



Hybrid Caffeic Acid-Based DHFR Inhibitors as Novel Antimicrobial and Anticancer Agents
 ,
,  and
and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Molecular Modeling
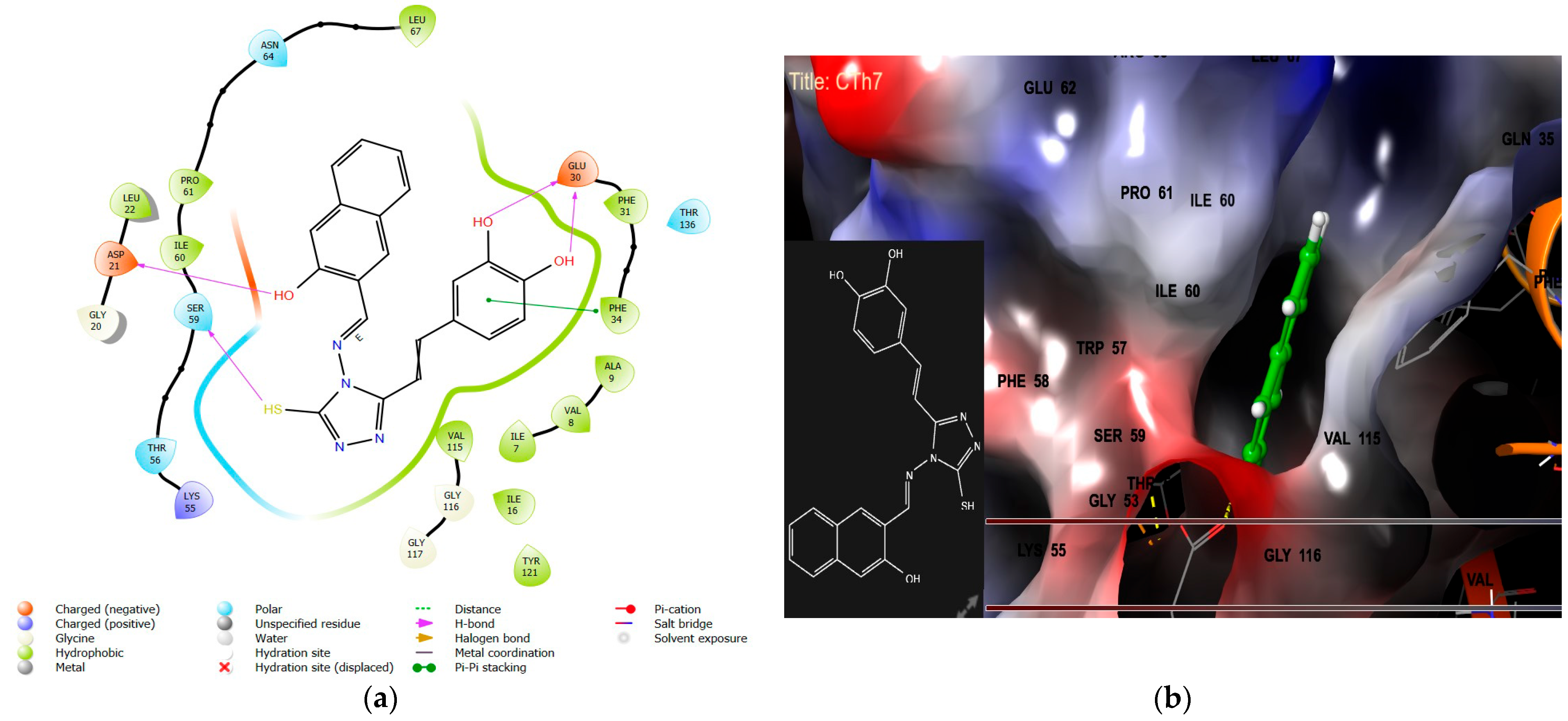
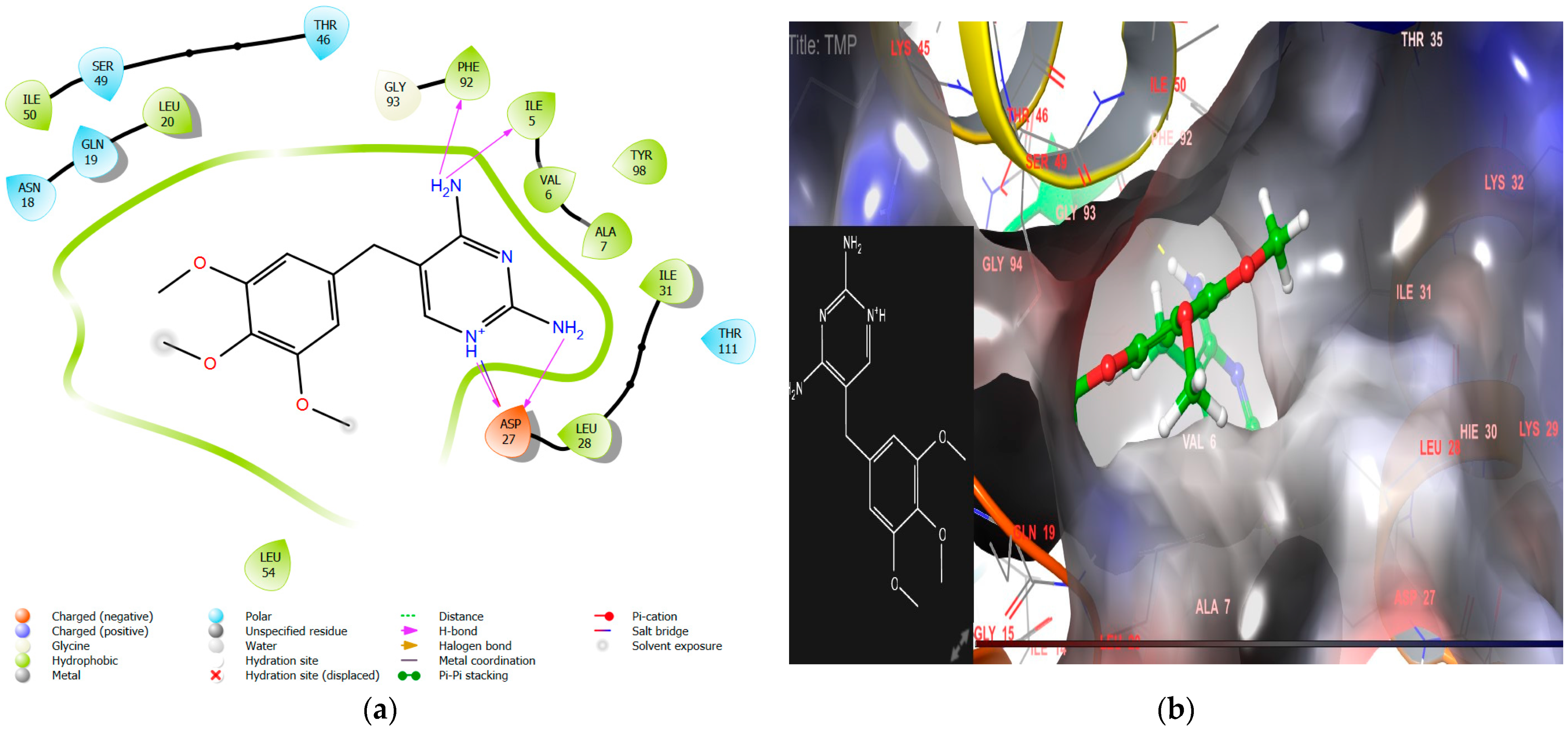
2.1.1. Molecular Docking Interaction Analysis of Homo Sapiens DHFR (PDB ID 1U72)
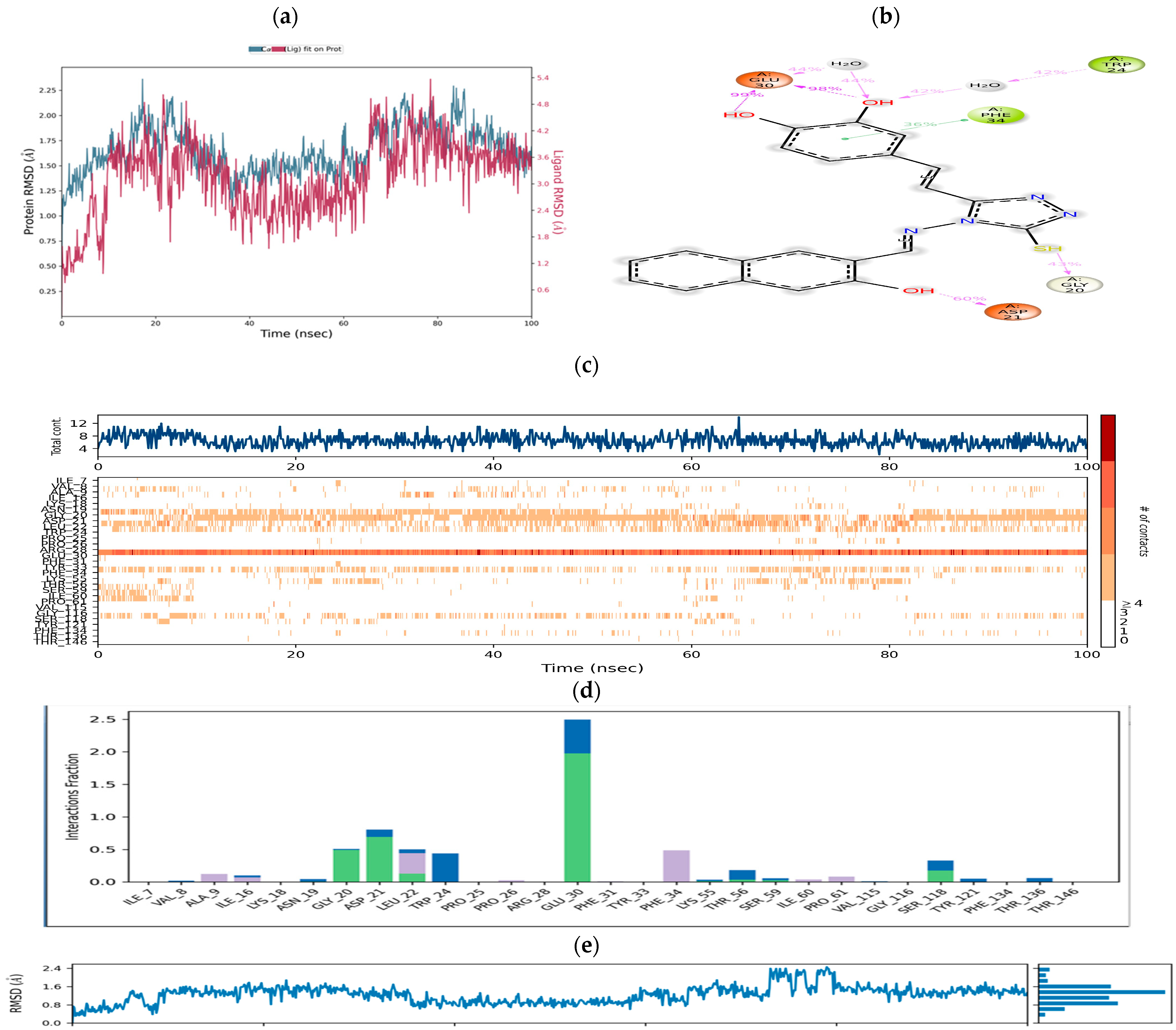
Molecular Dynamics Simulation Studies
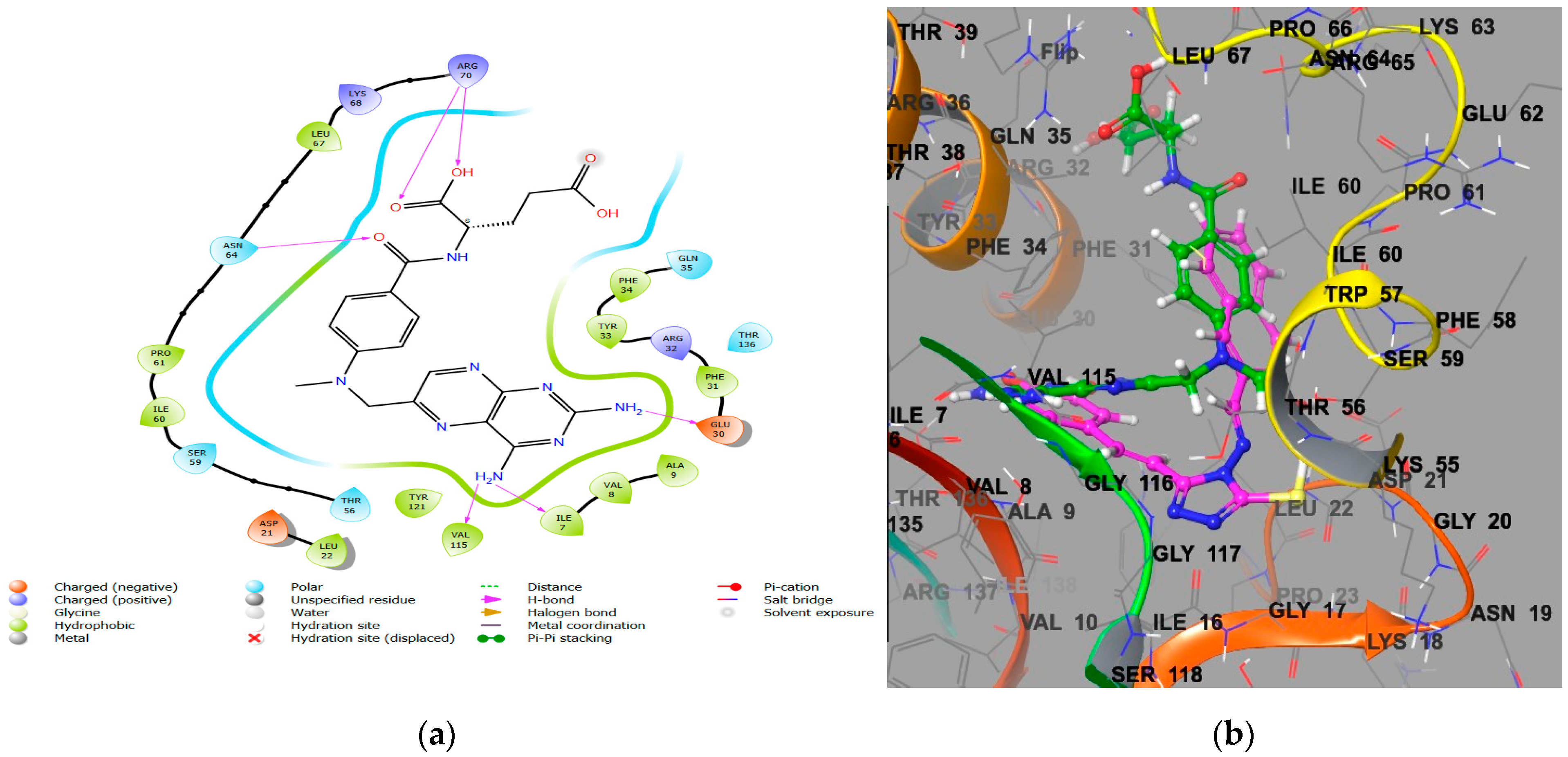
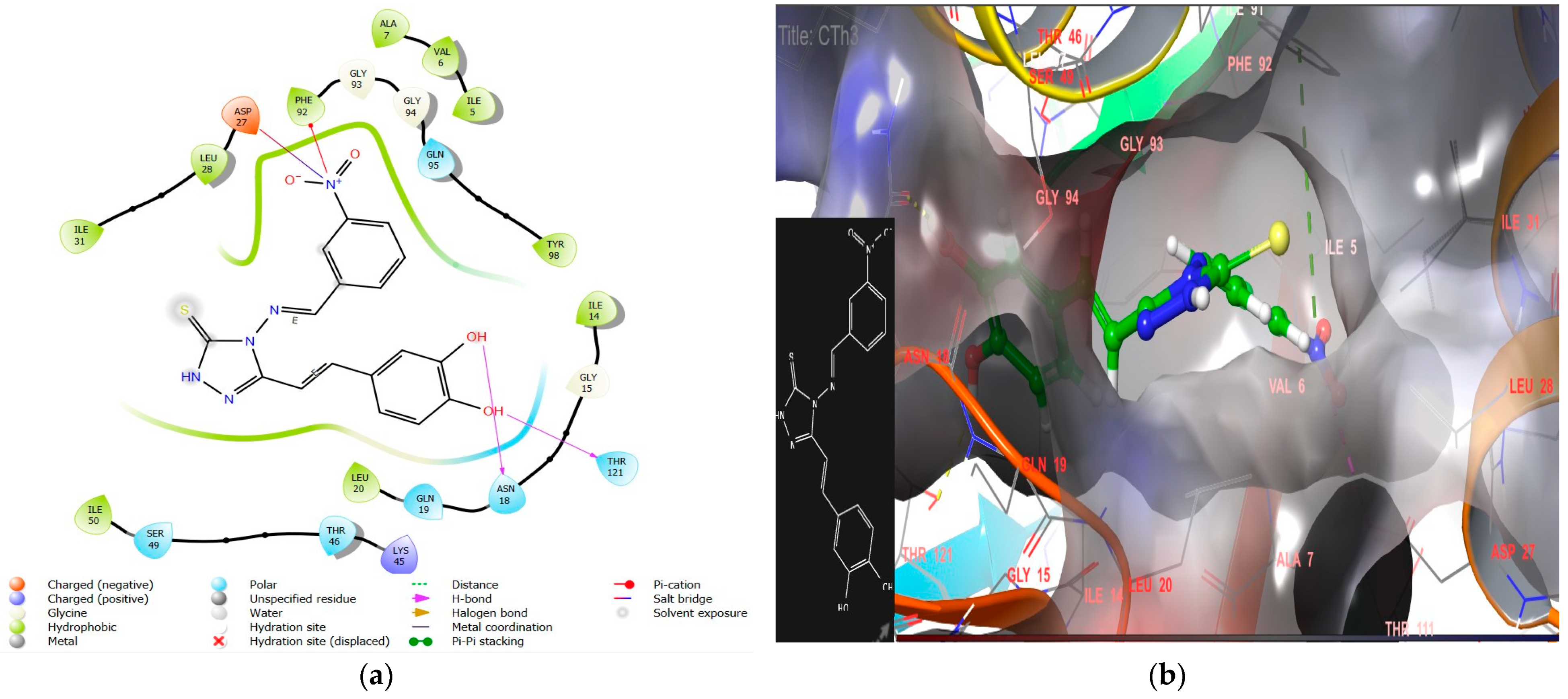
2.1.2. Molecular Docking Interaction Analysis of Microbe DHFR (PDB ID 2W9S)
2.1.3. ADMET Evaluation
2.2. Chemistry
Characterization of Synthesized Compounds
2.3. Biological Activity
2.3.1. DHFR Inhibition Assay
2.3.2. Assessment of Anticancer Activity
2.3.3. Antimicrobial Activity
3. Structure–Activity Relationship
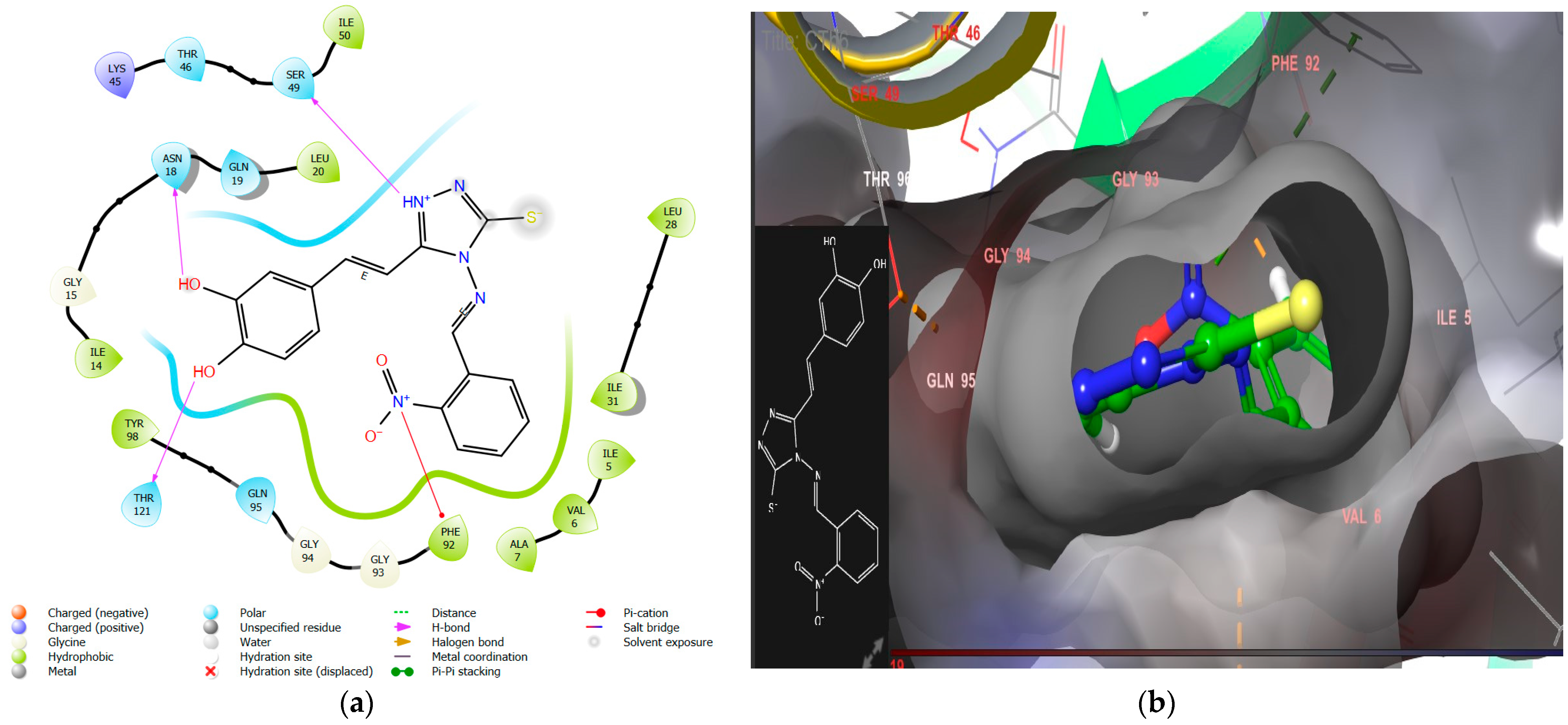
- The introduction of nitro substitution at the ortho and meta positions of an aromatic ring has been found to enhance both DHFR inhibitory activity and antimicrobial activity in derivatives. For instance, compounds CTh3 and CTh6 exhibit good DHFR inhibitory and excellent antimicrobial activity, with CTh3 showing an MIC of 5 µM against S. aureus.
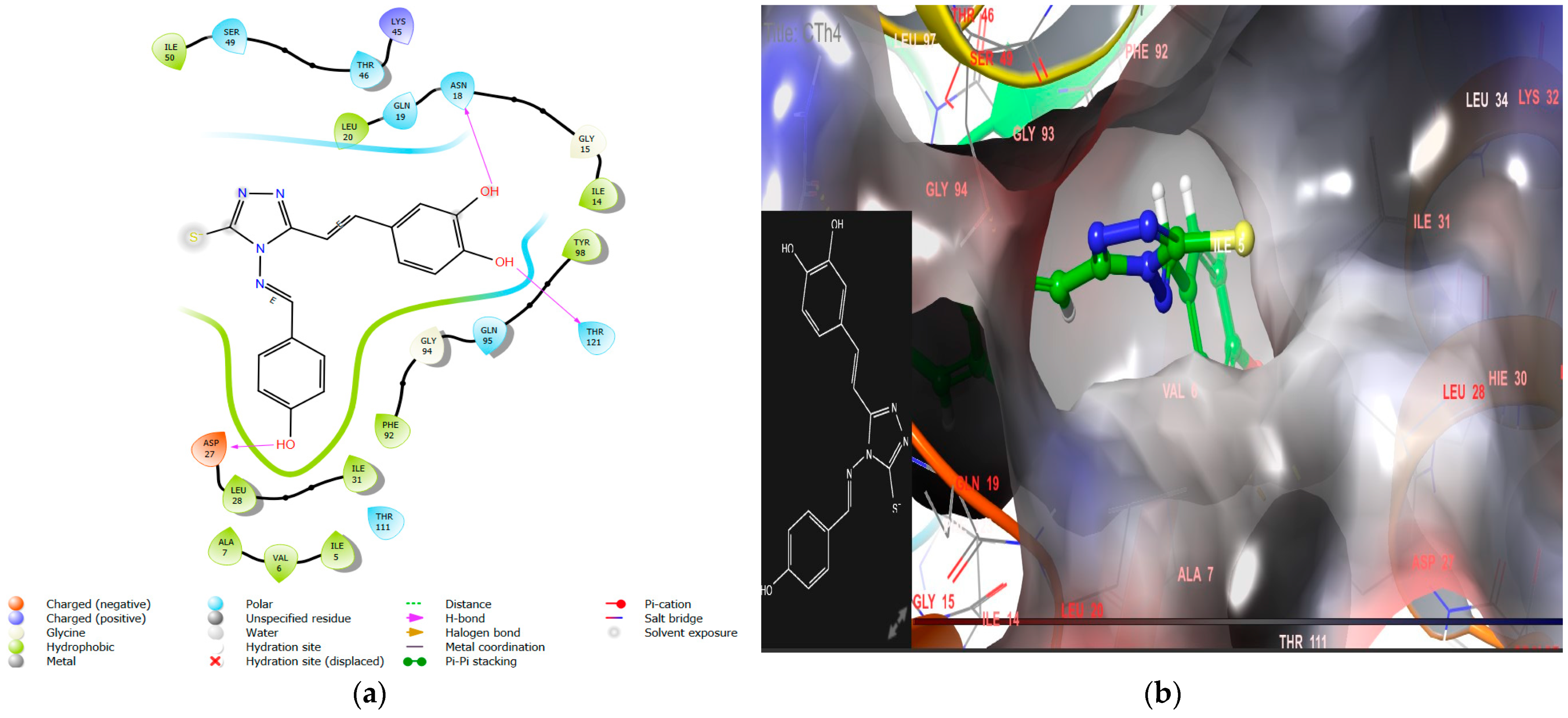
- Incorporating a hydroxyl group, as seen in CTh4, significantly increases the DHFR inhibitory activity, likely due to the hydrogen bond formation with the essential negatively charged amino acid residue Asp27. This compound also demonstrates excellent antimicrobial activity with MICs of 5 µM, 5 µM, and 11 µM against E. coli, P. aeruginosa, and C. albicans, respectively.
- Furthermore, when a chloro group is substituted on the aromatic ring of a Schiff base, the antifungal activity is further enhanced, particularly against A. niger. For example, compound CTh10 exhibits strong activity against A. niger with an MIC of 39 µM.
- The introduction of a bulky aromatic group leads to a sharp increase in DHFR inhibitory activity and remarkable anticancer activity. Compound CTh7, for example, displays an IC50 value of 8.53 µM against the MCF-7 cell line.
4. Materials and Methods
4.1. Instruments and Chemicals
4.2. Molecular Modelling
4.2.1. Molecular Docking
4.2.2. Binding Free Energies and MM/GBSA
4.2.3. ADMET Evaluation
4.2.4. Molecular Dynamics Simulations
4.3. Synthesis of Caffeic Acid Derivatives
4.3.1. Synthesis of 4-(3,4-Dihydroxyphenyl)but-3-en-2-one (C1)
4.3.2. Synthesis of 3-(3,4-Dihroxyphenyl)acrylohydrazide (C2)
4.3.3. Synthesis of 4-[2-(4-Amino-5-mercapto-4H-triazol-3-yl)vinyl] benzene-1,2-diol (CTh)
4.3.4. General Synthetic Procedure for the Synthesis of Schiff Bases
4.4. Assessment of Biological Activity
4.4.1. In Vitro DHFR Inhibition Assessment
4.4.2. Anticancer Activity (MTT Assay)
4.4.3. Antimicrobial Activity
5. Limitation and Future Scope
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Vo, G.; Nguyen, T.P.; Nguyen, H.T.; Nguyen, T.T. In silico and in vitro studies on the anti-cancer activity of artemetin, vitexicarpin and penduletin compounds from Vitex negundo. Saudi Pharm. J. 2022, 30, 1301–1314. [Google Scholar]
- World Health Organization. Cancer. 2022. Available online: https://www.who.int/health-topics/cancer#tab=tab_1,2018 (accessed on 19 November 2022).
- World Health Organization. Antibiotic Resistance. 2022. Available online: https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance (accessed on 19 November 2022).
- Salam, M.A.; Al-Amin, M.Y.; Salam, M.T.; Pawar, J.S.; Akhter, N.; Rabaan, A.A.; Alqumber, M.A. Antimicrobial resistance: A growing serious threat for global public health. Healthcare 2023, 11, 1946. [Google Scholar] [CrossRef]
- Majumder, M.A.A.; Rahman, S.; Cohall, D.; Bharatha, A.; Singh, K.; Haque, M.; Gittens-St Hilaire, M. Antimicrobial stewardship: Fighting antimicrobial resistance and protecting global public health. Infect. Drug Resist. 2020, 13, 4713–4738. [Google Scholar] [CrossRef] [PubMed]
- Aragaw, W.W.; Negatu, D.A.; Bungard, C.J.; Dartois, V.A.; Marrouni, A.E.; Nickbarg, E.B.; Olsen, D.B.; Warrass, R.; Dick, T. Pharmacological validation of dihydrofolate reductase as a drug target in Mycobacterium abscessus. Antimicrob. Agents Chemother. 2024, 68, e00717–e00723. [Google Scholar] [CrossRef] [PubMed]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.M.; Pratt, A.G.; Isaacs, J.D. Mechanism of action of methotrexate in rheumatoid arhritis, and the search for biomarkers. Nat. Rev. Rheumatol. 2016, 12, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Sehrawat, R.; Rathee, P.; Khatkar, S.; Küpeli Akkol, E.; Khayatkashani, M.; Nabavi, S.M.; Khatkar, A. Dihydrofolate Reductase (DHFR) Inhibitors: A Comprehensive Review. Curr. Med. Chem. 2023, 31, 799–824. [Google Scholar] [CrossRef]
- Foye, W.O.; Lemke, T.L.; Williams, D.A. Principles of Medicinal Chemistry, 4th ed.; Lea & Febiger: Philadelphia, PA, USA, 1974. [Google Scholar]
- He, J.; Qiao, W.; An, Q.; Yang, T.; Luo, Y. Dihydrofolate reductase inhibitors for use as antimicrobial agents. Eur. J. Med. Chem. 2020, 195, 112268. [Google Scholar] [CrossRef]
- Gibson, M.W.; Dewar, S.; Ong, H.B.; Sienkiewicz, N.; Fairlamb, A.H. Trypanosoma brucei DHFR-TS Revisited: Characterisation of a Bifunctional and Highly Unstable Recombinant Dihydrofolate Reductase-Thymidylate Synthase. PLoS Negl. Trop. Dis. 2016, 10, e0004714. [Google Scholar] [CrossRef]
- El-Gazzar, Y.I.; Georgey, H.H.; El-Messery, S.M.; Ewida, H.A.; Hassan, G.S.; Raafat, M.M.; Ewida, M.A.; El-Subbagh, H.I. Synthesis, biological evaluation and molecular modeling study of new (1,2,4-triazole or 1,3,4-thiadiazole)-methylthio-derivatives of quinazolin-4(3H)-one as DHFR inhibitors. Bioorg. Chem. 2017, 72, 282–292. [Google Scholar] [CrossRef]
- Banerjee, D.; Mayer-Kuckuk, P.; Capiaux, G.; Budak-Alpdogan, T.; Gorlick, R.; Bertino, J.R. Novel aspects of resistance to drugs targeted to dihydrofolate reductase and thymidylate synthase. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2002, 1587, 164–173. [Google Scholar] [CrossRef]
- McIvor, R.S. Drug-resistant dihydrofolate reductases: Generation, expression and therapeutic application. Bone Marrow Transplant. 1996, 18 (Suppl. S3), S50–S54. [Google Scholar] [PubMed]
- Wróbel, A.; Arciszewska, K.; Maliszewski, D.; Drozdowska, D. Trimethoprim and other nonclassical antifolates an excellent template for searching modifications of dihydrofolate reductase enzyme inhibitors. J. Antibiot. 2020, 73, 5–27. [Google Scholar] [CrossRef] [PubMed]
- Reeve, S.M.; Si, D.; Krucinska, J.; Yan, Y.; Viswanathan, K.; Wang, S.; Holt, G.T.; Frenkel, M.S.; Ojewole, A.A.; Estrada, A.; et al. Toward broad spectrum dihydrofolate reductase inhibitors targeting trimethoprim resistant enzymes identified in clinical isolates of methicillin resistant Staphylococcus aureus. ACS Infect. Dis. 2019, 5, 1896–1906. [Google Scholar] [CrossRef]
- Sehrawat, R.; Rathee, P.; Khatkar, S.; Küpeli Akkol, E.; Khatkar, A. In silico and in vitro analysis of phenolic acids for identification of potential dhfr inhibitors as antimicrobial and anticancer agents. Curr. Protein Pept. Sci. 2024, 25, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Sehrawat, R.; Rathee, P.; Küpeli Akkol, E.; Khatkar, S.; Lather, A.; Redhu, N.; Khatkar., A. Phenolic acids-versatile natural moiety with numerous biological applications. Curr. Top. Med. Chem. 2022, 22, 1472–1484. [Google Scholar] [CrossRef]
- Espíndola, K.M.M.; Ferreira, R.G.; Narvaez, L.E.M.; Silva Rosario, A.C.R.; Da Silva, A.H.M.; Silva, A.G.B.; Vieira, A.P.O.; Monteiro, M.C. Chemical and pharmacological aspects of caffeic acid and its activity in hepatocarcinom. Front. Oncol. 2019, 9, 541. [Google Scholar] [CrossRef] [PubMed]
- Pavlíková, N. Caffeic acid and diseases—Mechanisms of action. Int. J. Mol. Sci. 2022, 24, 588. [Google Scholar] [CrossRef]
- Kępa, M.; Miklasińska-Majdanik, M.; Wojtyczka, R.D.; Idzik, D.; Korzeniowski, K.; Smoleń-Dzirba, J.; Wąsik, T.J. Antimicrobial potential of caffeic acid against Staphylococcus aureus clinical strains. BioMed Res. Int. 2018, 2018, 7413504. [Google Scholar] [CrossRef]
- Khan, F.; Bamunuarachchi, N.I.; Tabassum, N.; Kim, Y.M. Caffeic acid and its derivatives: Antimicrobial drugs toward microbial pathogens. J. Agric. Food Chem. 2021, 69, 2979–3004. [Google Scholar] [CrossRef]
- Doshi, H.; Thakkar, S.; Khirsariya, P.; Thakur, M.; Ray, A. 6-Tosyl-4,5,6,7-tetrahydrothieno [2,3-c] pyridine-3-carboxamide analogues: Synthesis, characterization, MO calculation, and antibacterial activity. Appl. Biochem. Biotechnol. 2015, 175, 1700–1709. [Google Scholar] [CrossRef] [PubMed]
- Sztanke, K.; Tuzimski, T.; Rzymowska, J.; Pasternak, K.; Kandefer-Szerszen, M. Synthesis, determination of the lipophilicity, anticancer and antimicrobial properties of some fused 1,2,4-triazole derivatives. Eur. J. Med. Chem. 2008, 43, 404–419. [Google Scholar] [CrossRef] [PubMed]
- Popiolek, L.; Kosikowska, U.; Mazur, L.; Dobosz, M.; Malm, A. Synthesis and antimicrobial evaluation of some novel 1,2,4-triazole and 1,3,4-thiadiazole derivatives. Med. Chem. Res. 2013, 23, 3134–3147. [Google Scholar] [CrossRef] [PubMed]
- Vijesh, A.; Isloor, A.; Shetty, P.; Sundershan, S.; Kun Fun, H. New pyrazole derivatives containing 1,2,4-triazoles and benzoxazoles as potent antimicrobial and analgesic agents. Eur. J. Med. Chem. 2013, 62, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Salerno, L.; Pittala, V.; Romeo, G. Evaluation of novel aryloxyalkyl derivatives of imidazole and 1,2,4-triazole as heme oxygenase-1 (HO-1) inhibitors and their antitumor properties. Bioorg. Med. Chem. 2013, 21, 5145–5153. [Google Scholar] [CrossRef] [PubMed]
- Kulabas, N.; Tatar, E.; Ozakpınar, O.B. Synthesis and antiproliferative evaluation of novel 2-(4H–1,2,4- triazole-3-ylthio)acetamide derivatives as inducers of apoptosis in cancer cells. Eur. J. Med. Chem. 2016, 121, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Al-Abdullah, E.; Asiri, H.; Lahsasni, S.; Habib, E.; Ibrahim, T.; El-Emam, A. Synthesis, antimicrobial, and anti-inflammatory activity, of novel S-substituted and Nsubstituted 5-(1-adamantyl)-1,2,4-triazole-3-thiols. Drug Des. Dev. Ther. 2014, 8, 505–518. [Google Scholar]
- Liu, J.; Liu, Q.; Yang, X. Design, synthesis, and biological evaluation of 1,2,4-triazole bearing 5-substituted biphenyl-2-sulfonamide derivatives as potential antihypertensive candidates. Bioorg. Med. Chem. 2013, 21, 7742–7775. [Google Scholar] [CrossRef]
- Nair, V.P.; Anang, S.; Subramani, C. Endoplasmic reticulum stress induced synthesis of a novel viral factor mediates efficient replication of genotype-1 hepatitis E virus. PLoS Pathog. 2016, 12, e1005521. [Google Scholar] [CrossRef]
- Fischer, J.; Ganellin, C.R.; Rotella, D.P. Analogue-Based Drug Discovery III, 3rd ed.; Wiley: Hoboken, NJ, USA, 2012. [Google Scholar]
- Shi, L.; Ge, H.; Tan, S. Synthesis and antimicrobial activities of Schiff bases derived from 5-chloro-salicylaldehyde. Eur. J. Med. Chem. 2007, 42, 558–564. [Google Scholar] [CrossRef]
- Sinha, D.; Tiwari, A.; Singh, S. Synthesis, characterization and biological activity of Schiff base analogues of indole-3-carboxaldehyde. Eur. J. Med. Chem. 2008, 43, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Karthikeyan, M.S.; Prasad, D.J.; Poojary, B.; Bhat, K.S.; Holla, B.S.; Kumari, N.S. Synthesis and biological activity of Schiff and Mannich bases bearing 2,4-dichloro-5-fluorophenyl moiety. Bioorg. Med. Chem. 2006, 14, 7482–7489. [Google Scholar] [CrossRef] [PubMed]
- Patole, J.; Shingnapurkar, D.; Padhyea, S.; Ratledge, C. Schiff base conjugates of paminosalicylic acid as antimycobacterial agents. Bioorg. Med. Chem. Lett. 2006, 16, 1514–1517. [Google Scholar] [CrossRef] [PubMed]
- Hearn, M.J.; Cynamon, M.H. Design and synthesis of antituberculars: Preparation and evaluation against Mycobacterium tuberculosis of an isoniazid Schiff base. J. Antimicrob. Chemoter. 2004, 53, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Vicini, P.; Geronikaki, A.; Incerti, M.; Busonera, B.; Poni, G.; Cabras, C.A.; La Colla, P. Synthesis and biological evaluation of benzo[d]isothiazole, benzothiazole and thiazole Schiff bases. Bioorg. Med. Chem. 2003, 11, 4785–4789. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, S.V.; Bothara, K.G.; Raut, M.K.; Patil, A.A.; Sarkate, A.P.; Mokale, V.J. Design, synthesis and evaluation of antiinflammatory, analgesic and ulcerogenicity studies of novel S-substituted phenacyl-1,3,4-oxadiazole-2-thiol and Schiff bases of diclofenac acid as nonulcerogenic derivatives. Bioorg. Med. Chem. 2008, 16, 1822–1831. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, S.K.; Pandeya, S.N.; Stables, J.P.; Ramesh, A. Anticonvulsant activity of hydrazones, Schiff and Mannich bases of isatin derivatives. Eur. J. Pharm. Sci. 2002, 16, 129–132. [Google Scholar] [CrossRef]
- Riyadh, S.M.; El-Motairi, S.A.; Ahmed, H.E.; Khalil, K.D.; Habib, E.S.E. Synthesis, biological evaluation, and molecular docking of novel thiazoles and [1,3,4] thiadiazoles incorporating sulfonamide group as DHFR Inhibitors. Chem. Biodivers. 2018, 15, e1800231. [Google Scholar] [CrossRef] [PubMed]
- Katsila, T.; Spyroulias, G.A.; Patrinos, G.P.; Matsoukas, M.T. Computational approaches in target identification and drug discovery. Comput. Struct. Biotechnol. J. 2016, 14, 177–184. [Google Scholar] [CrossRef]
- Mali, G.; Shaikh, B.A.; Garg, S.; Kumar, A.; Bhattacharyya, S.; Erande, R.D.; Chate, A.V. Design, synthesis, and biological evaluation of densely substituted dihydropyrano [2, 3-c] pyrazoles via a taurine-catalyzed green multicomponent approach. ACS Omega 2021, 6, 30734–30742. [Google Scholar] [CrossRef]
- Thakkar, S.S.; Thakor, P.; Doshi, H.; Ray, A. 1,2,4-Triazole and 1,3,4-oxadiazole analogues: Synthesis, MO studies, in silico molecular docking studies, antimalarial as DHFR inhibitor and antimicrobial activities. Bioorg. Med. Chem. 2017, 25, 4064–4075. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yang, M.; Yuan, M.; Xue, L.; Li, H.; Tian, C.; Wang, X.; Liu, J.; Zhang, Z. Synthesis and antiproliferative activity of a series of novel 6-substituted pyrido[3,2-d]pyrimidines as potential nonclassical lipophilic antifolates targeting dihydrofolate reductase. Eur. J. Med. Chem. 2017, 128, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Ikram, M.; Butt, A.R.; Fatima, A.; Shahzadi, I.; Haider, A.; Ul-Hamid, A.; Nabgan, W. Graphitic-carbon nitride and poly acrylic acid doped vanadium oxide for efficient catalytic and antimicrobial activity: In silico molecular docking studies. J. Photochem. Photobiol. A Chem. 2023, 443, 114835. [Google Scholar] [CrossRef]
- Aouidate, A.; Ghaleb, A.; Ghamali, M.; Chtita, S.; Choukrad, M.; Sbai, A.; Bouachrine, M.; Lakhlifi, T. Combined 3D-QSAR and Molecular Docking Study on 7,8-dialkyl-1,3-diaminopyrrolo-[3,2-f] Quinazoline Series Compounds to understand the binding mechanism of DHFR Inhibitors. J. Mol. Struct. 2017, 1139, 319–327. [Google Scholar] [CrossRef]
- Bouhidel, A.; Cherouana, P.; Durand, A.; Doudouh, F.; Morini, B.; Guillot, S.; Dahaoui, S. Synthesis, spectroscopic characterization, crystal structure, Hirshfeld surface analysis and antimicrobial activities of two triazole Schiff bases and their silver complexes. Inorganica Chim. Acta 2018, 482, 34–47. [Google Scholar] [CrossRef]
- Kumar, S.; Dhankhar, S.; Arya, V.P.; Yadav, S.; Yadav, J.P. Antimicrobial activity of Salvadora oleoides Decne. against some microorganisms. J. Med. Plants Res. 2012, 6, 2754–2760. [Google Scholar]
- Maestro, version 12; Schrodinger, LLC.: New York, NY, USA, 2020.
- Available online: https://www.rcsb.org/structure/2W9S (accessed on 11 November 2021).
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Glide, version 6.6; Schrödinger, LLC.: New York, NY, USA, 2015.
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Available online: https://admetmesh.scbdd.com/ (accessed on 5 February 2021).
- Sharma, K.; Tanwar, O.; Sharma, S.; Ali, S.; Alam, M.M.; Zaman, M.S. Structural comparison of Mtb-DHFR and h-DHFR for design, synthesis and evaluation of selective non-pteridine analogues as antitubercular agents. Bioorg. Chem. 2018, 80, 319–333. [Google Scholar] [CrossRef] [PubMed]
- Azzam, R.A.; Elsayed, R.E.; Elgemeie, G.H. Design, synthesis, and antimicrobial evaluation of a new series of N-sulfonamide 2-pyridones as dual inhibitors of DHPS and DHFR enzymes. ACS Omega 2020, 5, 10401–10414. [Google Scholar] [CrossRef] [PubMed]
- DHFR Assay. Available online: https://www.sigmaaldrich.com/deepweb/assets/sigmaaldrich/product/documents/427/408/cs0340bul.pdf (accessed on 13 March 2023).
- Francesconi, V.; Giovannini, L.; Santucci, M.; Cichero, E.; Costi, M.P.; Naesens, L.; Tonelli, M. Synthesis, biological evaluation and molecular modeling of novel azaspiro dihydrotriazines as influenza virus inhibitors targeting the host factor dihydrofolate reductase (DHFR). Eur. J. Med. Chem. 2018, 155, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.M.L. Tetrazolium (MTT) assay for cellular viability and activity. Methods Mol. Biol. 1998, 79, 179–184. [Google Scholar] [PubMed]
- Van Meerloo, J.; Kaspers, G.J.L.; Cloos, J. Cell sensitivity assays: The MTT assay. Methods Mol. Biol. 2011, 731, 237–245. [Google Scholar]
- Fotakis, G.; Timbrell, J.A. In vitro cytotoxicity assays: Comparison of LDH, neutral red, MTT and protein assay in hepatoma cell lines following exposure to cadmium chloride. Toxicol. Lett. 2006, 160, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Tihauan, B.; Berca, L.M.; Adascălului, M.; Sanmartin, A.; Nica, S.; Cimponeriu, D.; Duta, D. Experimental in vitro cytotoxicity evaluation of plant bioactive compounds and phytoagents: A review. Rom. Biotechnol. Lett. 2020, 25, 1832–1842. [Google Scholar]
- Indian Pharmacopoeia, Vol-I. Indian Pharmacopoeia Commission. The Controller of Publications, New Delhi. 2018; p. 37. Available online: https://ipc.gov.in/shop/index.php?route=product/category&path=59 (accessed on 21 March 2023).
- Narasimhan, B.; Narang, R.; Judge, V.; Ohlan, S.; Ohlan, R. Synthesis, antimicrobial and QSAR studies of substituted anilides. Arkivoc 2007, 15, 112–126. [Google Scholar] [CrossRef]
- Lather, A.; Sharma, S.; Khatkar, A. Naringin derivatives as glucosamine-6-phosphate synthase inhibitors based preservatives and their biological evaluation. Sci. Rep. 2020, 10, 20477. [Google Scholar] [CrossRef]
- Pasrija, R.; Kundu, D. Interaction of curcumin with azoles and polyenes against aspergillus infections. Res. J. Life Sci. Bioinform. Pharm. Chem. Sci. 2018, 4, 271–279. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Docking Parameter/Biological Assessment | CTh1 | CTh4 | CTh6 | CTh7 | Methotrexate |
|---|---|---|---|---|---|
| Docking score | −8.8 | −8.2 | −8.8 | −9.9 | −9.8 |
| Binding energy [▲G (kcal/mol)] | −69.80 | −60.61 | −65.34 | −71.38 | −73.93 |
| Hydrogen bond | 2-hydrogen-bond with Glu30 | 2-hydrogen-bond with Glu30 | 2-hydrogen-bond with Glu30 | Asp21, Ser59, and 2-hydrogen-bond with Glu30 | Glu30, Ile7, Val115, Asn64, and 2-hydrogen-bond with Arg70 |
| Hydrophobic interaction | Pro61, Ile60, Leu22, Tyr121, Ile16, Val115, Phe34, Phe31, Ala9, Val8, Ile7 | Phe31, Phe34, Ile7, Val8, Ala9, Val115, Tyr121, Ile16, Le22, Ile60, Pro61 | Leu67, Pro61, Ile60, Tyr121, Ile16, Val115, Leu22, Ile7, Val8, Ala9, Phe31, Phe34 | Leu67, Pro61, Ile60, Tyr121, Ile16, Val115, Leu22, Ile7, Val8, Ala9, Phe31, Phe34 | Leu67, Phe34, Phe31, Tyr33, Ala9, Val8, Ile7, Tyr121, Val115, Leu22, Pro61, Ile60 |
| π–π stacking | Phe34 | Phe34 | - | Phe34 | - |
| π–cation bond | - | - | Phe34 | - | - |
| Salt bridge | - | - | Lys55 | - | - |
| DHFR Inhibition IC50 (µM) ± SD, n = 3 | 17.03 ± 1.10 | 2.04 ± 0.12 | 10.54 ± 0.7 | 0.15 ± 0.02 | 0.088 ± 0.001 |
| Anticancer IC50 (µM), n = 3 | 147.74 ± 11.4 | 122.17 ± 7.3 | 89.25 ± 4.17 | 8.53 ± 0.19 | * |
| Compound | Docking Score | Binding Energy [▲G (kcal/mol)] | Hydrogen Bond Interaction | Hydrophobic Interaction | Other Interaction | DHFR Inhibition [IC50 (µM) ± SD, n = 3] |
|---|---|---|---|---|---|---|
| CTh1 | −8.2 | −45.28 | Asn18, Thr121 | Ile50, Leu20, Ile14, Tyr98, Phe92, Tyr109, Ile5, Val6, Ala7, Leu28, Ile31 | - | 17.03 ± 1.10 |
| CTh3 | −9.8 | −71.12 | Asn18, Thr121 | Tyr98, Ile50, Ile31, leu 28, Ala7, Val 6, Ile5, Phe 92, Leu20, Ile 14 | π–cation with Phe92, salt bridge with Asp27 | 8.88 ± 0.4 |
| CTh4 | −9.5 | −72.84 | Asn18, Thr 121, Asp 27 | Tyr98, Ile50, Ile31, leu 28, Ala7, Val 6, Ile5, Phe 92, Leu20, Ile 14 | - | 2.04 ± 0.12 |
| CTh6 | −9.4 | −73.77 | Asn18, Thr 121, Ser 49 | Tyr98, Ile50, Ile31, Ala7, Val 6, Ile5, Phe 92, Leu20, Ile 14 | π–cation Phe92 | 10.54 ± 0.7 |
| CTh7 | −9.6 | −59.68 | Asp27, Ser 49 | Tyr98, Ile50, Ile31, leu 28, Ala7, Val 6, Ile5, Phe 92, Leu20, Ile 14, Trp 22 | - | 0.15 ± 0.02 |
| CTh10 | −9.3 | −57.23 | Asn 18, Ile5, Thr 121 | Tyr98, Ile50, Ile31, leu 28, Ala7, Val 6, Ile5, Phe 92, Leu20, Ile 14 | - | 11.30 ± 0.92 |
| CTh | −9.2 | −61.45 | Ala 7, Phe 92, 2-H-bond interaction with Asn18 | Tyr98, Ile50, Ile14, Ile31, Ala7, Val 6, Ile5, Phe 92, Leu20, Ile 14 | - | 16.92 ± 1.2 |
| TMP | −9.3 | −70.12 | 2-H-bond with Asp 27, Phe 92, Ile5 | Ala7, Ile 31, Leu 20, Leu28, Phe 92, Tyr 98, Ile 50, Leu 54, Ile 5, and Val 6 | Salt bridge with Asp27 | 15.33 ± 1.9 |
| Properties | CTh1 | CTh3 | CTh4 | CTh6 | CTh7 | CTh10 | CTh |
|---|---|---|---|---|---|---|---|
| Lipinski rule | Accepted | Accepted | Accepted | Accepted | Accepted | Accepted | Accepted |
| Absorption | |||||||
| Caco-2 permeability | −4.971 | −5.046 | −5.055 | −5.064 | −5.12 | −4.947 | −5.332 |
| MDCK permeability | 8.5 × 10−6 | 1.28 × 10−5 | 7.2 × 10−6 | 9.3 × 10−6 | 7.68 × 10−6 | 1.9 × 10−5 | 3.3 × 10−6 |
| Pgp-inhibitor | --- | --- | --- | --- | --- | --- | --- |
| HIA | --- | --- | --- | --- | --- | --- | --- |
| F20% | -- | --- | +++ | --- | +++ | --- | +++ |
| F30% | +++ | -- | +++ | - | +++ | --- | +++ |
| Distribution | |||||||
| PPB | 97.807% | 98.39% | 97.023% | 98.224% | 99.004 | 99.41% | 50.853% |
| VD | 0.455 | 0.22 | 0.404 | 0.273 | 0.51 | 0.924 | 0.365 |
| BBB penetration | --- | --- | --- | --- | --- | --- | --- |
| Metabolism | |||||||
| CYP1A2 inhibitor | ++ | ++ | ++ | ++ | ++ | ++ | - |
| CYP1A2 substrate | -- | --- | --- | --- | --- | --- | --- |
| CYP2C19 inhibitor | - | --- | -- | - | - | -- | --- |
| CYP2C9 substrate | +++ | --- | +++ | --- | --- | -- | ++ |
| CYP2D6 inhibitor | -- | -- | - | --- | - | -- | --- |
| CYP2D6 substrate | ++ | - | + | - | + | ++ | - |
| CYP3A4 inhibitor | ++ | ++ | +++ | ++ | ++ | + | - |
| CYP3A4 substrate | -- | -- | -- | -- | -- | -- | --- |
| Excretion | |||||||
| CL | 9.676 | 8.233 | 10.334 | 8.609 | 9.194 | 8.044 | 13.595 |
| T1/2 | 0.746 | 0.738 | 0.885 | 0.790 | 0.711 | 0.75 | 0.909 |
| Toxicity | |||||||
| hERG blockers | --- | --- | --- | --- | --- | --- | --- |
| AMES toxicity | ++ | +++ | + | ++ | ++ | +++ | +++ |
| Rat oral acute toxicity | --- | --- | --- | --- | --- | --- | --- |
| Skin sensitization | - | - | - | - | ++ | ++ | -- |
| Respiratory toxicity | --- | --- | --- | --- | --- | ++ | --- |
| Compound | MIC (µM) | ||||
|---|---|---|---|---|---|
| Bacterial Strain | Fungal Strain | ||||
| S. aureus | P. aeruginosa | E. coli | C. albicans | A. niger | |
| CTh1 | 43 | 86 | 86 | 173 | - |
| CTh3 | 5 | 10 | 10 | 20 | 83 |
| CTh4 | 22 | 5 | 5 | 11 | - |
| CTh6 | 10 | 10 | 20 | 41 | 83 |
| CTh7 | 19 | 19 | 39 | - | - |
| CTh10 | 19 | 39 | 39 | 39 | 39 |
| CTh | 63 | 127 | 255 | - | 127 |
| Trimethoprim | 63 | 31 | 31 | 255 | 127 |
| Ampicillin | 11 | 22 | 11 | NT | NT |
| Tetracycline | 144 | 36 | 36 | NT | NT |
| Fluconazole | NT | NT | NT | 0.81 | 260.12 |
| Voriconazole | NT | NT | NT | 0.34 | 5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sehrawat, R.; Pasrija, R.; Rathee, P.; Kumari, D.; Khatkar, A.; Küpeli Akkol, E.; Sobarzo-Sánchez, E. Hybrid Caffeic Acid-Based DHFR Inhibitors as Novel Antimicrobial and Anticancer Agents. Antibiotics 2024, 13, 479. https://doi.org/10.3390/antibiotics13060479
Sehrawat R, Pasrija R, Rathee P, Kumari D, Khatkar A, Küpeli Akkol E, Sobarzo-Sánchez E. Hybrid Caffeic Acid-Based DHFR Inhibitors as Novel Antimicrobial and Anticancer Agents. Antibiotics. 2024; 13(6):479. https://doi.org/10.3390/antibiotics13060479
Chicago/Turabian StyleSehrawat, Renu, Ritu Pasrija, Priyanka Rathee, Deepika Kumari, Anurag Khatkar, Esra Küpeli Akkol, and Eduardo Sobarzo-Sánchez. 2024. "Hybrid Caffeic Acid-Based DHFR Inhibitors as Novel Antimicrobial and Anticancer Agents" Antibiotics 13, no. 6: 479. https://doi.org/10.3390/antibiotics13060479
APA StyleSehrawat, R., Pasrija, R., Rathee, P., Kumari, D., Khatkar, A., Küpeli Akkol, E., & Sobarzo-Sánchez, E. (2024). Hybrid Caffeic Acid-Based DHFR Inhibitors as Novel Antimicrobial and Anticancer Agents. Antibiotics, 13(6), 479. https://doi.org/10.3390/antibiotics13060479