
Structure–Activity Relationship of Pyrrolidine Pentamine Derivatives as Inhibitors of the Aminoglycoside 6′-N-Acetyltransferase Type Ib


 , , ,
, , , 

Abstract

1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Small Molecule Compounds
4.2. Initial Growth Inhibition Assays
4.3. Checkerboard Assays
4.4. Molecular Docking
4.5. Molecular Dynamics (MD) Simulations
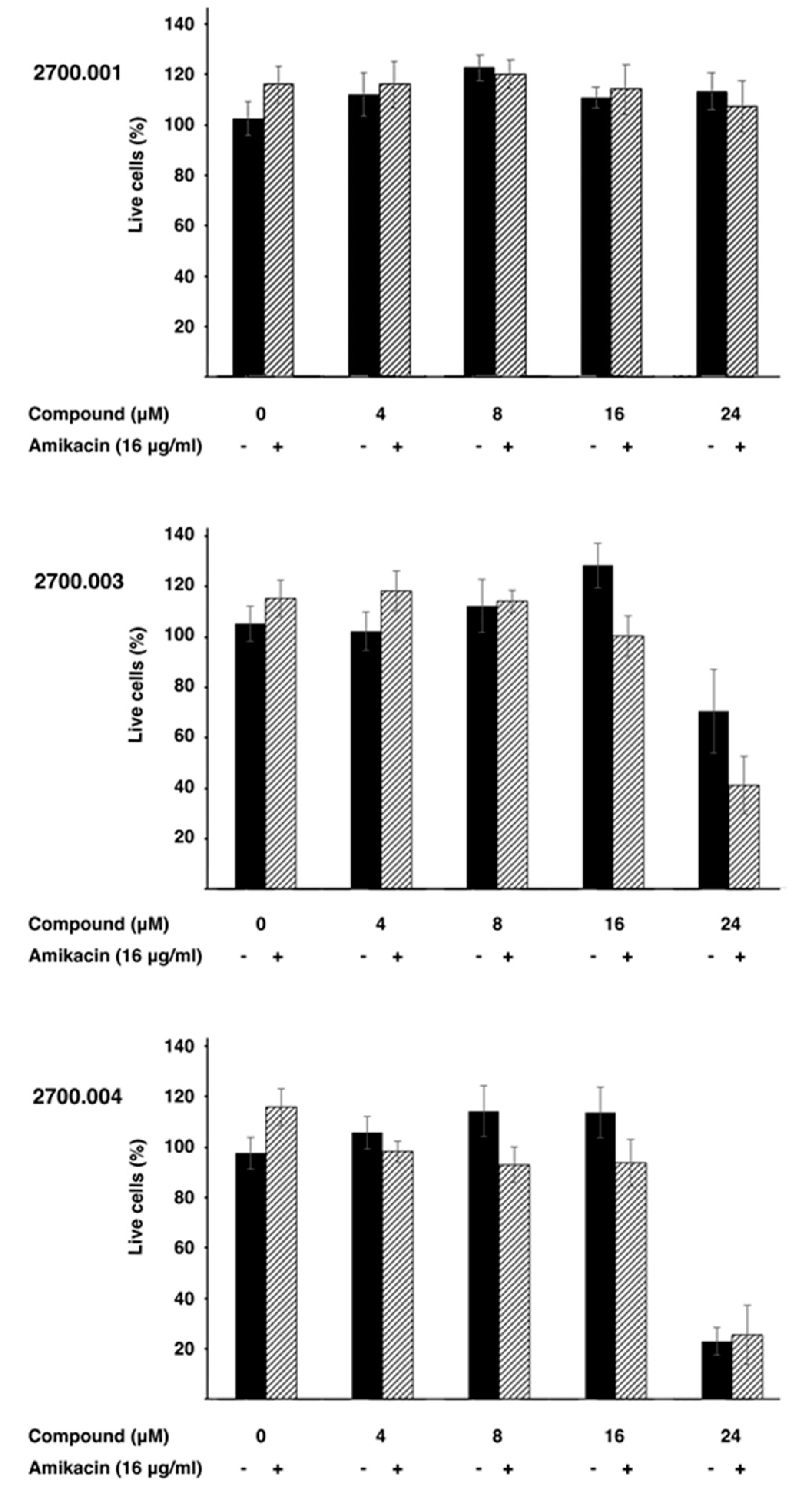
4.6. Cytotoxicity Assays
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boucher, H.W. Bad bugs, no drugs 2002–2020: Progress, challenges, and call to action. Trans. Am. Clin. Climatol. Assoc. 2020, 131, 65–71. [Google Scholar] [PubMed]
- Tamma, P.D.; Aitken, S.L.; Bonomo, R.A.; Mathers, A.J.; van Duin, D.; Clancy, C.J. Infectious Diseases Society of America guidance on the treatment of extended-spectrum beta-lactamase producing Enterobacterales (ESBL-E), carbapenem-resistant Enterobacterales (CRE), and Pseudomonas aeruginosa with difficult-to-treat resistance (DTR-P. aeruginosa). Clin. Infect. Dis. 2021, 72, 1109–1116. [Google Scholar] [PubMed]
- Centers for Disease Control and Prevention (U.S.). Antibiotic Resistance Threats in the United States, 2019; CDC: Atlanta, GA, USA, 2019.
- World-Health-Organization. WHO Bacterial Priority Pathogens List, 2024: Bacterial Pathogens of Public Health Importance to Guide Research, Development and Strategies to Prevent and Control Antimicrobial Resistance; World Health Organization: Geneva, Switzerland, 2024. [Google Scholar]
- McCreary, E.K.; Heil, E.L.; Tamma, P.D. New perspectives on antimicrobial agents:cefiderocol. Antimicrob. Agents Chemother. 2021, 65, e0217120. [Google Scholar] [CrossRef] [PubMed]
- Zampaloni, C.; Mattei, P.; Bleicher, K.; Winther, L.; Thate, C.; Bucher, C.; Adam, J.M.; Alanine, A.; Amrein, K.E.; Baidin, V.; et al. A novel antibiotic class targeting the lipopolysaccharide transporter. Nature 2024, 625, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Tolmasky, M.E. Strategies to prolong the useful life of existing antibiotics and help overcoming the antibiotic resistance crisis. In Frontiers in Clinical Drug Research-Anti Infectives; Atta-ur-Rhaman, Ed.; Bentham Books: Sharjah, United Arab Emirates, 2017; Volume 1, pp. 1–27. [Google Scholar]
- Krause, K.M.; Serio, A.W.; Kane, T.R.; Connolly, L.E. Aminoglycosides: An overview. Cold Spring Harb. Perspect. Med. 2016, 6, a027029. [Google Scholar] [CrossRef] [PubMed]
- Vakulenko, S.B.; Mobashery, S. Versatility of aminoglycosides and prospects for their future. Clin. Microbiol. Rev. 2003, 16, 430–450. [Google Scholar] [CrossRef] [PubMed]
- Houghton, J.L.; Green, K.D.; Chen, W.; Garneau-Tsodikova, S. The future of aminoglycosides: The end or renaissance? Chembiochem 2010, 11, 880–902. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.S.; Tolmasky, M.E. Amikacin: Uses, resistance, and prospects for inhibition. Molecules 2017, 22, 2267. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.S.; Tolmasky, M.E. Aminoglycoside modifying enzymes. Drug Resist. Updat. 2010, 13, 151–171. [Google Scholar] [CrossRef]
- Alfieri, A.; Di Franco, S.; Donatiello, V.; Maffei, V.; Fittipaldi, C.; Fiore, M.; Coppolino, F.; Sansone, P.; Pace, M.C.; Passavanti, M.B. Plazomicin against multidrug-resistant bacteria: A scoping review. Life 2022, 12, 1949. [Google Scholar] [CrossRef]
- Labby, K.J.; Garneau-Tsodikova, S. Strategies to overcome the action of aminoglycoside-modifying enzymes for treating resistant bacterial infections. Future Med. Chem. 2013, 5, 1285–1309. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.D. Antibiotic adjuvants: Rescuing antibiotics from resistance. Trends Microbiol. 2016, 24, 862–871. [Google Scholar] [CrossRef]
- Ramirez, M.S.; Nikolaidis, N.; Tolmasky, M.E. Rise and dissemination of aminoglycoside resistance: The aac(6′)-Ib paradigm. Front. Microbiol. 2013, 4, 121. [Google Scholar] [CrossRef]
- Kawaguchi, H. Discovery, chemistry, and activity of amikacin. J. Infect. Dis. 1976, 134, S242–S248. [Google Scholar] [CrossRef]
- Lin, D.L.; Tran, T.; Alam, J.Y.; Herron, S.R.; Ramirez, M.S.; Tolmasky, M.E. Inhibition of aminoglycoside 6′-N-acetyltransferase type Ib by zinc: Reversal of amikacin resistance in Acinetobacter baumannii and Escherichia coli by a zinc ionophore. Antimicrob. Agents Chemother. 2014, 58, 4238–4241. [Google Scholar] [CrossRef]
- Soler Bistue, A.J.; Martin, F.A.; Vozza, N.; Ha, H.; Joaquin, J.C.; Zorreguieta, A.; Tolmasky, M.E. Inhibition of aac(6′)-Ib-mediated amikacin resistance by nuclease-resistant external guide sequences in bacteria. Proc. Natl. Acad. Sci. USA 2009, 106, 13230–13235. [Google Scholar] [CrossRef] [PubMed]
- Magallon, J.; Vu, P.; Reeves, C.; Kwan, S.; Phan, K.; Oakley-Havens, C.; Ramirez, M.S.; Tolmasky, M.E. Amikacin in combination with zinc pyrithione prevents growth of a carbapenem-resistant/multidrug-resistant Klebsiella pneumoniae isolate. Int. J. Antimicrob. Agents 2021, 58, 106442. [Google Scholar] [CrossRef] [PubMed]
- Reeves, C.M.; Magallon, J.; Rocha, K.; Tran, T.; Phan, K.; Vu, P.; Yi, Y.; Oakley-Havens, C.L.; Cedano, J.; Jimenez, V.; et al. Aminoglycoside 6′-N-acetyltransferase type Ib [AAC(6′)-Ib]-mediated aminoglycoside resistance: Phenotypic conversion to susceptibility by silver ions. Antibiotics 2020, 10, 29. [Google Scholar] [CrossRef]
- Tran, T.; Chiem, K.; Jani, S.; Arivett, B.A.; Lin, D.L.; Lad, R.; Jimenez, V.; Farone, M.B.; Debevec, G.; Santos, R.; et al. Identification of a small molecule inhibitor of the aminoglycoside 6′-N-acetyltransferase type Ib [AAC(6′)-Ib] using mixture-based combinatorial libraries. Int. J. Antimicrob. Agents 2018, 51, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Rocha, K.; Magallon, J.; Reeves, C.; Phan, K.; Vu, P.; Oakley-Havens, C.L.; Kwan, S.; Ramirez, M.S.; LaVoi, T.; Donow, H.; et al. Inhibition of aminoglycoside 6′-N-acetyltransferase type Ib (AAC(6′)-Ib): Structure-activity relationship of substituted pyrrolidine pentamine derivatives as inhibitors. Biomedicines 2021, 9, 1218. [Google Scholar] [CrossRef]
- Vong, K.; Auclair, K. Understanding and overcoming aminoglycoside resistance caused by N-6′-acetyltransferase. Medchemcomm 2012, 3, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Magana, A.J.; Sklenicka, J.; Pinilla, C.; Giulianotti, M.; Chapagain, P.; Santos, R.; Ramirez, M.S.; Tolmasky, M.E. Restoring susceptibility to aminoglycosides: Identifying small molecule inhibitors of enzymatic inactivation. RSC Med. Chem. 2023, 14, 1591–1602. [Google Scholar] [CrossRef] [PubMed]
- Ngo, H.X.; Garneau-Tsodikova, S. What are the drugs of the future? Medchemcomm 2018, 9, 757–758. [Google Scholar] [CrossRef] [PubMed]
- Shakya, T.; Stogios, P.J.; Waglechner, N.; Evdokimova, E.; Ejim, L.; Blanchard, J.E.; McArthur, A.G.; Savchenko, A.; Wright, G.D. A small molecule discrimination map of the antibiotic resistance kinome. Chem. Biol. 2011, 18, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Blondelle, S.E.; Pinilla, C.; Boggiano, C. Synthetic combinatorial libraries as an alternative strategy for the development of novel treatments for infectious diseases. Methods Enzymol. 2003, 369, 322–344. [Google Scholar] [PubMed]
- d’Acoz, O.D.; Hue, F.; Ye, T.; Wang, L.; Leroux, M.; Rajngewerc, L.; Tran, T.; Phan, K.; Ramirez, M.S.; Reisner, W.; et al. Dynamics and quantitative contribution of the aminoglycoside 6′-N-acetyltransferase type Ib [AAC(6′)-Ib] to amikacin resistance. mSphere 2024, 9, e00789-23. [Google Scholar]
- Boucher, H.W.; Ambrose, P.G.; Chambers, H.F.; Ebright, R.H.; Jezek, A.; Murray, B.E.; Newland, J.G.; Ostrowsky, B.; Rex, J.H. White paper: Developing antimicrobial drugs for resistant pathogens, narrow-spectrum indications, and unmet needs. J. Infect. Dis. 2017, 216, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Naylor, N.R.; Evans, S.; Pouwels, K.B.; Troughton, R.; Lamagni, T.; Muller-Pebody, B.; Knight, G.M.; Atun, R.; Robotham, J.V. Quantifying the primary and secondary effects of antimicrobial resistance on surgery patients: Methods and data sources for empirical estimation in England. Front. Public. Health 2022, 10, 803943. [Google Scholar] [CrossRef] [PubMed]
- Papp-Wallace, K.M. The latest advances in beta-lactam/beta-lactamase inhibitor combinations for the treatment of Gram-negative bacterial infections. Expert. Opin. Pharmacother. 2019, 20, 2169–2184. [Google Scholar] [CrossRef]
- Boggiano, C.; Reixach, N.; Pinilla, C.; Blondelle, S.E. Successful identification of novel agents to control infectious diseases from screening mixture-based peptide combinatorial libraries in complex cell-based bioassays. Biopolymers 2003, 71, 103–116. [Google Scholar] [CrossRef]
- Pang, A.H.; Green, K.D.; Chandrika, N.T.; Garzan, A.; Punetha, A.; Holbrook, S.Y.L.; Willby, M.J.; Posey, J.E.; Tsodikov, O.V.; Garneau-Tsodikova, S. Discovery of substituted benzyloxy-benzylamine inhibitors of acetyltransferase Eis and their anti-mycobacterial activity. Eur. J. Med. Chem. 2022, 242, 114698. [Google Scholar] [CrossRef]
- Punetha, A.; Ngo, H.X.; Holbrook, S.Y.L.; Green, K.D.; Willby, M.J.; Bonnett, S.A.; Krieger, K.; Dennis, E.K.; Posey, J.E.; Parish, T.; et al. Structure-guided optimization of inhibitors of acetyltransferase Eis from Mycobacterium tuberculosis. ACS Chem. Biol. 2020, 15, 1581–1594. [Google Scholar] [CrossRef] [PubMed]
- Garzan, A.; Willby, M.J.; Green, K.D.; Tsodikov, O.V.; Posey, J.E.; Garneau-Tsodikova, S. Discovery and optimization of two Eis inhibitor families as kanamycin adjuvants against drug-resistant M. tuberculosis. ACS Med. Chem. Lett. 2016, 7, 1219–1221. [Google Scholar] [CrossRef] [PubMed]
- Arivett, B.A.; Fiester, S.E.; Ream, D.C.; Centron, D.; Ramirez, M.S.; Tolmasky, M.E.; Actis, L.A. Draft genome of the multidrug-resistant Acinetobacter baumannii strain A155 clinical isolate. Genome Announc. 2015, 3, 10–1128. [Google Scholar] [CrossRef]
- Hoel, D.G. Statistical aspects of chemical mixtures. In Methods for Assessing the Effects of Mixtures of Chemicals; Vouk, V.B., Butler, G.C., Upton, A.C., Parke, D.V., Asher, S.C., Eds.; Wiley: New York, NY, USA, 1987; pp. 369–377. [Google Scholar]
- Vetting, M.W.; Park, C.H.; Hegde, S.S.; Jacoby, G.A.; Hooper, D.C.; Blanchard, J.S. Mechanistic and structural analysis of aminoglycoside N-acetyltransferase AAC(6′)-Ib and its bifunctional, fluoroquinolone-active AAC(6′)-Ib-cr variant. Biochemistry 2008, 47, 9825–9835. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Schrodinger, LLC. The PyMOL Molecular Graphics System, Version 1.8; Schrödinger, LLC.: New York, NY, USA, 2015. [Google Scholar]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmuller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Nose, S.; Klein, M. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Essman, U.; Perera, L.; Berkowitz, M.; Darden, T.; Lee, H.; Pedersen, L. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Ryckaert, J.; Ciccotti, G.; Berendsen, H. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1997, 23, 327–341. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Graham, F.L.; Smiley, J.; Russell, W.C.; Nairn, R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 1977, 36, 59–74. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Checkerboard Analysis Compound Concentration for Potentiation (μM) | |||||||
|---|---|---|---|---|---|---|---|
| 2-Fold | 3-Fold | ||||||
| Compound | Growth Inhibition (%) 1 | SEM | 50% | 80% | 50% | 80% | |
| Structure | ID | ||||||
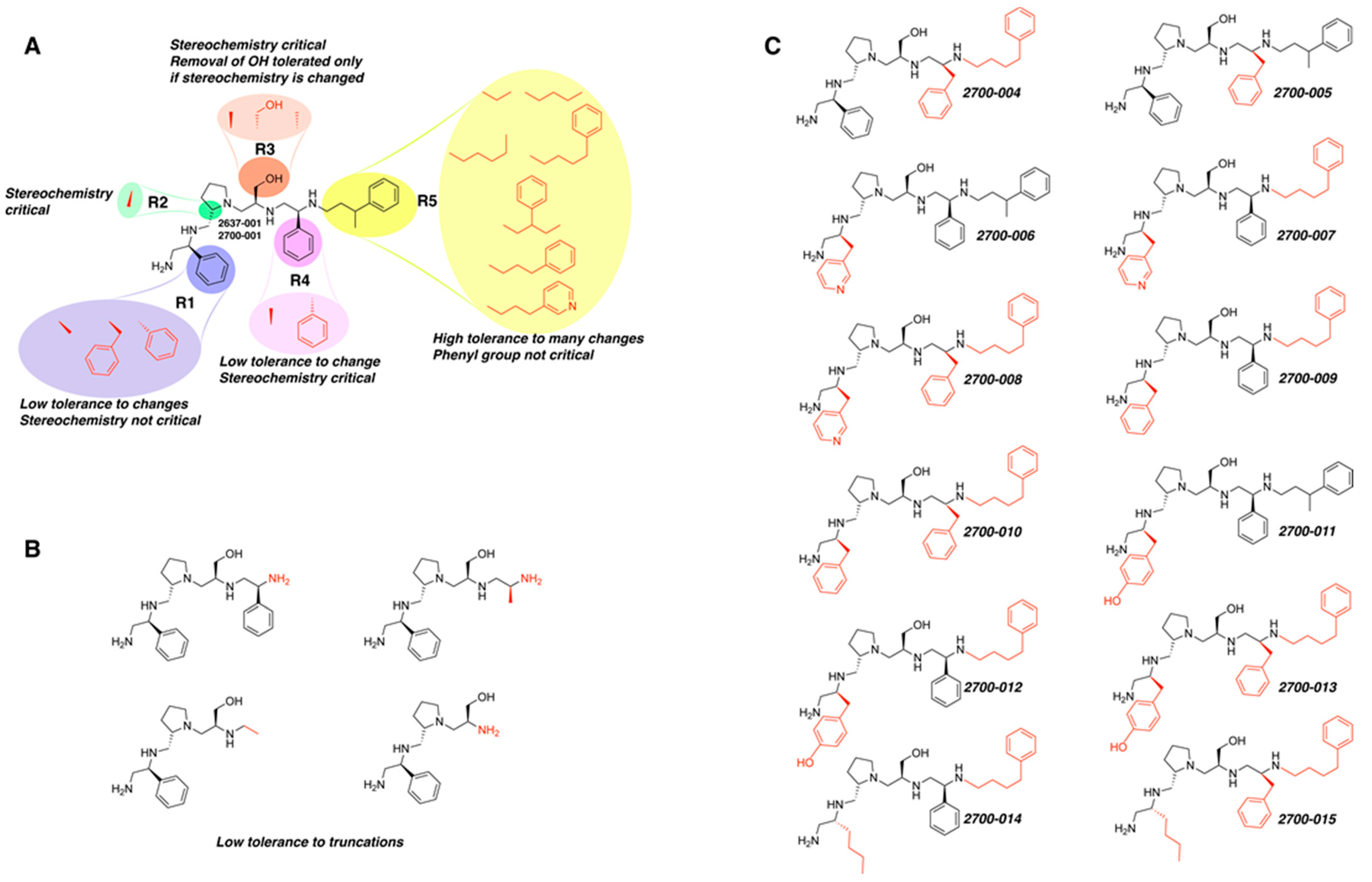
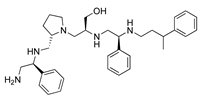 R1: S-phenyl R2: S-pyrrolidine R3: S-hydroxymethyl R4: S-phenyl R5: 3-phenylbutyl | 2637.001 | 60 | 4 | 3.0 | 4.2 | 6.7 | 9.4 |
| 2700.001 | 52 | 4 | 6.5 | 10.4 | 8.8 | 12.9 | |
 R1: S-benzyl R2: S-pyrrolidine R3: S-hydroxymethyl R4: S-phenyl R5: 3-phenylbutyl | 2637.003 | 20 | 3 | N.D. | N.D. | N.D. | N.D. |
| 2700.002 | 19 | 4 | N.D. | N.D. | N.D. | N.D. | |
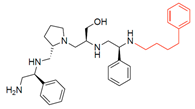 R1: S-phenyl R2: S-pyrrolidine R3: S-hydroxymethyl R4: S-phenyl R5: 4-phenylbutyl | 2637.011 | 66 | 6 | 8.3 | 8.9 | 11.6 | 12.5 |
| 2700.003 | 53 | 9 | 7.2 | 10.0 | 8.9 | 12.2 | |
| Compound | Functionalities | %Inhibition (Average, n = 8) | Standard Error | |||||
|---|---|---|---|---|---|---|---|---|
| ID | Structure | R1 | R2 | R3 | R4 | R5 | ||
| 2700.001 2637.001 |  | S-phenyl | S-pyrrolidine | S-hydroxymethyl | S-phenyl | 3-phenylbutyl | 50 | 4 |
| 2700.002 2637.003 | 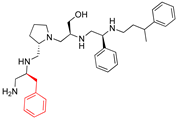 | S-benzyl | S-pyrrolidine | S-hydroxymethyl | S-phenyl | 3-phenylbutyl | 19 ** | 4 |
| 2700.003 2637.011 |  | S-phenyl | S-pyrrolidine | S-hydroxymethyl | S-phenyl | 4-phenylbutyl | 53 | 9 |
| 2700.004 |  | S-phenyl | S-pyrrolidine | S-hydroxymethyl | S-benzyl | 4-phenylbutyl | 46 | 14 |
| 2700.005 | 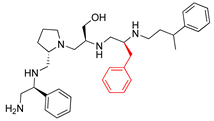 | S-phenyl | S-pyrrolidine | S-hydroxymethyl | S-benzyl | 3-phenylbutyl | 21 ** | 3 |
| 2700.006 |  | S-3-methylpyridine | S-pyrrolidine | S-hydroxymethyl | S-phenyl | 3-phenylbutyl | 14 ** | 2 |
| 2700.007 | 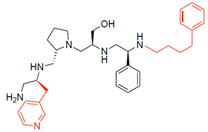 | S-3-methylpyridine | S-pyrrolidine | S-hydroxymethyl | S-phenyl | 4-phenylbutyl | 14 ** | 2 |
| 2700.008 | 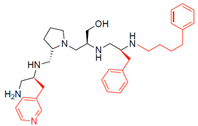 | S-3-methylpyridine | S-pyrrolidine | S-hydroxymethyl | S-benzyl | 4-phenylbutyl | 11 * | 3 |
| 2700.009 | 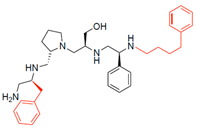 | S-benzyl | S-pyrrolidine | S-hydroxymethyl | S-phenyl | 4-phenylbutyl | 12 ** | 3 |
| 2700.010 | 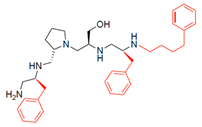 | S-benzyl | S-pyrrolidine | S-hydroxymethyl | S-benzyl | 4-phenylbutyl | 23 * | 7 |
| 2700.011 |  | S-4-hydroxybutyl | S-pyrrolidine | S-hydroxymethyl | S-phenyl | 3-phenylbutyl | 10 ** | 3 |
| 2700.012 | 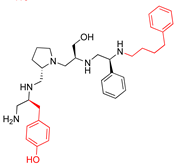 | S-4-hydroxybutyl | S-pyrrolidine | S-hydroxymethyl | S-phenyl | 4-phenylbutyl | 12 ** | 3 |
| 2700.013 | 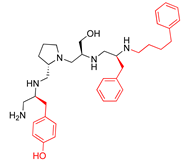 | S-4-hydroxybutyl | S-pyrrolidine | S-hydroxymethyl | S-benzyl | 4-phenylbutyl | 19 ** | 4 |
| 2700.014 | 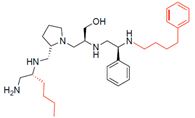 | R-butyl | S-pyrrolidine | S-hydroxymethyl | S-phenyl | 4-phenylbutyl | 19 ** | 4 |
| 2700.015 | 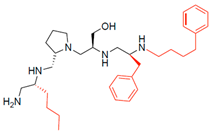 | R-butyl | S-pyrrolidine | S-hydroxymethyl | S-benzyl | 4-phenylbutyl | 17 ** | 3 |
| Compound ID | Inhibition (μM) | |||
|---|---|---|---|---|
| 2-Fold | 3-Fold | |||
| 50% | 80% | 50% | 80% | |
| 2700.001 | 6.5 | 10.4 | 8.8 | 12.9 |
| 2700.003 | 7.2 | 10.0 | 8.9 | 12.2 |
| 2700.004 | 19.3 | 23.4 | >24 | >24 |
| 2700.005 | 14.2 | 15.0 | 19.9 | 19.5 |
| 2700.007 | >24 | >24 | >24 | >24 |
| 2700.010 | 16.2 | 15.5 | 19.9 | 19.6 |
| 2700.013 | >24 | >24 | >24 | >24 |
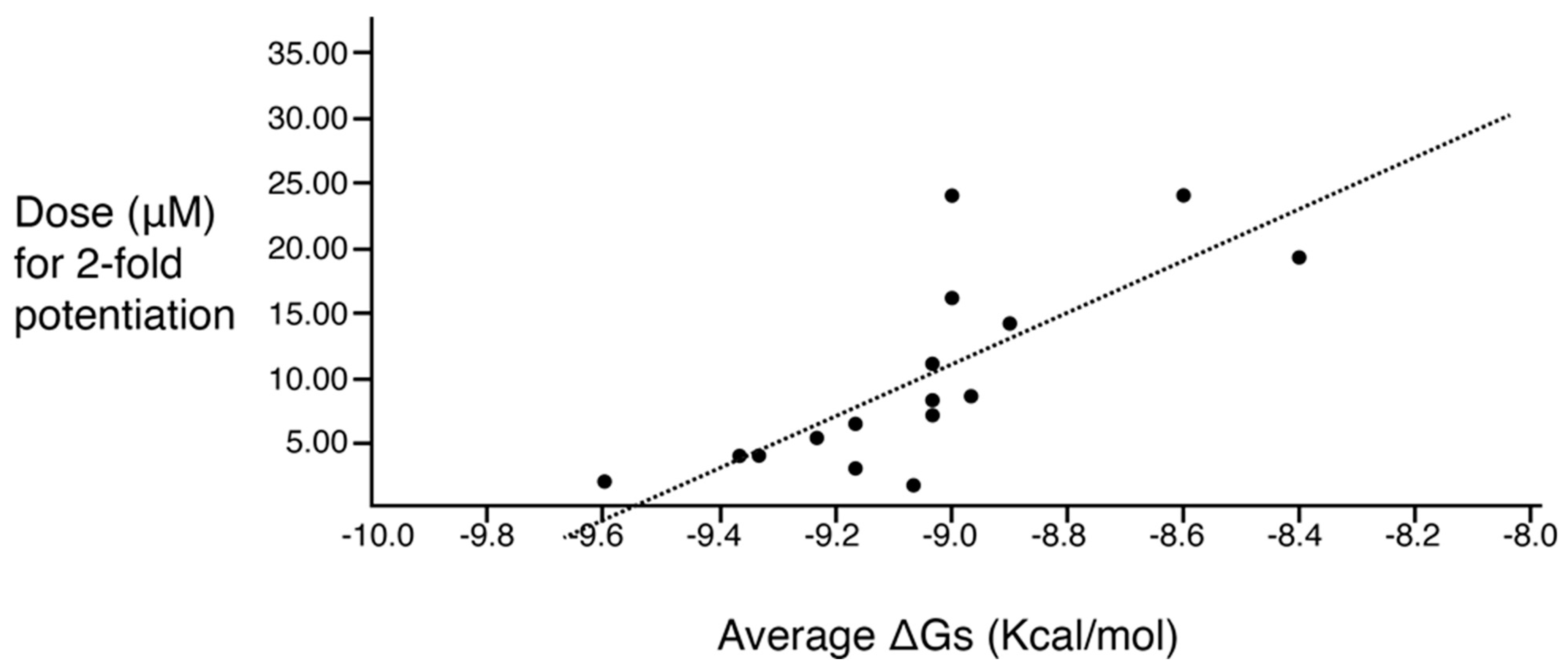
| Compound | Functionalities | Delta G (Kcal/mol) | Dose for 2-Fold Potentiation (µM) | |||||
|---|---|---|---|---|---|---|---|---|
| ID | Structure | R1 | R2 | R3 | R4 | R5 | ||
| Series 2700 | ||||||||
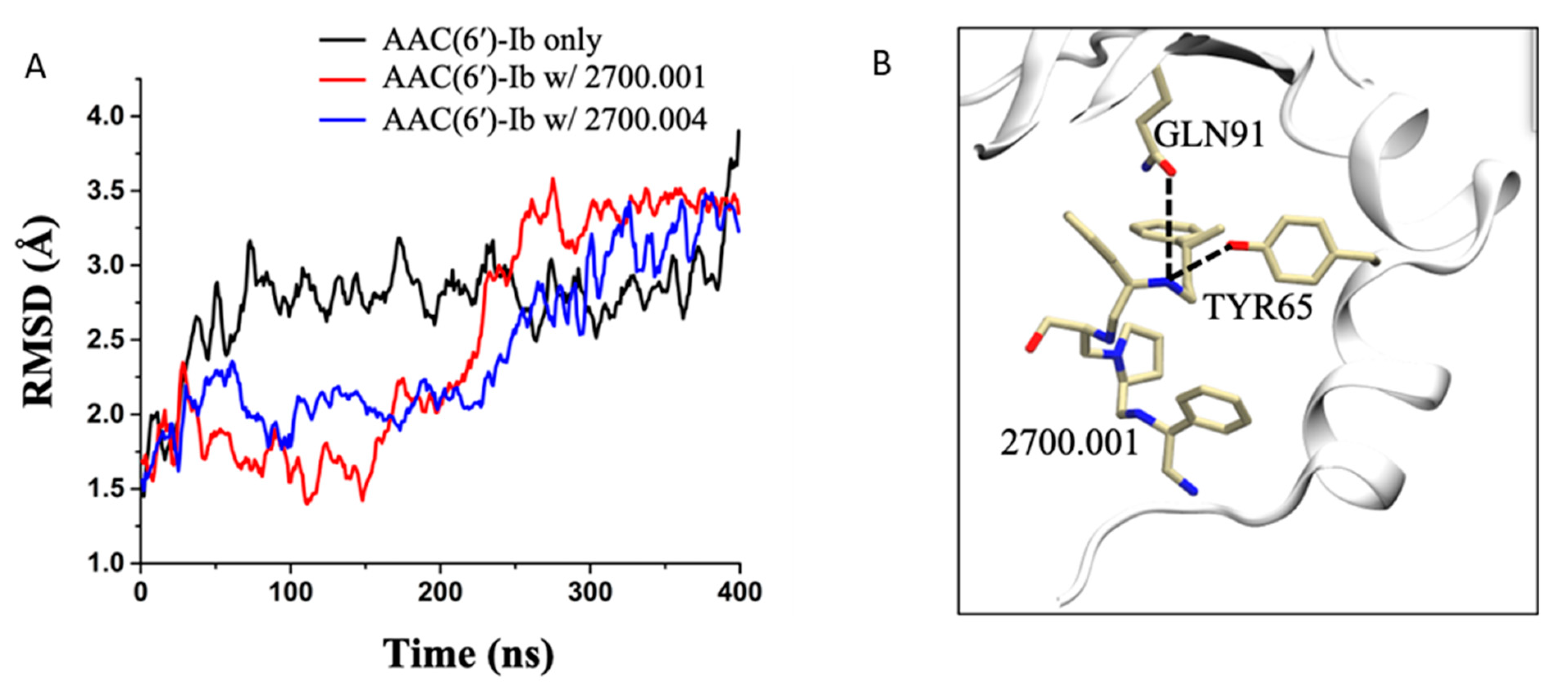
| 2700.001 | 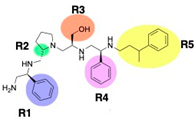 | S-phenyl | S-pyrrolidine | S-hydroxymethyl | S-phenyl | 3-phenylbutyl | −9.2 ± 0.6 | 6.5 |
| 2700.003 | 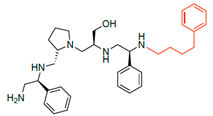 | S-phenyl | S-pyrrolidine | S-hydroxymethyl | S-phenyl | 4-phenylbutyl | −9.0 ± 0.6 | 7.2 |
| 2700.004 |  | S-phenyl | S-pyrrolidine | S-hydroxymethyl | S-benzyl | 4-phenylbutyl | −8.4 ± 0.2 | 19.3 |
| 2700.005 | 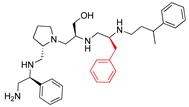 | S-phenyl | S-pyrrolidine | S-hydroxymethyl | S-benzyl | 3-phenylbutyl | −8.9 ± 0.5 | 14.2 |
| 2700.007 | 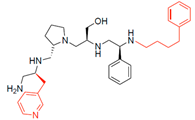 | S-3-methylpyridine | S-pyrrolidine | S-hydroxymethyl | S-phenyl | 4-phenylbutyl | −9.0 ± 0.4 | 24.0 |
| 2700.010 |  | S-benzyl | S-pyrrolidine | S-hydroxymethyl | S-benzyl | 4-phenylbutyl | −9.0 ± 0.9 | 16.2 |
| 2700.013 |  | S-4-hydroxybutyl | S-pyrrolidine | S-hydroxymethyl | S-benzyl | 4-phenylbutyl | −8.6 ± 0.2 | 24.0 |
| Series 2637 | ||||||||
| 2637.001 | 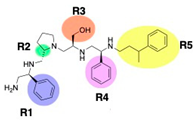 | S-phenyl | S-pyrrolidine | S-hydroxymethyl | S-phenyl | 3-phenylbutyl | −9.2 ± 0.6 | 3.0 |
| 2637.004 | 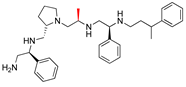 | S-phenyl | S-pyrrolidine | S-methyl | S-phenyl | 3-phenylbutyl | −9.0 ± 0.3 | 8.7 |
| 2637.007 |  | S-phenyl | S-pyrrolidine | S-hydroxymethyl | S-phenyl | 3-ethyl | −9.6 ± 0.0 | 2.0 |
| 2637.010 | 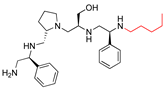 | S-phenyl | S-pyrrolidine | S-hydroxymethyl | S-phenyl | pentyl | −9.1 ± 0.5 | 1.7 |
| 2637.011 |  | S-phenyl | S-pyrrolidine | S-hydroxymethyl | S-phenyl | phenylbutyl | −9.0 ± 0.6 | 8.3 |
| 2637.012 | 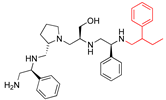 | S-phenyl | S-pyrrolidine | S-hydroxymethyl | S-phenyl | 2-phenylbutyl | −9.0 ± 0.4 | 11.1 |
| 2637.013 | 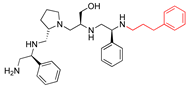 | S-phenyl | S-pyrrolidine | S-hydroxymethyl | S-phenyl | phenylpropyl | −9.3 ± 0.5 | 4.1 |
| 2637.014 | 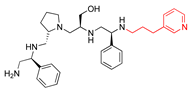 | S-phenyl | S-pyrrolidine | S-hydroxymethyl | S-phenyl | (pyridin-3-yl)propyl | −9.4 ± 0.2 | 4.0 |
| 2637.019 | 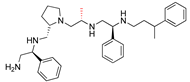 | S-phenyl | S-pyrrolidine | R-methyl | S-phenyl | 3-phenylbutyl | −9.2 ± 0.1 | 5.4 |
| 2637.020 | 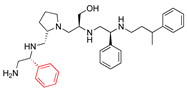 | R-phenyl | S-pyrrolidine | S-hydroxymethyl | S-phenyl | 3-phenylbutyl | −9.2 ± 0.5 | 5.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sklenicka, J.; Tran, T.; Ramirez, M.S.; Donow, H.M.; Magaña, A.J.; LaVoi, T.; Mamun, Y.; Jimenez, V.; Chapagain, P.; Santos, R.; et al. Structure–Activity Relationship of Pyrrolidine Pentamine Derivatives as Inhibitors of the Aminoglycoside 6′-N-Acetyltransferase Type Ib. Antibiotics 2024, 13, 672. https://doi.org/10.3390/antibiotics13070672
Sklenicka J, Tran T, Ramirez MS, Donow HM, Magaña AJ, LaVoi T, Mamun Y, Jimenez V, Chapagain P, Santos R, et al. Structure–Activity Relationship of Pyrrolidine Pentamine Derivatives as Inhibitors of the Aminoglycoside 6′-N-Acetyltransferase Type Ib. Antibiotics. 2024; 13(7):672. https://doi.org/10.3390/antibiotics13070672
Chicago/Turabian StyleSklenicka, Jan, Tung Tran, Maria S. Ramirez, Haley M. Donow, Angel J. Magaña, Travis LaVoi, Yasir Mamun, Verónica Jimenez, Prem Chapagain, Radleigh Santos, and et al. 2024. "Structure–Activity Relationship of Pyrrolidine Pentamine Derivatives as Inhibitors of the Aminoglycoside 6′-N-Acetyltransferase Type Ib" Antibiotics 13, no. 7: 672. https://doi.org/10.3390/antibiotics13070672
APA StyleSklenicka, J., Tran, T., Ramirez, M. S., Donow, H. M., Magaña, A. J., LaVoi, T., Mamun, Y., Jimenez, V., Chapagain, P., Santos, R., Pinilla, C., Giulianotti, M. A., & Tolmasky, M. E. (2024). Structure–Activity Relationship of Pyrrolidine Pentamine Derivatives as Inhibitors of the Aminoglycoside 6′-N-Acetyltransferase Type Ib. Antibiotics, 13(7), 672. https://doi.org/10.3390/antibiotics13070672