ and PI3K-γ Inhibition by IPI-145 Abrogates Immune Responses and Suppresses Activity in Autoimmune and Inflammatory Disease Models. Chem. Biol. 2013, 20, 1364–1374. [Google Scholar] [CrossRef]
- Yang, Q.; Modi, P.; Newcomb, T.; Quéva, C.; Gandhi, V. Idelalisib: First-in-Class PI3K Delta Inhibitor for the Treatment of Chronic Lymphocytic Leukemia, Small Lymphocytic Leukemia, and Follicular Lymphoma. Clin. Cancer Res. 2015, 21, 1537–1542. [Google Scholar] [CrossRef]
- Burris, H.A., 3rd; Flinn, I.W.; Patel, M.R.; Fenske, T.S.; Deng, C.; Brander, D.M.; Gutierrez, M.; Essell, J.H.; Kuhn, J.G.; Miskin, H.P.; et al. Umbralisib, a Novel PI3Kδ and Casein Kinase-1ε Inhibitor, in Relapsed or Refractory Chronic Lymphocytic Leukaemia and Lymphoma: An Open-Label, Phase 1, Dose-Escalation, First-in-Human Study. Lancet Oncol. 2018, 19, 486–496. [Google Scholar] [CrossRef]
- GBD 2019 Antimicrobial Resistance Collaborators. Global Mortality Associated with 33 Bacterial Pathogens in 2019: A Systematic Analysis for the Global Burden of Disease Study 2019. Lancet 2022, 400, 2221–2248. [Google Scholar] [CrossRef] [PubMed]
- Mulani, M.S.; Kamble, E.E.; Kumkar, S.N.; Tawre, M.S.; Pardesi, K.R. Emerging Strategies to Combat ESKAPE Pathogens in the Era of Antimicrobial Resistance: A Review. Front. Microbiol. 2019, 10, 539. [Google Scholar] [CrossRef]
- Shinu, P.; Mouslem, A.K.A.; Nair, A.B.; Venugopala, K.N.; Attimarad, M.; Singh, V.A.; Nagaraja, S.; Alotaibi, G.; Deb, P.K. Progress Report: Antimicrobial Drug Discovery in the Resistance Era. Pharmaceuticals 2022, 15, 413. [Google Scholar] [CrossRef]
- Munguia, J.; Nizet, V. Pharmacological Targeting of the Host-Pathogen Interaction: Alternatives to Classical Antibiotics to Combat Drug-Resistant Superbugs. Trends Pharmacol. Sci. 2017, 38, 473–488. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Baby, D.; Rajguru, J.P.; Patil, P.B.; Thakkannavar, S.S.; Pujari, V.B. Inflammation and Cancer. Ann. Afr. Med. 2019, 18, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, V.; Chen, Y.-S.; Dolat, L.; Valdivia, R.H. The Effector TepP Mediates Recruitment and Activation of Phosphoinositide 3-Kinase on Early Vacuoles. mSphere 2017, 2, e00207-17. [Google Scholar] [CrossRef]
- García-Gil, A.; Galán-Enríquez, C.S.; Pérez-López, A.; Nava, P.; Alpuche-Aranda, C.; Ortiz-Navarrete, V. SopB Activates the Akt-YAP Pathway to Promote Salmonella Survival within B Cells. Virulence 2018, 9, 1390–1402. [Google Scholar] [CrossRef]
- Cano, V.; March, C.; Insua, J.L.; Aguiló, N.; Llobet, E.; Moranta, D.; Regueiro, V.; Brennan, G.P.; Millán-Lou, M.I.; Martín, C.; et al. Klebsiella Pneumoniae Survives within Macrophages by Avoiding Delivery to Lysosomes. Cell. Microbiol. 2015, 17, 1537–1560. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Welham, M.J.; Kotani, K.; Stein, R.; Warne, P.H.; Zvelebil, M.J.; Higashi, K.; Volinia, S.; Downward, J.; Waterfield, M.D. P110delta, a Novel Phosphoinositide 3-Kinase in Leukocytes. Proc. Natl. Acad. Sci. USA 1997, 94, 4330–4335. [Google Scholar] [CrossRef]
- Hirsch, E.; Katanaev, V.L.; Garlanda, C.; Azzolino, O.; Pirola, L.; Silengo, L.; Sozzani, S.; Mantovani, A.; Altruda, F.; Wymann, M.P. Central Role for G Protein-Coupled Phosphoinositide 3-Kinase Gamma in Inflammation. Science 2000, 287, 1049–1053. [Google Scholar] [CrossRef]
- Schmid, M.C.; Avraamides, C.J.; Dippold, H.C.; Franco, I.; Foubert, P.; Ellies, L.G.; Acevedo, L.M.; Manglicmot, J.R.E.; Song, X.; Wrasidlo, W.; et al. Receptor Tyrosine Kinases and TLR/IL1Rs Unexpectedly Activate Myeloid Cell PI3kγ, a Single Convergent Point Promoting Tumor Inflammation and Progression. Cancer Cell 2011, 19, 715–727. [Google Scholar] [CrossRef]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kγ Is a Molecular Switch That Controls Immune Suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, M.M.; Cappello, P.; Nguyen, A.V.; Ralainirina, N.; Hardamon, C.R.; Foubert, P.; Schmid, M.C.; Sun, P.; Mose, E.; Bouvet, M.; et al. Macrophage PI3Kγ Drives Pancreatic Ductal Adenocarcinoma Progression. Cancer Discov. 2016, 6, 870–885. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.L.; Souza, D.G.; Fagundes, C.T.; Amaral, F.A.; Assenzio, B.; Puntorieri, V.; Del Sorbo, L.; Fanelli, V.; Bosco, M.; Delsedime, L.; et al. Phosphoinositide-3 Kinase Gamma Activity Contributes to Sepsis and Organ Damage by Altering Neutrophil Recruitment. Am. J. Respir. Crit. Care Med. 2010, 182, 762–773. [Google Scholar] [CrossRef]
- De Henau, O.; Rausch, M.; Winkler, D.; Campesato, L.F.; Liu, C.; Cymerman, D.H.; Budhu, S.; Ghosh, A.; Pink, M.; Tchaicha, J.; et al. Overcoming Resistance to Checkpoint Blockade Therapy by Targeting PI3Kγ in Myeloid Cells. Nature 2016, 539, 443–447. [Google Scholar] [CrossRef]
- Kalla, C.; Ott, G.; Finotello, F.; Niewola-Staszkowska, K.; Conza, G.D.; Lahn, M.; van der Veen, L.; Schüler, J.; Falkenstern-Ge, R.; Kopecka, J.; et al. The Highly Selective and Oral Phosphoinositide 3-Kinase Delta (PI3K-δ) Inhibitor Roginolisib Induces Apoptosis in Mesothelioma Cells and Increases Immune Effector Cell Composition. Transl. Oncol. 2024, 43, 101857. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Jenkins, S.A.; Gu, C.; Smeds, E.; Liu, Q.; Vasan, R.; Russell, B.H.; Xu, Y. Bacillus Anthracis Spore Entry into Epithelial Cells Is an Actin-Dependent Process Requiring c-Src and PI3K. PLoS ONE 2010, 5, e11665. [Google Scholar] [CrossRef]
- Verbeke, P.; Welter-Stahl, L.; Ying, S.; Hansen, J.; Häcker, G.; Darville, T.; Ojcius, D.M. Recruitment of BAD by the Chlamydia Trachomatis Vacuole Correlates with Host-Cell Survival. PLoS Pathog. 2006, 2, e45. [Google Scholar] [CrossRef]
- Burnham, C.-A.D.; Shokoples, S.E.; Tyrrell, G.J. Invasion of HeLa Cells by Group B Streptococcus Requires the Phosphoinositide-3-Kinase Signalling Pathway and Modulates Phosphorylation of Host-Cell Akt and Glycogen Synthase Kinase-3. Microbiology 2007, 153, 4240–4252. [Google Scholar] [CrossRef]
- Oviedo-Boyso, J.; Cortés-Vieyra, R.; Huante-Mendoza, A.; Yu, H.B.; Valdez-Alarcón, J.J.; Bravo-Patiño, A.; Cajero-Juárez, M.; Finlay, B.B.; Baizabal-Aguirre, V.M. The Phosphoinositide-3-Kinase-Akt Signaling Pathway Is Important for Staphylococcus Aureus Internalization by Endothelial Cells. Infect. Immun. 2011, 79, 4569–4577. [Google Scholar] [CrossRef]
- Kierbel, A.; Gassama-Diagne, A.; Mostov, K.; Engel, J.N. The Phosphoinositol-3-Kinase-Protein Kinase B/Akt Pathway Is Critical for Pseudomonas Aeruginosa Strain PAK Internalization. Mol. Biol. Cell 2005, 16, 2577–2585. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Yi, J.; Lu, J.; Nie, M.; Huang, M.; Rong, J.; Zhu, Z.; Chen, J.; Zhou, X.; Li, B.; et al. Acetylcysteine Reduces ROS-Mediated Oxidative DNA Damage and PI3K/Akt Pathway Activation Induced by Infection. Oxid. Med. Cell. Longev. 2018, 2018, 1874985. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.K.; Deng, Q.; Cavnar, P.J.; Wu, Y.I.; Hahn, K.M.; Huttenlocher, A. Differential Regulation of Protrusion and Polarity by PI3K during Neutrophil Motility in Live Zebrafish. Dev. Cell 2010, 18, 226–236. [Google Scholar] [CrossRef]
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed Cell Death as a Defence against Infection. Nat. Rev. Immunol. 2017, 17, 151–164. [Google Scholar] [CrossRef]
- Yang, K.-Y.; Arcaroli, J.; Kupfner, J.; Pitts, T.M.; Park, J.S.; Strasshiem, D.; Perng, R.-P.; Abraham, E. Involvement of Phosphatidylinositol 3-Kinase Gamma in Neutrophil Apoptosis. Cell. Signal. 2003, 15, 225–233. [Google Scholar] [CrossRef]
- Kinkead, L.C.; Krysa, S.J.; Allen, L.-A.H. Neutrophil Survival Signaling During Infection. Front. Cell. Infect. Microbiol. 2022, 12, 889290. [Google Scholar] [CrossRef]
- Sarkar, A.; Möller, S.; Bhattacharyya, A.; Behnen, M.; Rupp, J.; van Zandbergen, G.; Solbach, W.; Laskay, T. Mechanisms of Apoptosis Inhibition in Chlamydia Pneumoniae-Infected Neutrophils. Int. J. Med. Microbiol. 2015, 305, 493–500. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Xiao, J.; Wang, J.; Lu, S.; Zheng, K.; Yu, M.; Liu, J.; Wang, C.; Ding, N.; Liang, M.; et al. The Inclusion Membrane Protein 0556 Inhibits Human Neutrophils Apoptosis Through PI3K/AKT and NF-κB Signaling Pathways. Front. Immunol. 2021, 12, 694573. [Google Scholar] [CrossRef]
- Sun, X.; Liu, B.; Sartor, R.B.; Jobin, C. Phosphatidylinositol 3-Kinase-γ Signaling Promotes Campylobacter Jejuni-Induced Colitis through Neutrophil Recruitment in Mice. J. Immunol. 2013, 190, 357–365. [Google Scholar] [CrossRef]
- Deng, Q.; Harvie, E.A.; Huttenlocher, A. Distinct Signalling Mechanisms Mediate Neutrophil Attraction to Bacterial Infection and Tissue Injury. Cell. Microbiol. 2012, 14, 517–528. [Google Scholar] [CrossRef]
- Lai, Y.; Wang, M.; Cheng, A.; Mao, S.; Ou, X.; Yang, Q.; Wu, Y.; Jia, R.; Liu, M.; Zhu, D.; et al. Regulation of Apoptosis by Enteroviruses. Front. Microbiol. 2020, 11, 1145. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Cohen, J.I. The Role of PI3K/Akt in Human Herpesvirus Infection: From the Bench to the Bedside. Virology 2015, 479–480, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Diehl, N.; Schaal, H. Make Yourself at Home: Viral Hijacking of the PI3K/Akt Signaling Pathway. Viruses 2013, 5, 3192–3212. [Google Scholar] [CrossRef]
- Weisberg, E.; Parent, A.; Yang, P.L.; Sattler, M.; Liu, Q.; Liu, Q.; Wang, J.; Meng, C.; Buhrlage, S.J.; Gray, N.; et al. Repurposing of Kinase Inhibitors for Treatment of COVID-19. Pharm. Res. 2020, 37, 167. [Google Scholar] [CrossRef] [PubMed]
- Fritch, E.J.; Mordant, A.L.; Gilbert, T.S.K.; Wells, C.I.; Yang, X.; Barker, N.K.; Madden, E.A.; Dinnon, K.H., 3rd; Hou, Y.J.; Tse, L.V.; et al. Investigation of the Host Kinome Response to Coronavirus Infection Reveals PI3K/mTOR Inhibitors as Betacoronavirus Antivirals. J. Proteome Res. 2023, 22, 3159–3177. [Google Scholar] [CrossRef]
- Schmid, M.C.; Kang, S.W.; Chen, H.; Paradise, M.; Ghebremedhin, A.; Kaneda, M.M.; Chin, S.-M.; Do, A.; Watterson, D.M.; Varner, J.A. PI3Kγ Stimulates a High Molecular Weight Form of Myosin Light Chain Kinase to Promote Myeloid Cell Adhesion and Tumor Inflammation. Nat. Commun. 2022, 13, 1768. [Google Scholar] [CrossRef]
- Shepard, R.M.; Ghebremedhin, A.; Pratumchai, I.; Robinson, S.R.; Betts, C.; Hu, J.; Sasik, R.; Fisch, K.M.; Zak, J.; Chen, H.; et al. PI3Kγ Inhibition Circumvents Inflammation and Vascular Leak in SARS-CoV-2 and Other Infections. Sci. Transl. Med. 2024, 16, eadi6887. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, C.-C. Inflammatory Response of Macrophages in Infection. Hepatobiliary Pancreat. Dis. Int. 2014, 13, 138–152. [Google Scholar] [CrossRef]
- Lu, Y.-C.; Yeh, W.-C.; Ohashi, P.S. LPS/TLR4 Signal Transduction Pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef]
- Troutman, T.D.; Bazan, J.F.; Pasare, C. Toll-like Receptors, Signaling Adapters and Regulation of the pro-Inflammatory Response by PI3K. Cell Cycle 2012, 11, 3559–3567. [Google Scholar] [CrossRef]
- Opal, S.M. Endotoxins and Other Sepsis Triggers. Contrib. Nephrol. 2010, 167, 14–24. [Google Scholar]
- Park, Y.C.; Lee, C.H.; Kang, H.S.; Chung, H.T.; Kim, H.D. Wortmannin, a Specific Inhibitor of Phosphatidylinositol-3-Kinase, Enhances LPS-Induced NO Production from Murine Peritoneal Macrophages. Biochem. Biophys. Res. Commun. 1997, 240, 692–696. [Google Scholar] [CrossRef]
- Guha, M.; Mackman, N. The Phosphatidylinositol 3-Kinase-Akt Pathway Limits Lipopolysaccharide Activation of Signaling Pathways and Expression of Inflammatory Mediators in Human Monocytic Cells. J. Biol. Chem. 2002, 277, 32124–32132. [Google Scholar] [CrossRef]
- Luyendyk, J.P.; Schabbauer, G.A.; Tencati, M.; Holscher, T.; Pawlinski, R.; Mackman, N. Genetic Analysis of the Role of the PI3K-Akt Pathway in Lipopolysaccharide-Induced Cytokine and Tissue Factor Gene Expression in Monocytes/macrophages. J. Immunol. 2008, 180, 4218–4226. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Jayasooriya, R.G.P.T.; Lee, K.-T.; Kang, C.-H.; Dilshara, M.G.; Lee, H.-J.; Choi, Y.H.; Choi, I.-W.; Kim, G.-Y. Isobutyrylshikonin Inhibits Lipopolysaccharide-Induced Nitric Oxide and Prostaglandin E2 Production in BV2 Microglial Cells by Suppressing the PI3K/Akt-Mediated Nuclear Transcription Factor-κB Pathway. Nutr. Res. 2014, 34, 1111–1119. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.; Xin, Y.; Guo, Y.; Diao, Y.; Kou, X.; Luo, L.; Yin, Z. Ampelopsin Reduces Endotoxic Inflammation via Repressing ROS-Mediated Activation of PI3K/Akt/NF-κB Signaling Pathways. Int. Immunopharmacol. 2012, 12, 278–287. [Google Scholar] [CrossRef]
- Gajanayaka, N.; Dong, S.X.M.; Ali, H.; Iqbal, S.; Mookerjee, A.; Lawton, D.A.; Caballero, R.E.; Cassol, E.; Cameron, D.W.; Angel, J.B.; et al. TLR-4 Agonist Induces IFN-γ Production Selectively in Proinflammatory Human M1 Macrophages through the PI3K-mTOR- and JNK-MAPK-Activated p70S6K Pathway. J. Immunol. 2021, 207, 2310–2324. [Google Scholar] [CrossRef]
- Tau, G.; Rothman, P. Biologic Functions of the IFN-Gamma Receptors. Allergy 1999, 54, 1233–1251. [Google Scholar] [CrossRef]
- Saponaro, C.; Cianciulli, A.; Calvello, R.; Dragone, T.; Iacobazzi, F.; Panaro, M.A. The PI3K/Akt Pathway Is Required for LPS Activation of Microglial Cells. Immunopharmacol. Immunotoxicol. 2012, 34, 858–865. [Google Scholar] [CrossRef]
- Zhou, L.-T.; Wang, K.-J.; Li, L.; Li, H.; Geng, M. Pinocembrin Inhibits Lipopolysaccharide-Induced Inflammatory Mediators Production in BV2 Microglial Cells through Suppression of PI3K/Akt/NF-κB Pathway. Eur. J. Pharmacol. 2015, 761, 211–216. [Google Scholar] [CrossRef]
- Guo, C.; Yang, L.; Luo, J.; Zhang, C.; Xia, Y.; Ma, T.; Kong, L. Sophoraflavanone G from Sophora Alopecuroides Inhibits Lipopolysaccharide-Induced Inflammation in RAW264.7 Cells by Targeting PI3K/Akt, JAK/STAT and Nrf2/HO-1 Pathways. Int. Immunopharmacol. 2016, 38, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Fukao, T.; Koyasu, S. PI3K and Negative Regulation of TLR Signaling. Trends Immunol. 2003, 24, 358–363. [Google Scholar] [CrossRef]
- Allen, L.-A.H.; Allgood, J.A.; Han, X.; Wittine, L.M. Phosphoinositide3-Kinase Regulates Actin Polymerization during Delayed Phagocytosis of Helicobacter Pylori. J. Leukoc. Biol. 2005, 78, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Isberg, R.R.; O’Connor, T.J.; Heidtman, M. The Legionella Pneumophila Replication Vacuole: Making a Cosy Niche inside Host Cells. Nat. Rev. Microbiol. 2009, 7, 13–24. [Google Scholar] [CrossRef]
- Tachado, S.D.; Samrakandi, M.M.; Cirillo, J.D. Non-Opsonic Phagocytosis of Legionella Pneumophila by Macrophages Is Mediated by Phosphatidylinositol 3-Kinase. PLoS ONE 2008, 3, e3324. [Google Scholar] [CrossRef]
- De-Leon-Lopez, Y.S.; Thompson, M.E.; Kean, J.J.; Flaherty, R.A. The PI3K-Akt Pathway Is a Multifaceted Regulator of the Macrophage Response to Diverse Group B Isolates. Front. Cell. Infect. Microbiol. 2023, 13, 1258275. [Google Scholar] [CrossRef] [PubMed]
- Lovewell, R.R.; Hayes, S.M.; O’Toole, G.A.; Berwin, B. Pseudomonas Aeruginosa Flagellar Motility Activates the Phagocyte PI3K/Akt Pathway to Induce Phagocytic Engulfment. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L698–L707. [Google Scholar] [CrossRef]
- Maus, U.A.; Backi, M.; Winter, C.; Srivastava, M.; Schwarz, M.K.; Rückle, T.; Paton, J.C.; Briles, D.; Mack, M.; Welte, T.; et al. Importance of Phosphoinositide 3-Kinase Gamma in the Host Defense against Pneumococcal Infection. Am. J. Respir. Crit. Care Med. 2007, 175, 958–966. [Google Scholar] [CrossRef]
- Lv, Y.; Fang, L.; Ding, P.; Liu, R. PI3K/Akt-Beclin1 Signaling Pathway Positively Regulates Phagocytosis and Negatively Mediates NF-κB-Dependent Inflammation in Staphylococcus Aureus-Infected Macrophages. Biochem. Biophys. Res. Commun. 2019, 510, 284–289. [Google Scholar] [CrossRef]
- Novosad, S.A.; Sapiano, M.R.P.; Grigg, C.; Lake, J.; Robyn, M.; Dumyati, G.; Felsen, C.; Blog, D.; Dufort, E.; Zansky, S.; et al. Vital Signs: Epidemiology of Sepsis: Prevalence of Health Care Factors and Opportunities for Prevention. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 864–869. [Google Scholar] [CrossRef]
- Srisuwan, S.; Tongtawe, P.; Srimanote, P.; Voravuthikunchai, S.P. Rhodomyrtone Modulates Innate Immune Responses of THP-1 Monocytes to Assist in Clearing Methicillin-Resistant Staphylococcus Aureus. PLoS ONE 2014, 9, e110321. [Google Scholar] [CrossRef]
- Brinch, K.S.; Sandberg, A.; Baudoux, P.; Van Bambeke, F.; Tulkens, P.M.; Frimodt-Møller, N.; Høiby, N.; Kristensen, H.-H. Plectasin Shows Intracellular Activity against Staphylococcus Aureus in Human THP-1 Monocytes and in a Mouse Peritonitis Model. Antimicrob. Agents Chemother. 2009, 53, 4801–4808. [Google Scholar] [CrossRef]
- Miricescu, D.; Totan, A.; Stanescu-Spinu, I.-I.; Badoiu, S.C.; Stefani, C.; Greabu, M. PI3K/AKT/mTOR Signaling Pathway in Breast Cancer: From Molecular Landscape to Clinical Aspects. Int. J. Mol. Sci. 2020, 22, 173. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, A.S. PI3K/Akt/mTOR Inhibitors in Cancer: At the Bench and Bedside. Semin. Cancer Biol. 2019, 59, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Ellis, H.; Ma, C.X. PI3K Inhibitors in Breast Cancer Therapy. Curr. Oncol. Rep. 2019, 21, 110. [Google Scholar] [CrossRef]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt Signal Transduction for Cancer Therapy. Signal Transduct. Target. Ther. 2021, 6, 425. [Google Scholar] [CrossRef]
- Pahan, K.; Raymond, J.R.; Singh, I. Inhibition of Phosphatidylinositol 3-Kinase Induces Nitric-Oxide Synthase in Lipopolysaccharide- or Cytokine-Stimulated C6 Glial Cells. J. Biol. Chem. 1999, 274, 7528–7536. [Google Scholar] [CrossRef] [PubMed]
- Soromou, L.W.; Chu, X.; Jiang, L.; Wei, M.; Huo, M.; Chen, N.; Guan, S.; Yang, X.; Chen, C.; Feng, H.; et al. In Vitro and in Vivo Protection Provided by Pinocembrin against Lipopolysaccharide-Induced Inflammatory Responses. Int. Immunopharmacol. 2012, 14, 66–74. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, Y.; Qin, Y.; He, W.; Benlahrech, A.; Zhang, Q.; Jiang, X.; Lu, Z.; Ji, G.; Zheng, Y. Micheliolide Provides Protection of Mice against Staphylococcus Aureus and MRSA Infection by down-Regulating Inflammatory Response. Sci. Rep. 2017, 7, 41964. [Google Scholar] [CrossRef]
- Zhong, W.; Qian, K.; Xiong, J.; Ma, K.; Wang, A.; Zou, Y. Curcumin Alleviates Lipopolysaccharide Induced Sepsis and Liver Failure by Suppression of Oxidative Stress-Related Inflammation via PI3K/AKT and NF-κB Related Signaling. Biomed. Pharmacother. 2016, 83, 302–313. [Google Scholar] [CrossRef]
- Zhang, H.; Li, F.; Pan, Z.; Wu, Z.; Wang, Y.; Cui, Y. Activation of PI3K/Akt Pathway Limits JNK-Mediated Apoptosis during EV71 Infection. Virus Res. 2014, 192, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Mazzon, M.; Castro, C.; Thaa, B.; Liu, L.; Mutso, M.; Liu, X.; Mahalingam, S.; Griffin, J.L.; Marsh, M.; McInerney, G.M. Alphavirus-Induced Hyperactivation of PI3K/AKT Directs pro-Viral Metabolic Changes. PLoS Pathog. 2018, 14, e1006835. [Google Scholar] [CrossRef]
- Khadem, F.; Jia, P.; Mou, Z.; Feiz Barazandeh, A.; Liu, D.; Keynan, Y.; Uzonna, J.E. Pharmacological Inhibition of p110δ Subunit of PI3K Confers Protection against Experimental Leishmaniasis. J. Antimicrob. Chemother. 2017, 72, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-M.; Splinter, P.L.; Tietz, P.S.; Huang, B.Q.; Billadeau, D.D.; LaRusso, N.F. Phosphatidylinositol 3-Kinase and Frabin Mediate Cryptosporidium Parvum Cellular Invasion via Activation of Cdc42. J. Biol. Chem. 2004, 279, 31671–31678. [Google Scholar] [CrossRef] [PubMed]
- Liao, F.; Gu, W.; Fu, X.; Yuan, B.; Zhang, Y. Community-Acquired Methicillin-Resistant Staphylococcus Aureus Provoked Cytokine Storm Causing Severe Infection on BALB/c Mice. Mol. Immunol. 2021, 140, 167–174. [Google Scholar] [CrossRef]
- Coutinho, A.E.; Chapman, K.E. The Anti-Inflammatory and Immunosuppressive Effects of Glucocorticoids, Recent Developments and Mechanistic Insights. Mol. Cell. Endocrinol. 2011, 335, 2–13. [Google Scholar] [CrossRef]
- Annane, D. Corticosteroids for Severe Sepsis: An Evidence-Based Guide for Physicians. Ann. Intensive Care 2011, 1, 7. [Google Scholar] [CrossRef]
- Clark, M.A.; Plank, L.D.; Connolly, A.B.; Streat, S.J.; Hill, A.A.; Gupta, R.; Monk, D.N.; Shenkin, A.; Hill, G.L. Effect of a Chimeric Antibody to Tumor Necrosis Factor-Alpha on Cytokine and Physiologic Responses in Patients with Severe Sepsis—A Randomized, Clinical Trial. Crit. Care Med. 1998, 26, 1650–1659. [Google Scholar] [CrossRef]
- Opal, S.M.; Fisher, C.J., Jr.; Dhainaut, J.F.; Vincent, J.L.; Brase, R.; Lowry, S.F.; Sadoff, J.C.; Slotman, G.J.; Levy, H.; Balk, R.A.; et al. Confirmatory Interleukin-1 Receptor Antagonist Trial in Severe Sepsis: A Phase III, Randomized, Double-Blind, Placebo-Controlled, Multicenter Trial. The Interleukin-1 Receptor Antagonist Sepsis Investigator Group. Crit. Care Med. 1997, 25, 1115–1124. [Google Scholar] [CrossRef]
- Fisher, C.J., Jr.; Dhainaut, J.F.; Opal, S.M.; Pribble, J.P.; Balk, R.A.; Slotman, G.J.; Iberti, T.J.; Rackow, E.C.; Shapiro, M.J.; Greenman, R.L. Recombinant Human Interleukin 1 Receptor Antagonist in the Treatment of Patients with Sepsis Syndrome. Results from a Randomized, Double-Blind, Placebo-Controlled Trial. Phase III rhIL-1ra Sepsis Syndrome Study Group. JAMA 1994, 271, 1836–1843. [Google Scholar] [CrossRef] [PubMed]
- Place, D.E.; Lee, S.; Kanneganti, T.-D. PANoptosis in Microbial Infection. Curr. Opin. Microbiol. 2021, 59, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Lacey, C.A.; Miao, E.A. Programmed Cell Death in the Evolutionary Race against Bacterial Virulence Factors. Cold Spring Harb. Perspect. Biol. 2020, 12, a036459. [Google Scholar] [CrossRef] [PubMed]
- Ashida, H.; Mimuro, H.; Ogawa, M.; Kobayashi, T.; Sanada, T.; Kim, M.; Sasakawa, C. Cell Death and Infection: A Double-Edged Sword for Host and Pathogen Survival. J. Cell Biol. 2011, 195, 931–942. [Google Scholar] [CrossRef]

| Drug Name | Class | Mode of Action | Clinical Trial Stage | Reference |
|---|---|---|---|---|
| Alpelisib | PI3k-α inhibitor | Selectively targets the mutated PI3k-α in many solid tumors to suppress increased activity | Approved | [25,26] |
| Copanlisib | Pan-PI3k inhibitor | Targets all class I PI3k isoforms to hinder B-cell proliferation and survival in follicular lymphomas | Approved | [25,27] |
| Duvelisib | PI3k-γ, δ inhibitor | Selectivity for γ and δ isoforms in treatment of CLL and inflammatory and autoimmune conditions | Approved | [25,28] |
| Idelalisib | PI3k-δ inhibitor | Inhibits δ isoform in hematopoietic cells to slow B-cell cancer proliferation | Approved | [25,29] |
| Umbralisib | PI3k-δ inhibitor | Inhibits δ isoform and casein kinase 1ε in treatment of CLL and other lymphomas | Approved | [25,30] |
| TL117 | Pan-PI3k inhibitor | Combination therapy with paclitaxel to treat head and neck squamous cell carcinoma | I/II | NCT04843098, [25] |
| GSK2636771 | PI3k-β | Blocks β isoform to treat cancers with PTEN mutations | II | NCT04439149 |
| Eganelisib (IPI-549) | PI3k-γ inhibitor | Used in combination with Tecentriq and Abraxane to treat triple-negative breast cancer or with Tecentriq and Avastin to treat renal cell carcinoma | II | NCT03961698 |
| AZD8186 | PI3k-β inhibitor | Combination therapy with docetaxel to treat solid tumors with PTEN or PIK3-β mutations | I | NCT03218826 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, D.; Hoffman, A.; Askarian, F.; Bjånes, E.; Lin, E.X.; Varner, J.; Nizet, V. The Role of PI3k-Gamma Modulation in Bacterial Infection: A Review of the Literature and Selected Experimental Observations. Antibiotics 2025, 14, 315. https://doi.org/10.3390/antibiotics14030315
Sun D, Hoffman A, Askarian F, Bjånes E, Lin EX, Varner J, Nizet V. The Role of PI3k-Gamma Modulation in Bacterial Infection: A Review of the Literature and Selected Experimental Observations. Antibiotics. 2025; 14(3):315. https://doi.org/10.3390/antibiotics14030315
Chicago/Turabian StyleSun, Daniel, Alexandria Hoffman, Fatemeh Askarian, Elisabet Bjånes, Eric X. Lin, Judith Varner, and Victor Nizet. 2025. "The Role of PI3k-Gamma Modulation in Bacterial Infection: A Review of the Literature and Selected Experimental Observations" Antibiotics 14, no. 3: 315. https://doi.org/10.3390/antibiotics14030315
APA StyleSun, D., Hoffman, A., Askarian, F., Bjånes, E., Lin, E. X., Varner, J., & Nizet, V. (2025). The Role of PI3k-Gamma Modulation in Bacterial Infection: A Review of the Literature and Selected Experimental Observations. Antibiotics, 14(3), 315. https://doi.org/10.3390/antibiotics14030315