
Genomic Analysis of Kitasatospora setae to Explore Its Biosynthetic Potential Regarding Secondary Metabolites

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Genome Profile of the Strain K. setae
2.2. Genome Annotation of K. setae
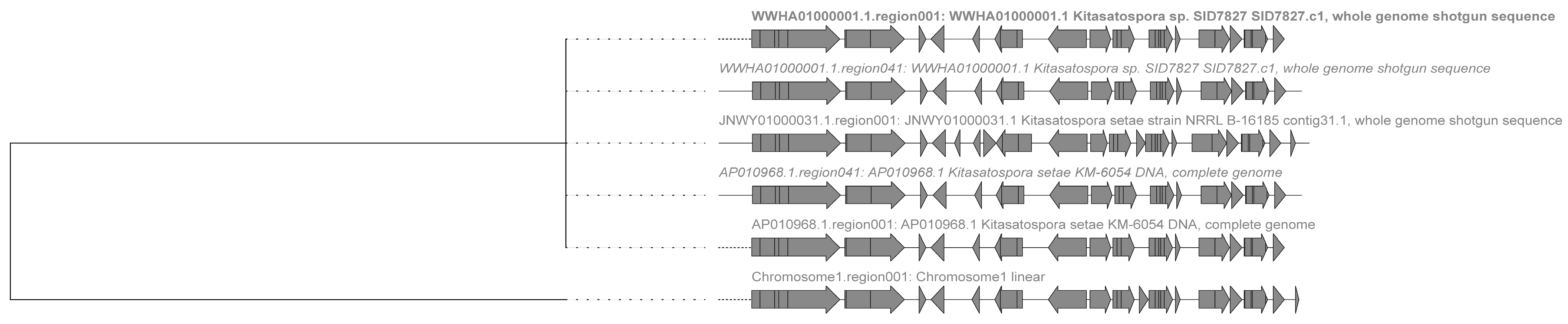
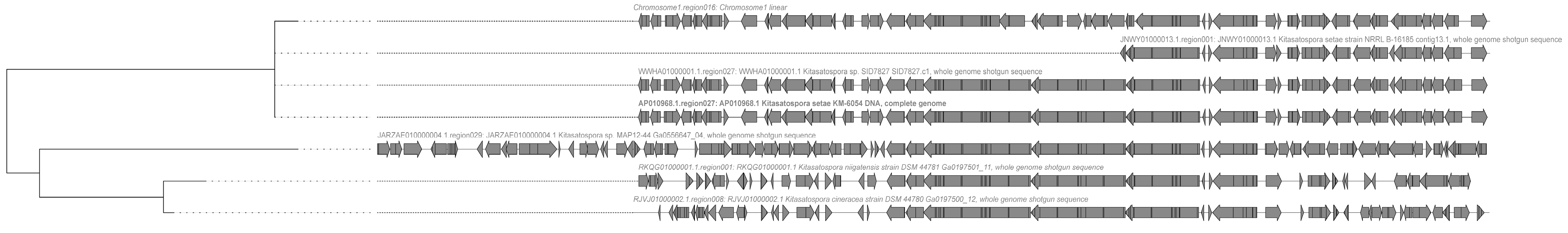
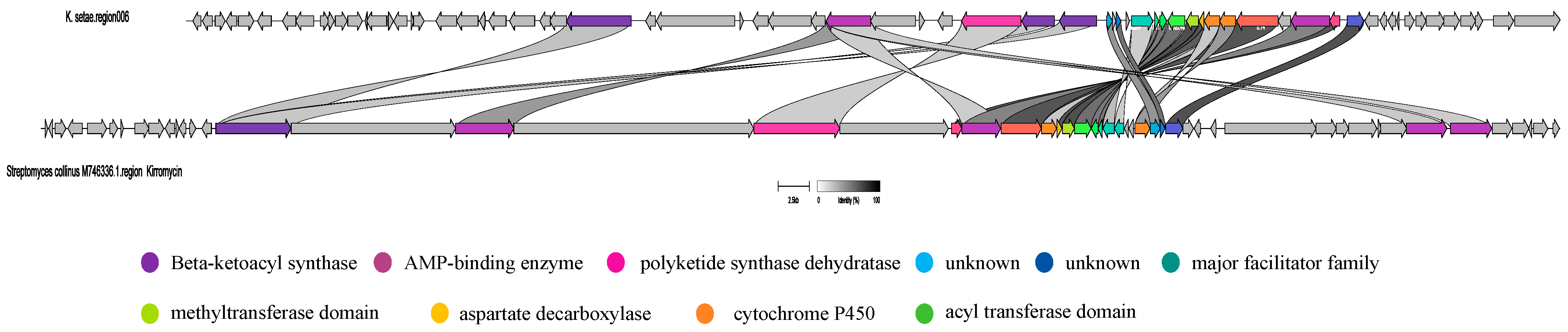
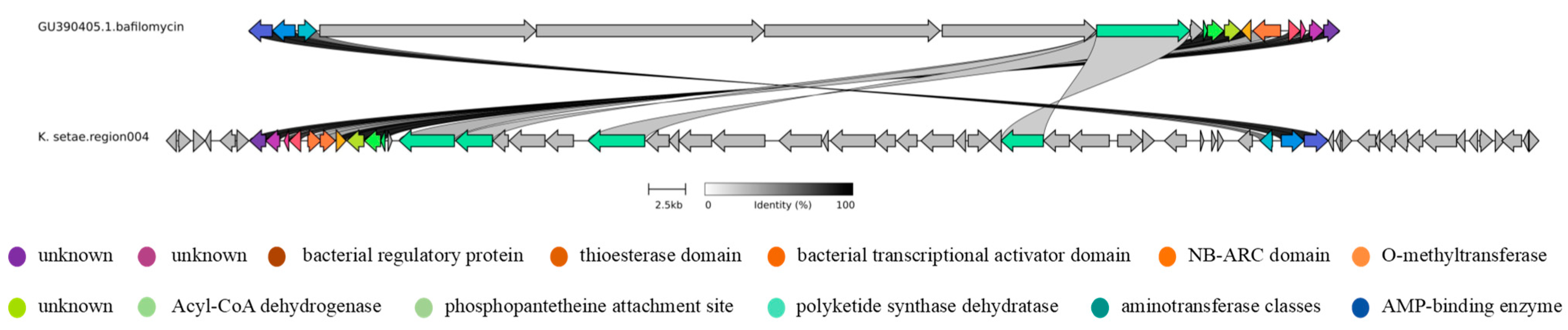
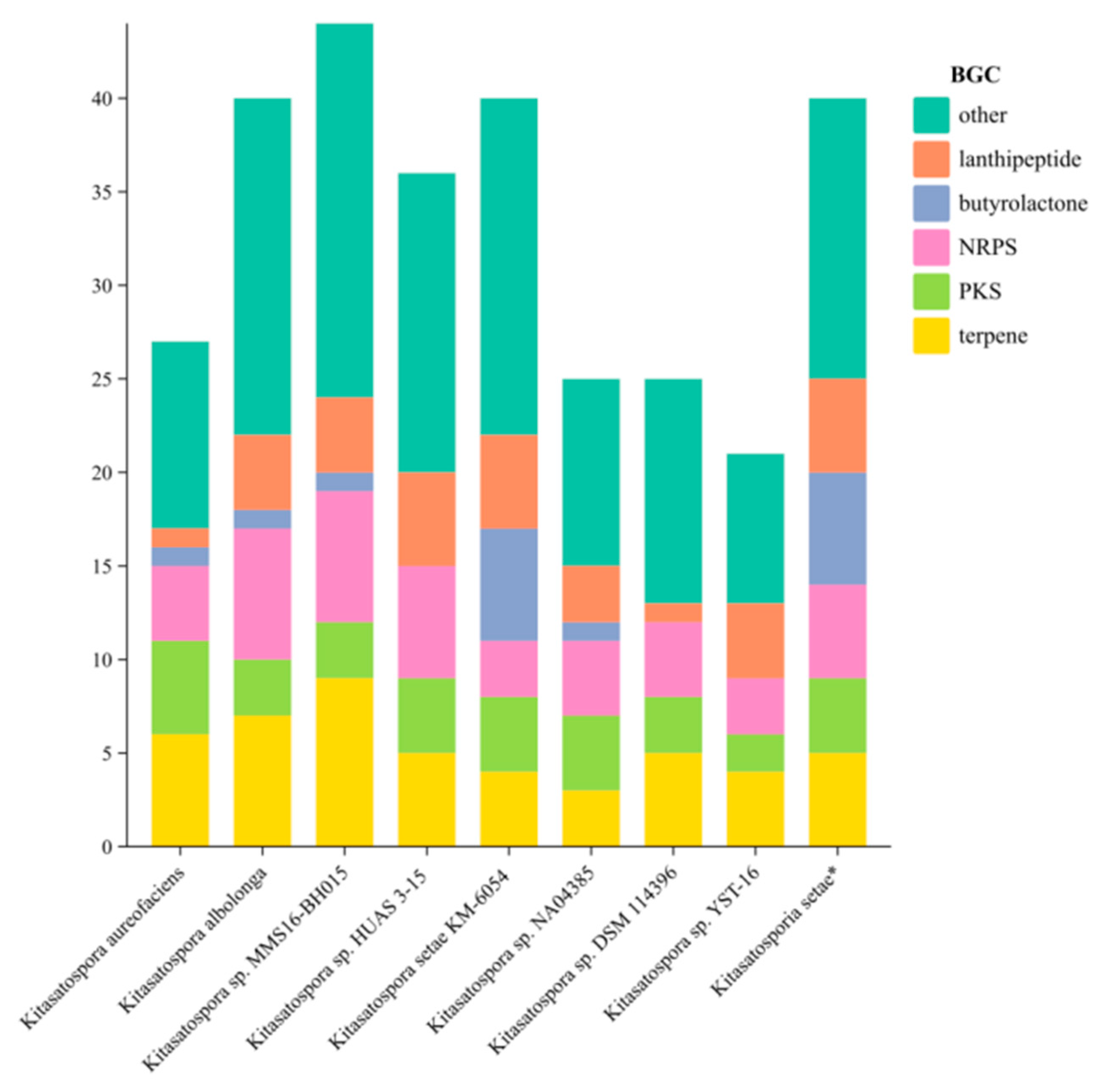
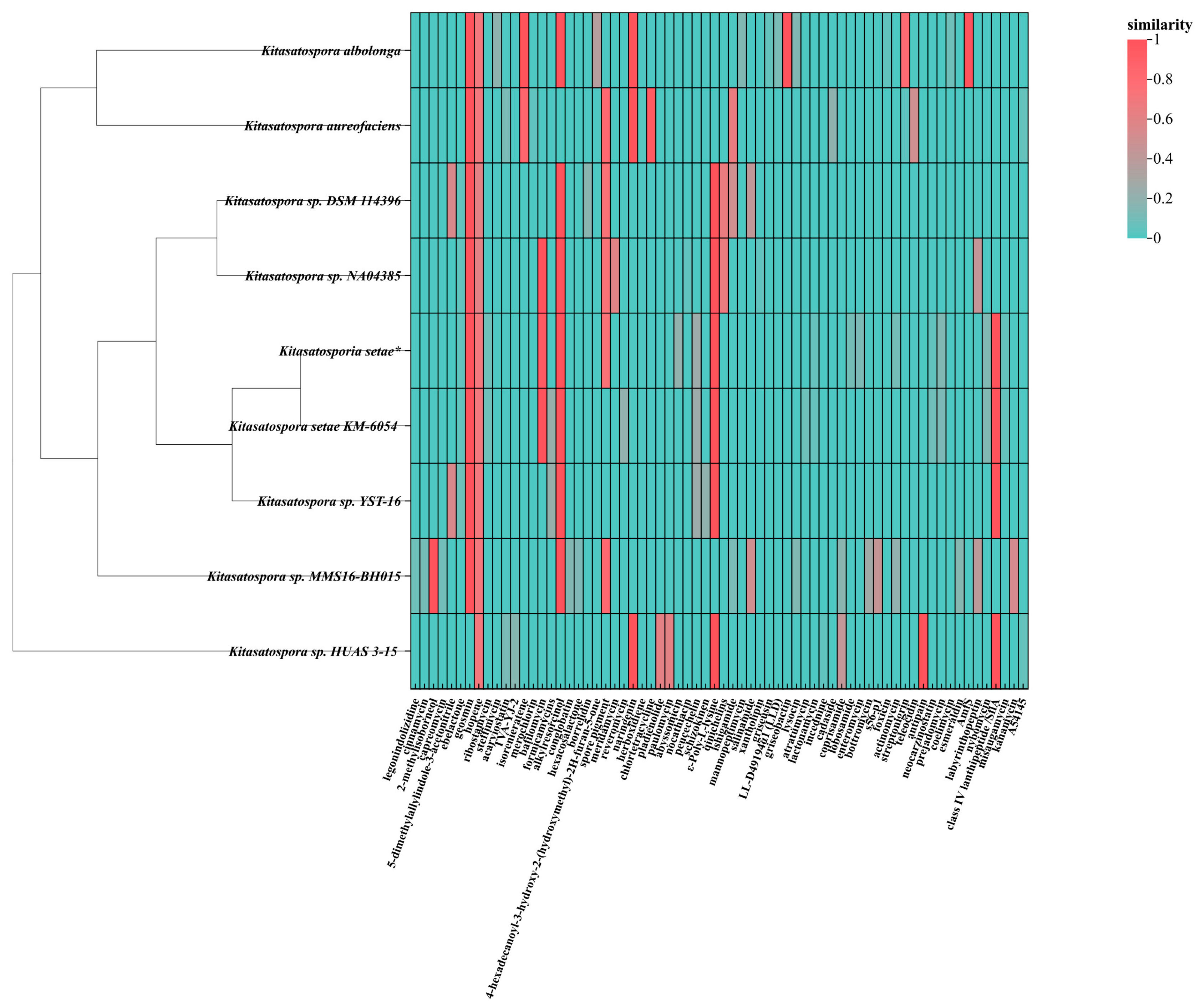
2.3. Analysis of Secondary Metabolite Biosynthetic Potential
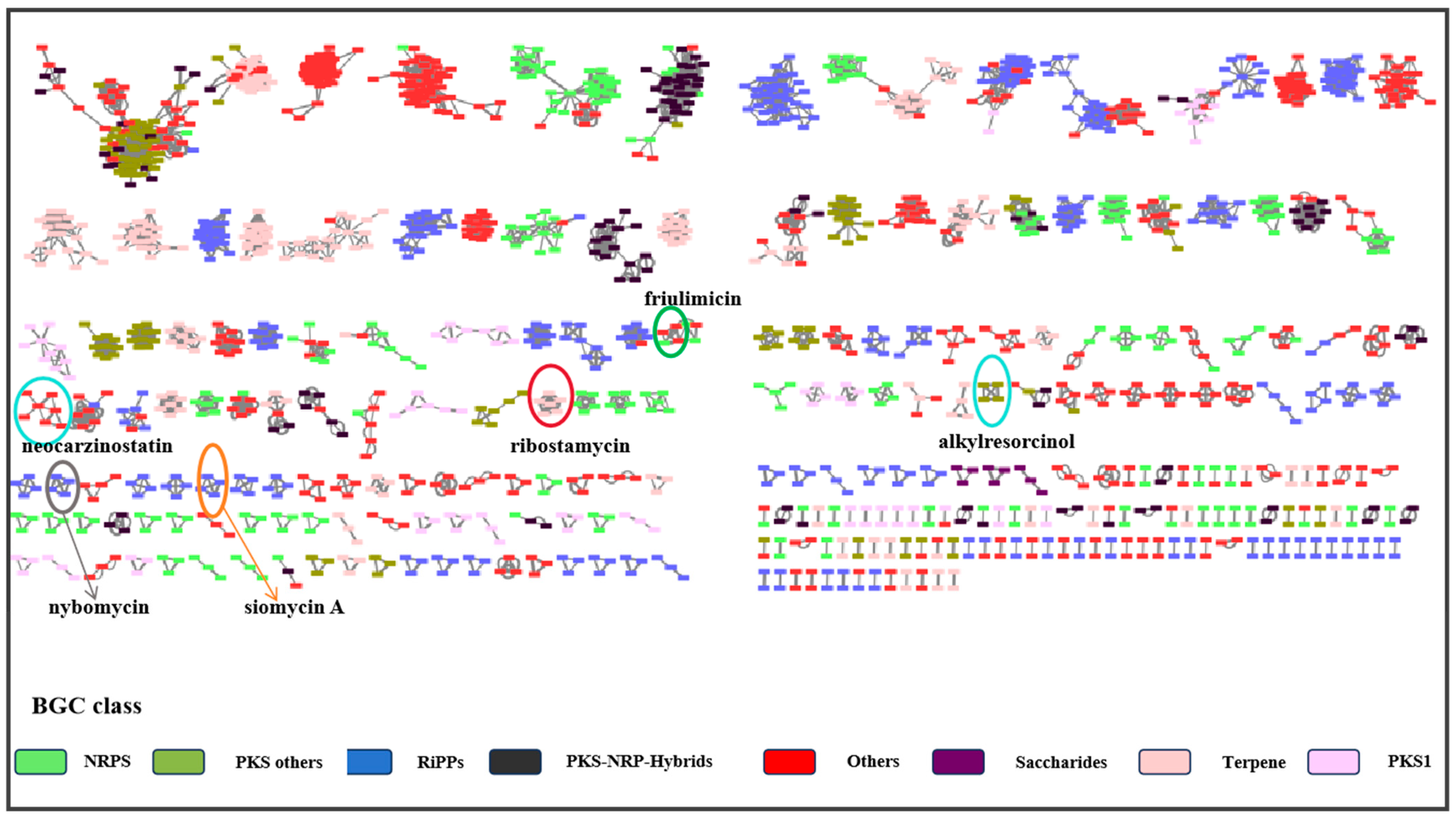
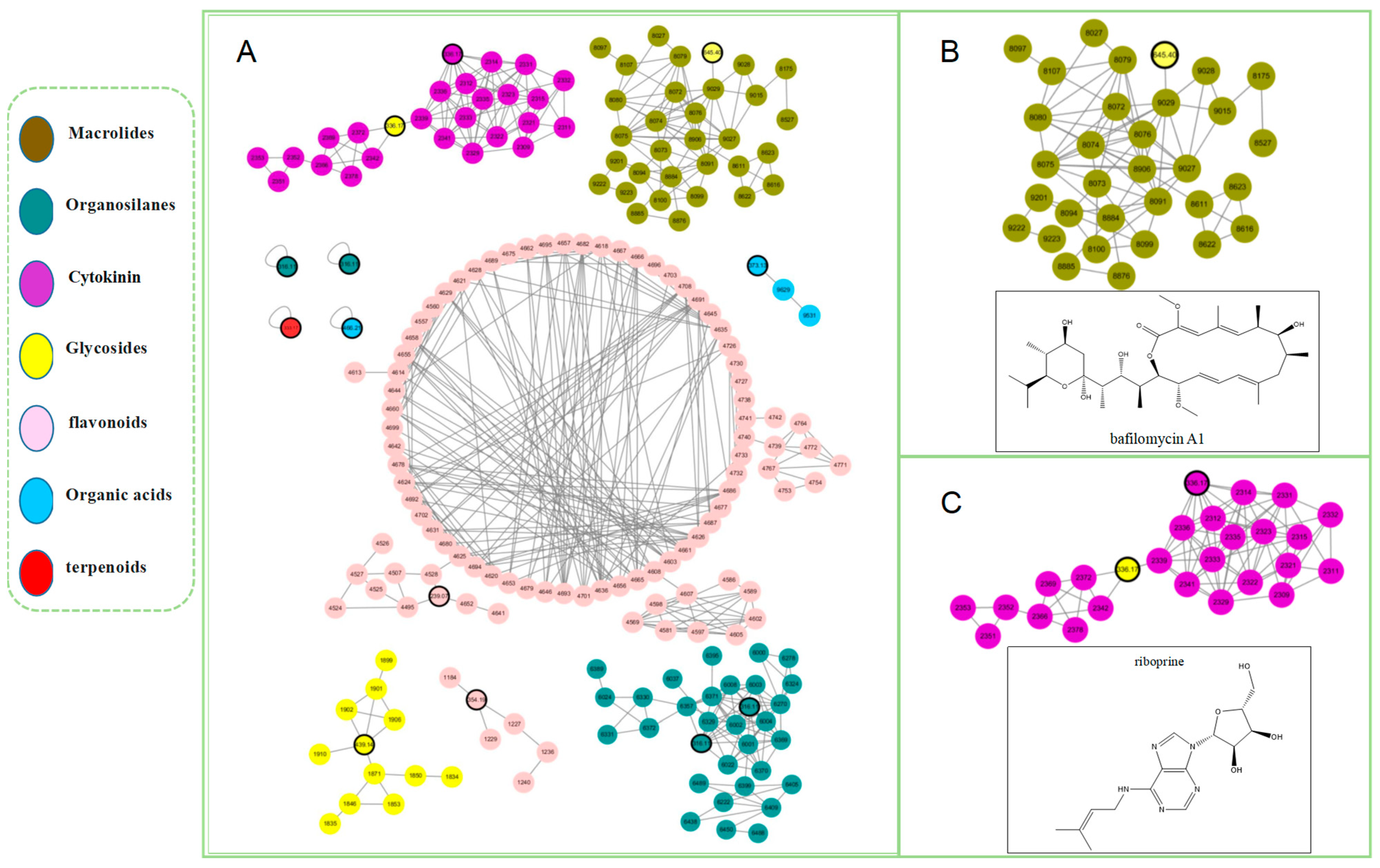
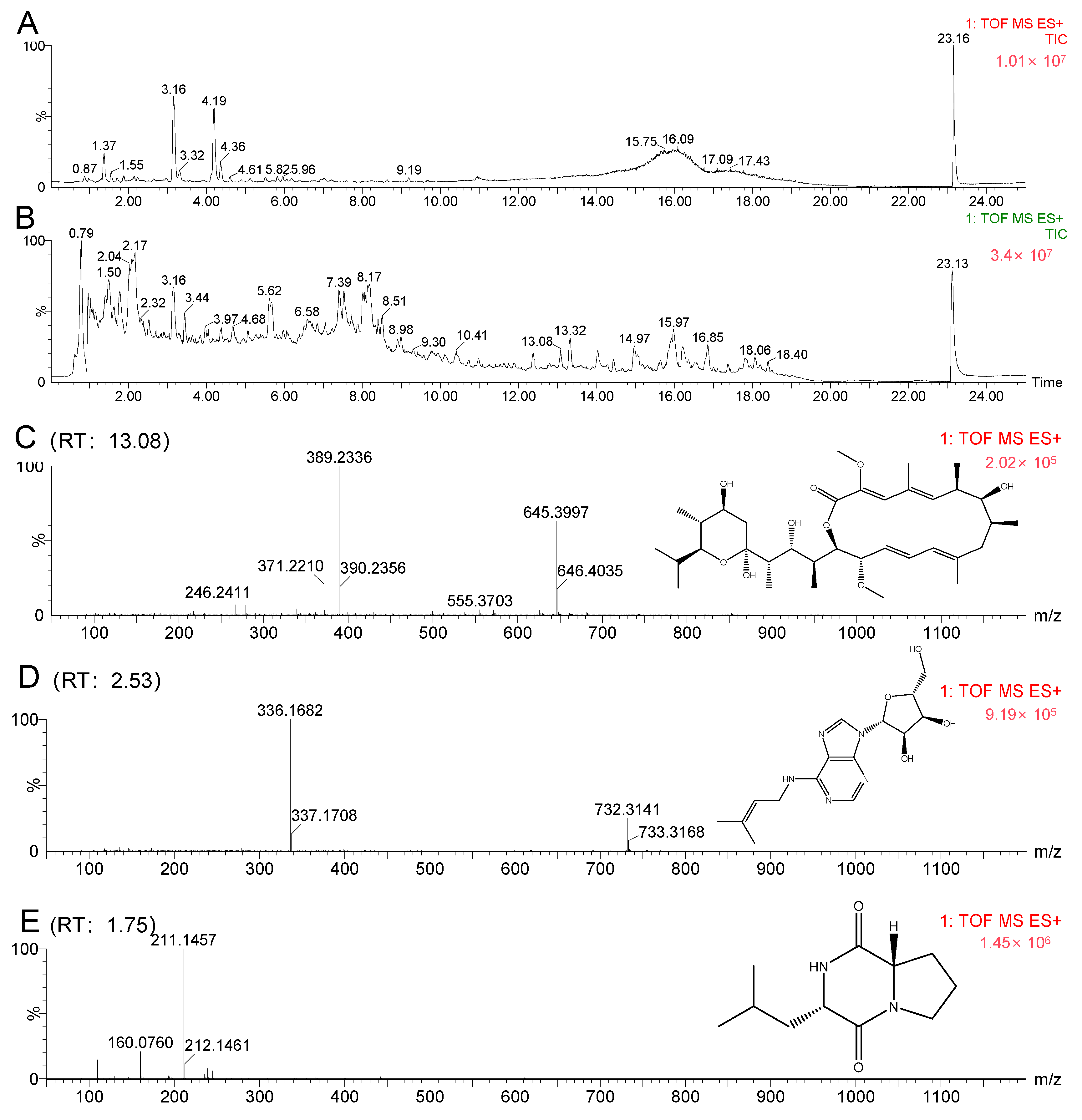
2.4. Molecular Networking Analysis
3. Discussion
4. Materials and Methods
4.1. Instruments Used in the Experiment
4.2. Rare Actinomycete Strains
4.3. Genomic DNA Preparation and Purification
4.4. High-Throughput Sequencing
4.5. Assembly and Genome Annotation
4.6. Prediction of Secondary Metabolite Gene Clusters
4.7. Chemical Analysis of Secondary Metabolites
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aslam, B.; Wang, W.; Arshad, M.I.; Khurshid, M.; Muzammil, S.; Rasool, M.H.; Nisar, M.A.; Alvi, R.F.; Aslam, M.A.; Qamar, M.U.; et al. Antibiotic resistance: A rundown of a global crisis. Infect. Drug Resist. 2018, 11, 1645–1658. [Google Scholar] [CrossRef] [PubMed]
- Van Bergeijk, D.A.; Terlouw, B.R.; Medema, M.H.; van Wezel, G.P. Ecology and genomics of Actinobacteria: New concepts for natural product discovery. Nat. Rev. Microbiol. 2020, 18, 546–558. [Google Scholar] [CrossRef]
- Vrancken, K.; Anné, J. Secretory production of recombinant proteins by Streptomyces. Future Microbiol. 2009, 4, 181–188. [Google Scholar] [CrossRef]
- Subramani, R.; Aalbersberg, W. Marine actinomycetes: An ongoing source of novel bioactive metabolites. Microbiol. Res. 2012, 167, 571–580. [Google Scholar] [CrossRef]
- Peng, D.; Li, S.; Wang, J.; Chen, C.; Zhou, M. Integrated biological and chemical control of rice sheath blight by Bacillus subtilis NJ-18 and jinggangmycin. Pest Manag. Sci. 2014, 70, 258–263. [Google Scholar] [CrossRef]
- Niu, G.; Li, W. Next-Generation Drug Discovery to Combat Antimicrobial Resistance. Trends Biochem. Sci. 2019, 44, 961–972. [Google Scholar] [CrossRef] [PubMed]
- Katz, L.; Baltz, R.H. Natural product discovery: Past, present, and future. J. Ind. Microbiol. Biotechnol. 2016, 43, 155–176. [Google Scholar] [CrossRef]
- Hao, X.; Yu, J.; Wang, Y.; Connolly, J.A.; Liu, Y.; Zhang, Y.; Yu, L.; Cen, S.; Goss, R.J.; Gan, M. Zelkovamycins B–E, Cyclic Octapeptides Containing Rare Amino Acid Residues from an Endophytic Kitasatospora sp. Org. Lett. 2020, 22, 9346–9350. [Google Scholar] [CrossRef]
- Watrous, J.; Roach, P.; Alexandrov, T.; Heath, B.S.; Yang, J.Y.; Kersten, R.D.; van der Voort, M.; Pogliano, K.; Gross, H.; Raaijmakers, J.M.; et al. Mass spectral molecular networking of living microbial colonies. Proc. Natl. Acad. Sci. USA 2012, 109, E1743–E1752. [Google Scholar] [CrossRef] [PubMed]
- Scherlach, K.; Hertweck, C. Mining and unearthing hidden biosynthetic potential. Nat. Commun. 2021, 12, 3864. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.M.; Qiao, Y.; Ang, E.L.; Zhao, H. Using natural products for drug discovery: The impact of the genomics era. Expert Opin. Drug Discov. 2017, 12, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Scherlach, K.; Hertweck, C. Triggering cryptic natural product biosynthesis in microorganisms. Org. Biomol. Chem. 2009, 7, 1753–1760. [Google Scholar] [CrossRef] [PubMed]
- Fenical, W.; Jensen, P.R. Developing a new resource for drug discovery: Marine actinomycete bacteria. Nat. Chem. Biol. 2006, 2, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.; Du, A.Q.; Zhou, Z.Y.; Lai, C.; Yu, W.C.; Yu, J.B.; Yu, Y.L.; Chen, J.W.; Zhang, H.W.; Xu, X.W.; et al. An atlas of bacterial secondary metabolite biosynthesis gene clusters. Environ. Microbiol. 2021, 23, 6981–6992. [Google Scholar] [CrossRef] [PubMed]
- Alam, K.; Hao, J.; Zhong, L.; Fan, G.; Ouyang, Q.; Islam, M.M.; Islam, S.; Sun, H.; Zhang, Y.; Li, R.; et al. Complete genome sequencing and in silico genome mining reveal the promising metabolic potential in Streptomyces strain CS-7. Front. Microbiol. 2022, 13, 939919. [Google Scholar] [CrossRef] [PubMed]
- Yun, B.R.; Malik, A.; Kim, S.B. Genome based characterization of Kitasatospora sp. MMS16-BH015, a multiple heavy metal resistant soil actinobacterium with high antimicrobial potential. Gene 2020, 733, 144379. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Zhang, H.; Wu, P.; Entwistle, S.; Li, X.; Yohe, T.; Yi, H.; Yang, Z.; Yin, Y. DbCAN-seq: A database of carbohydrate-active enzyme (CAZyme) sequence and annotation. Nucleic Acids Res. 2018, 46, D516–D521. [Google Scholar] [CrossRef] [PubMed]
- Ameri, R.; García, J.L.; Derenfed, A.B.; Pradel, N.; Neifar, S.; Mhiri, S.; Mezghanni, M.; Jaouadi, N.Z.; Barriuso, J.; Bejar, S. Genome sequence and Carbohydrate Active Enzymes (CAZymes) repertoire of the thermophilic Caldicoprobacter algeriensis TH7C1T. Microb. Cell Factories 2022, 21, 91. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Wu, Z.X.; Wei, L.; Chen, Z.S.; Yoganathan, S. Exploration of Antibiotic Activity of Aminoglycosides, in Particular Ribostamycin Alone and in Combination with Ethylenediaminetetraacetic Acid Against Pathogenic Bacteria. Front. Microbiol. 2020, 11, 1718. [Google Scholar] [CrossRef]
- Bemer, P.; Juvin, M.-E.; Bryskier, A.; Drugeon, H. In Vitro Activities of a New Lipopeptide, HMR 1043, against Susceptible and Resistant Gram-Positive Isolates. Antimicrob. Agents Chemother. 2003, 47, 3025–3029. [Google Scholar] [CrossRef]
- Nolden, S.; Wagner, N.; Biener, R.; Schwartz, D. Analysis of RegA, a pathway-specific regulator of the friulimicin biosynthesis in Actinoplanes friuliensis. J. Biotechnol. 2009, 140, 99–106. [Google Scholar] [CrossRef]
- Musiol, E.M.; Härtner, T.; Kulik, A.; Moldenhauer, J.; Piel, J.; Wohlleben, W.; Weber, T. Supramolecular Templating in Kirromycin Biosynthesis: The Acyltransferase KirCII Loads Ethylmalonyl-CoA Extender onto a Specific ACP of the trans-AT PKS. Chem. Biol. 2011, 18, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Laezza, C.; Caruso, M.G.; Gentile, T.; Notarnicola, M.; Malfitano, A.M.; Di Matola, T.; Messa, C.; Gazzerro, P.; Bifulco, M. N6-isopentenyladenosine inhibits cell proliferation and induces apoptosis in a human colon cancer cell line DLD1. Int. J. Cancer 2009, 124, 1322–1329. [Google Scholar] [CrossRef]
- Laezza, C.; Malfitano, A.M.; Di Matola, T.; Ricchi, P.; Bifulco, M. Involvement of Akt/NF-κB pathway in N6-isopentenyladenosine-induced apoptosis in human breast cancer cells. Mol. Carcinog. 2010, 49, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Castiglioni, S.; Casati, S.; Ottria, R.; Ciuffreda, P.; Maier, J.A. N6-isopentenyladenosine and its analogue N6-benzyladenosine induce cell cycle arrest and apoptosis in bladder carcinoma T24 cells. Anti-Cancer Agents Med. Chem. 2013, 13, 672–678. [Google Scholar] [CrossRef]
- Ranieri, R.; Ciaglia, E.; Amodio, G.; Picardi, P.; Proto, M.C.; Gazzerro, P.; Pisanti, S. N6-isopentenyladenosine dual targeting of AMPK and Rab7 prenylation inhibits melanoma growth through the impairment of autophagic flux. Cell Death Differ. 2018, 25, 353–367. [Google Scholar] [CrossRef]
- Dymarska, M.; Janeczko, T.; Kostrzewa-Susłow, E. Glycosylation of 3-Hydroxyflavone, 3-Methoxyflavone, Quercetin and Baicalein in Fungal Cultures of the Genus Isaria. Molecules 2018, 23, 2477. [Google Scholar] [CrossRef] [PubMed]
- Reghunath, S.; Siji, J.V.; Mohandas, C.; Nambisan, B. Isolation and Identification of Bioactive Molecules Produced by Entomopathogenic bacteria, Acinetobacter calcoaceticus. Appl. Microbiol. 2017, 3, 1000134. [Google Scholar] [CrossRef]
- Kononova, L.I.; Filatova, L.B.; Eroshenko, D.V.; Korobov, V.P. Suppression of development of vancomycin-resistant Staphylococcus epidermidis by low-molecular-weight cationic peptides of the lantibiotic family. Microbiology 2017, 86, 571–582. [Google Scholar] [CrossRef]
- Draper, L.A.; Cotter, P.D.; Hill, C.; Ross, R.P. Lantibiotic Resistance. Microbiol. Mol. Biol. Rev. 2015, 79, 171–191. [Google Scholar] [CrossRef]
- Tian, S.; Sun, Y.; Chen, Z.; Zhao, R. Bioavailability and Bioactivity of Alkylresorcinols from Different Cereal Products. J. Food Qual. 2020, 2020, 5781356. [Google Scholar] [CrossRef]
- Gerber, N.N.; Lechevalier, H.A. Geosmin, an earthly-smelling substance isolated from actinomycetes. Appl. Microbiol. 1965, 13, 935–938. [Google Scholar] [CrossRef] [PubMed]
- Abdulsalam, O.; Wagner, K.; Wirth, S.; Kunert, M.; David, A.; Kallenbach, M.; Boland, W.; Kothe, E.; Krause, K. Phytohormones and volatile organic compounds, like geosmin, in the ectomycorrhiza of Tricholoma vaccinum and Norway spruce (Picea abies). Mycorrhiza 2021, 31, 173–188. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Wang, F.; Tan, Z.; Cui, J.; Jia, S. Antifungal mechanisms of ε-poly-L-Lysine with different molecular weights on Saccharomyces cerevisiae. Korean J. Chem. Eng. 2020, 37, 482–492. [Google Scholar] [CrossRef]
- Yoshida, T.; Nagasawa, T. ε-Poly-l-lysine: Microbial production, biodegradation and application potential. Appl. Microbiol. 2003, 62, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Musiol, E.M.; Greule, A.; Härtner, T.; Kulik, A.; Wohlleben, W.; Weber, T. The AT2 domain of KirCI loads malonyl extender units to the ACPs of the kirromycin PKS. ChemBioChem 2013, 14, 1343–1352. [Google Scholar] [CrossRef] [PubMed]
- Nara, A.; Hashimoto, T.; Komatsu, M.; Nishiyama, M.; Kuzuyama, T.; Ikeda, H. Characterization of bafilomycin biosynthesis in Kitasatospora setae KM-6054 and comparative analysis of gene clusters in Actinomycetales microorganisms. J. Antibiot. 2017, 70, 616–624. [Google Scholar] [CrossRef]
- Zhang, W.; Fortman, J.L.; Carlson, J.C.; Yan, J.; Liu, Y.; Bai, F.; Guan, W.; Jia, J.; Matainaho, T.; Sherman, D.H.; et al. Characterization of the Bafilomycin Biosynthetic Gene Cluster from Streptomyces lohii. ChemBioChem 2013, 14, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Augustijn, H.E.; Reitz, Z.L.; Biermann, F.; Alanjary, M.; Fetter, A.; Terlouw, B.R.; Metcalf, W.W.; Helfrich, E.J.; et al. antiSMASH 7.0: New and improved predictions for detection, regulation, chemical structures and visualisation. Nucleic Acids Res. 2023, 51, 46–50. [Google Scholar] [CrossRef]
- Navarro-Muñoz, J.C.; Selem-Mojica, N.; Mullowney, M.W.; Kautsar, S.A.; Tryon, J.H.; Parkinson, E.I.; De Los Santos, E.L.; Yeong, M.; Cruz-Morales, P.; Abubucker, S.; et al. A computational framework to explore large-scale biosynthetic diversity. Nat. Chem. Biol. 2020, 16, 60. [Google Scholar] [CrossRef]
- Wei, B.; Du, A.Q.; Ying, T.T.; Hu, G.A.; Zhou, Z.Y.; Yu, W.C.; He, J.; Yu, Y.L.; Wang, H.; Xu, X.W. Secondary Metabolic Potential of Kutzneria. J. Nat. Prod. 2023, 86, 1120–1127. [Google Scholar] [CrossRef] [PubMed]
- Nothias, L.F.; Petras, D.; Schmid, R.; Dührkop, K.; Rainer, J.; Sarvepalli, A.; Protsyuk, I.; Ernst, M.; Tsugawa, H.; Fleischauer, M.; et al. Feature-based molecular networking in the GNPS analysis environment. Nat. Methods 2020, 17, 905–908. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, Y.; Zhou, Z.; Feng, F.; Zhao, H.; Tan, S.; Li, J.; Wu, S.; Ju, Z.; He, S.; Ding, L. Genomic Analysis of Kitasatospora setae to Explore Its Biosynthetic Potential Regarding Secondary Metabolites. Antibiotics 2024, 13, 459. https://doi.org/10.3390/antibiotics13050459
Xue Y, Zhou Z, Feng F, Zhao H, Tan S, Li J, Wu S, Ju Z, He S, Ding L. Genomic Analysis of Kitasatospora setae to Explore Its Biosynthetic Potential Regarding Secondary Metabolites. Antibiotics. 2024; 13(5):459. https://doi.org/10.3390/antibiotics13050459
Chicago/Turabian StyleXue, Yutong, Zhiyan Zhou, Fangjian Feng, Hang Zhao, Shuangling Tan, Jinling Li, Sitong Wu, Zhiran Ju, Shan He, and Lijian Ding. 2024. "Genomic Analysis of Kitasatospora setae to Explore Its Biosynthetic Potential Regarding Secondary Metabolites" Antibiotics 13, no. 5: 459. https://doi.org/10.3390/antibiotics13050459
APA StyleXue, Y., Zhou, Z., Feng, F., Zhao, H., Tan, S., Li, J., Wu, S., Ju, Z., He, S., & Ding, L. (2024). Genomic Analysis of Kitasatospora setae to Explore Its Biosynthetic Potential Regarding Secondary Metabolites. Antibiotics, 13(5), 459. https://doi.org/10.3390/antibiotics13050459