
The Impact of Urban Pollution on Plasmid-Mediated Resistance Acquisition in Enterobacteria from a Tropical River

 ,
,  , , and
, , and
Abstract
1. Introduction
2. Results
2.1. Genome Characterization and Taxonomic Identification
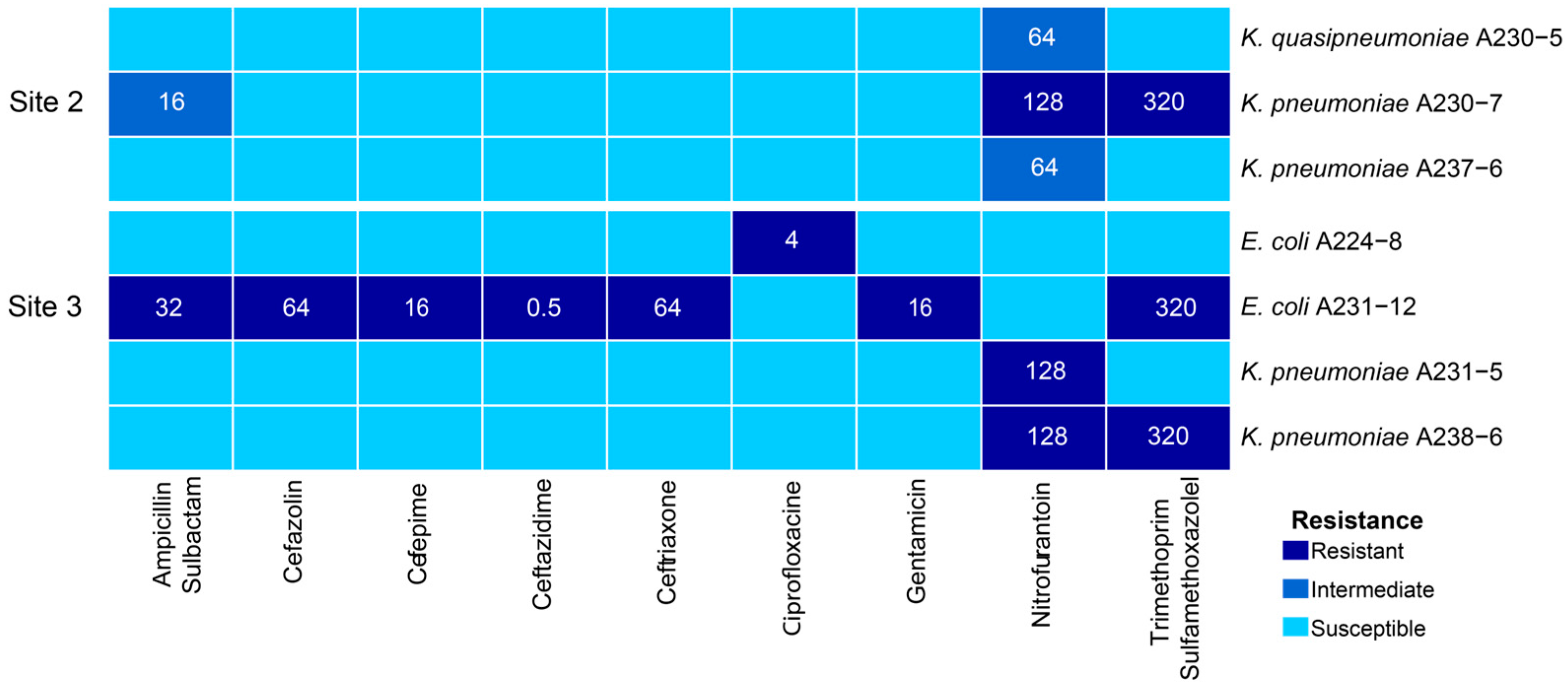
2.2. Phenotypic Characterization of Antibiotic Resistance
2.3. Genotypic Characterization of Antibiotic Resistance
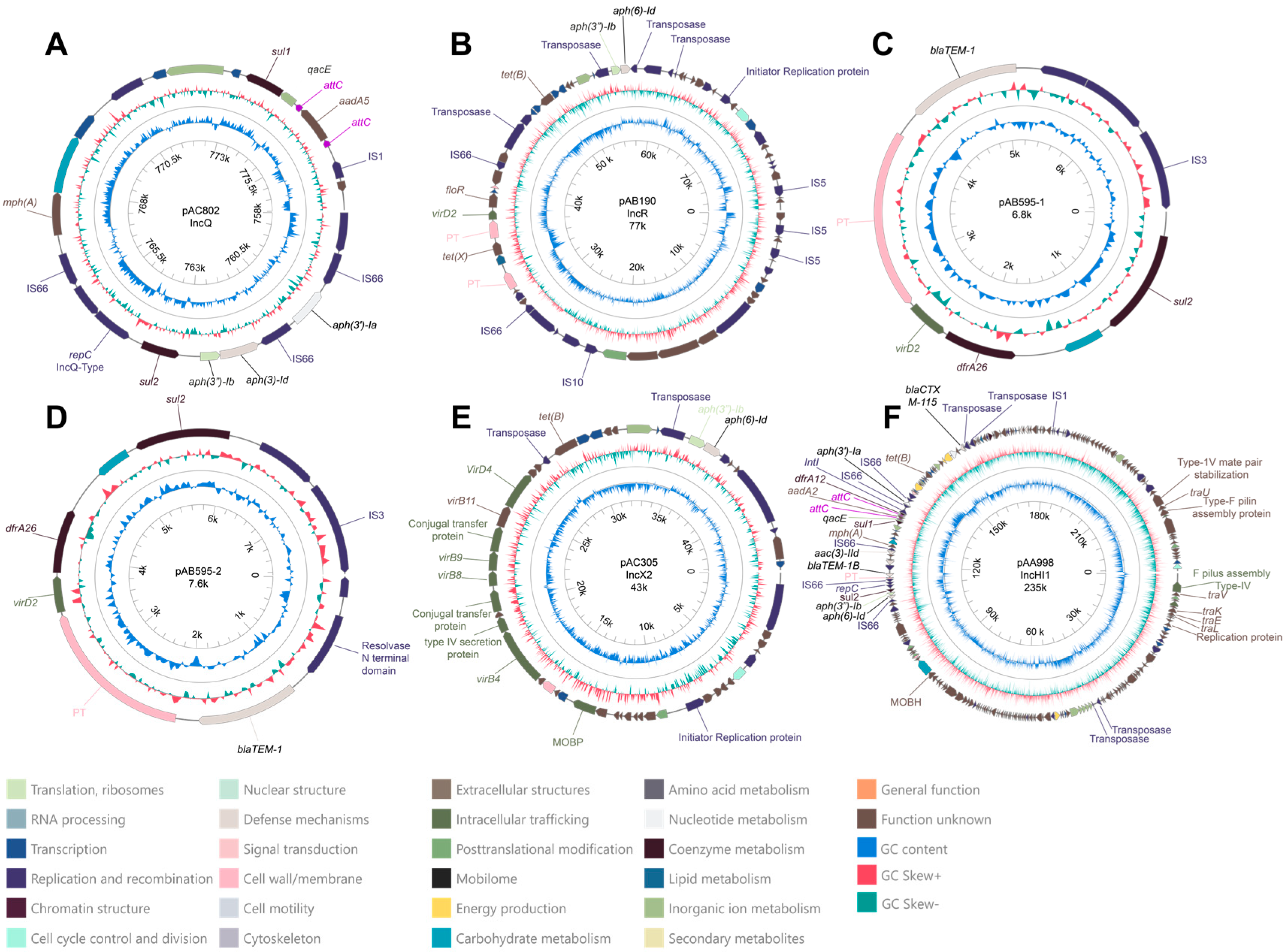
2.4. Description of Plasmids with Antibiotic-Resistance Genes
2.4.1. Plasmid pAC082
2.4.2. Plasmid pAB190
2.4.3. Plasmids pAB595-1 and pAB595-2
2.4.4. Plasmid pAC305
2.4.5. Description and Comparative Analysis of Plasmid pAA998
3. Discussion
4. Materials and Methods
4.1. Sampling Sites
4.2. Sample Processing and Bacterial Isolation
4.3. Biochemical and Serological Analysis
4.4. Chemical Analysis
4.5. Anthropogenic Contaminant Detection by Mass Spectrometry
4.6. DNA Extraction and Sequencing
4.7. Quality Control of Raw Data
4.8. Assembly and Quality Evaluation
4.9. Taxonomic Identity and Phylogenomic Analysis
4.10. Antibiotic Susceptibility Testing
4.11. Identification of Antibiotic-Resistance Genes (ARGs) and Mobile Genetic Elements (MGEs)
4.12. Search for Mutations Conferring an Antibiotic-Resistance Phenotype
4.13. Sequencing of A224-8 and A231-12 Isolates Through PacBio Technology for Plasmid Confirmation
4.14. Genomic Comparison of Plasmid pAA998
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Persson, L.; Carney Almroth, B.M.; Collins, C.D.; Cornell, S.; de Wit, C.A.; Diamond, M.L.; Fantke, P.; Hassellöv, M.; MacLeod, M.; Ryberg, M.W.; et al. Outside the Safe Operating Space of the Planetary Boundary for Novel Entities. Environ. Sci. Technol. 2022, 56, 1510–1521. [Google Scholar] [CrossRef] [PubMed]
- Qiu, D.; Ke, M.; Zhang, Q.; Zhang, F.; Lu, T.; Sun, L.; Qian, H. Response of Microbial Antibiotic Resistance to Pesticides: An Emerging Health Threat. Sci. Total Environ. 2022, 850, 158057. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lu, J.; Zhang, S.; Li, J.; Mao, L.; Yuan, Z.; Bond, P.L.; Guo, J. Non-Antibiotic Pharmaceuticals Promote the Transmission of Multidrug Resistance Plasmids through Intra- and Intergenera Conjugation. ISME J. 2021, 15, 2493–2508. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.; Huang, H.; Chen, Y. Effects of Emerging Pollutants on the Occurrence and Transfer of Antibiotic Resistance Genes: A Review. J. Hazard. Mater. 2021, 420, 126602. [Google Scholar] [CrossRef] [PubMed]
- Strokal, M.; Bai, Z.; Franssen, W.; Hofstra, N.; Koelmans, A.A.; Ludwig, F.; Ma, L.; van Puijenbroek, P.; Spanier, J.E.; Vermeulen, L.C.; et al. Urbanization: An Increasing Source of Multiple Pollutants to Rivers in the 21st Century. Npj Urban Sustain. 2021, 1, 1–13. [Google Scholar] [CrossRef]
- Wilkinson, J.L.; Boxall, A.B.A.; Kolpin, D.W.; Leung, K.M.Y.; Lai, R.W.S.; Galbán-Malagón, C.; Adell, A.D.; Mondon, J.; Metian, M.; Marchant, R.A.; et al. Pharmaceutical Pollution of the World’s Rivers. Proc. Natl. Acad. Sci. USA 2022, 119, e2113947119. [Google Scholar] [CrossRef]
- Buelow, E.; Ploy, M.-C.; Dagot, C. Role of Pollution on the Selection of Antibiotic Resistance and Bacterial Pathogens in the Environment. Curr. Opin. Microbiol. 2021, 64, 117–124. [Google Scholar] [CrossRef]
- Abe, K.; Nomura, N.; Suzuki, S. Biofilms: Hot Spots of Horizontal Gene Transfer (HGT) in Aquatic Environments, with a Focus on a New HGT Mechanism. FEMS Microbiol. Ecol. 2020, 96, fiaa031. [Google Scholar] [CrossRef]
- Metzger, G.A.; Ridenhour, B.J.; France, M.; Gliniewicz, K.; Millstein, J.; Settles, M.L.; Forney, L.J.; Stalder, T.; Top, E.M. Biofilms Preserve the Transmissibility of a Multi-Drug Resistance Plasmid. npj Biofilms Microbiomes 2022, 8, 95. [Google Scholar] [CrossRef]
- Iredell, J.; Brown, J.; Tagg, K. Antibiotic Resistance in Enterobacteriaceae: Mechanisms and Clinical Implications. BMJ 2016, 352, h6420. [Google Scholar] [CrossRef]
- Mena-Rivera, L.; Vásquez-Bolaños, O.; Gómez-Castro, C.; Fonseca-Sánchez, A.; Rodríguez-Rodríguez, A.; Sánchez-Gutiérrez, R. Ecosystemic Assessment of Surface Water Quality in the Virilla River: Towards Sanitation Processes in Costa Rica. Water 2018, 10, 845. [Google Scholar] [CrossRef]
- Herrera-Murillo, J.; Anchía-Leitón, D.; Rojas-Marín, J.F.; Mora-Campos, D.; Gamboa-Jiménez, A.A.; Chaves-Villalobos, M. Influencia de los patrones de uso de la tierra en la calidad de las aguas superfciales de la subcuenca del río Virilla, Costa Rica. Geogr. J. Cent. Am. 2018, 4, 11–35. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. TYGS Is an Automated High-Throughput Platform for State-of-the-Art Genome-Based Taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef]
- Nogrado, K.; Cho, A.; Han, D.; Ho, C.T.; Fredrickson, J.K.; Lee, J.-H. Complete Genome Sequence of Shigella Sonnei Strain SE6-1, Capable of Selenate Reduction. Microbiol. Resour. Announc. 2021, 10, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Magiorakos, A.-P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-Resistant, Extensively Drug-Resistant and Pandrug-Resistant Bacteria: An International Expert Proposal for Interim Standard Definitions for Acquired Resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef]
- Weinstein, M.P.; Lewis, J.S., II; Bobenchik, A.M.; Campeau, S.; Cullen, S.K.; Galas, M.F.; Gold, H.; Humphries, R.M.; Kirn, T.J.; Limbago, B.; et al. Performance Standards for Antimicrobial Susceptibility Testing; Clinical & Laboratory Standards Institute: Wayne, PA, USA, 2024. [Google Scholar]
- Ho, P.-L.; Ng, K.-Y.; Lo, W.-U.; Law, P.Y.; Lai, E.L.-Y.; Wang, Y.; Chow, K.-H. Plasmid-Mediated OqxAB Is an Important Mechanism for Nitrofurantoin Resistance in Escherichia coli. Antimicrob. Agents Chemother. 2015, 60, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Hansen, L.H.; Johannesen, E.; Burmølle, M.; Sørensen, A.H.; Sørensen, S.J. Plasmid-Encoded Multidrug Efflux Pump Conferring Resistance to Olaquindox in Escherichia coli. Antimicrob. Agents Chemother. 2004, 48, 3332–3337. [Google Scholar] [CrossRef] [PubMed]
- Varani, A.; He, S.; Siguier, P.; Ross, K.; Chandler, M. The IS6 Family, a Clinically Important Group of Insertion Sequences Including IS26. Mob. DNA 2021, 12, 11. [Google Scholar] [CrossRef]
- Razavi, M.; Kristiansson, E.; Flach, C.-F.; Larsson, D.G.J. The Association between Insertion Sequences and Antibiotic Resistance Genes. mSphere 2020, 5, e00418-20. [Google Scholar] [CrossRef]
- Rozwandowicz, M.; Brouwer, M.S.M.; Fischer, J.; Wagenaar, J.A.; Gonzalez-Zorn, B.; Guerra, B.; Mevius, D.J.; Hordijk, J. Plasmids Carrying Antimicrobial Resistance Genes in Enterobacteriaceae. J. Antimicrob. Chemother. 2018, 73, 1121–1137. [Google Scholar] [CrossRef]
- Huttner, A.; Verhaegh, E.M.; Harbarth, S.; Muller, A.E.; Theuretzbacher, U.; Mouton, J.W. Nitrofurantoin Revisited: A Systematic Review and Meta-Analysis of Controlled Trials. J. Antimicrob. Chemother. 2015, 70, 2456–2464. [Google Scholar] [CrossRef] [PubMed]
- Mujeeb Rahiman, K.M.; Mohamed Hatha, A.A.; Gnana Selvam, A.D.; Thomas, A.P. Relative Prevalence of Antibiotic Resistance among Heterotrophic Bacteria from Natural and Culture Environments of Freshwater Prawn, Macrobrachium rosenbergii. J. World Aquac. Soc. 2016, 47, 470–480. [Google Scholar] [CrossRef]
- de Souza, Z.N.; de Moura, D.F.; de Almeida Campos, L.A.; Córdula, C.R.; Cavalcanti, I.M.F. Antibiotic Resistance Profiles on Pathogenic Bacteria in the Brazilian Environments. Arch. Microbiol. 2023, 205, 185. [Google Scholar] [CrossRef]
- Osei Sekyere, J. Genomic Insights into Nitrofurantoin Resistance Mechanisms and Epidemiology in Clinical Enterobacteriaceae. Future Sci. OA 2018, 4, FSO293. [Google Scholar] [CrossRef]
- Sandegren, L.; Lindqvist, A.; Kahlmeter, G.; Andersson, D.I. Nitrofurantoin Resistance Mechanism and Fitness Cost in Escherichia coli. J. Antimicrob. Chemother. 2008, 62, 495–503. [Google Scholar] [CrossRef]
- dos Santos, C.; dos Santos, L.S.; Franco, O.L. Fosfomycin and Nitrofurantoin: Classic Antibiotics and Perspectives. J. Antibiot. 2021, 74, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Smilack, J.D. Trimethoprim-Sulfamethoxazole. Mayo Clin. Proc. 1999, 74, 730–734. [Google Scholar] [CrossRef]
- Shin, H.W.; Lim, J.; Kim, S.; Kim, J.; Kwon, G.C.; Koo, S.H. Characterization of Trimethoprim-Sulfamethoxazole Resistance Genes and Their Relatedness to Class 1 Integron and Insertion Sequence Common Region in Gram-Negative Bacilli. J. Microbiol. Biotechnol. 2015, 25, 137–142. [Google Scholar] [CrossRef] [PubMed]
- de los Santos, E.; Laviña, M.; Poey, M.E. Strict Relationship between Class 1 Integrons and Resistance to Sulfamethoxazole in Escherichia coli. Microb. Pathog. 2021, 161, 105206. [Google Scholar] [CrossRef]
- Lye, Y.L.; Bong, C.W.; Lee, C.W.; Zhang, R.J.; Zhang, G.; Suzuki, S.; Chai, L.C. Anthropogenic Impacts on Sulfonamide Residues and Sulfonamide Resistant Bacteria and Genes in Larut and Sangga Besar River, Perak. Sci. Total Environ. 2019, 688, 1335–1347. [Google Scholar] [CrossRef]
- Hoa, P.T.P.; Managaki, S.; Nakada, N.; Takada, H.; Shimizu, A.; Anh, D.H.; Viet, P.H.; Suzuki, S. Antibiotic Contamination and Occurrence of Antibiotic-Resistant Bacteria in Aquatic Environments of Northern Vietnam. Sci. Total Environ. 2011, 409, 2894–2901. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Cui, F.; Zeng, G.; Jiang, M.; Yang, Z.; Yu, Z.; Zhu, M.; Shen, L. Quaternary Ammonium Compounds (QACs): A Review on Occurrence, Fate and Toxicity in the Environment. Sci. Total Environ. 2015, 518–519, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Cantón, R.; Ruiz-Garbajosa, P. Co-Resistance: An Opportunity for the Bacteria and Resistance Genes. Curr. Opin. Pharmacol. 2011, 11, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Bush, K.; Jacoby, G.A. Updated Functional Classification of β-Lactamases. Antimicrob. Agents Chemother. 2010, 54, 969–976. [Google Scholar] [CrossRef] [PubMed]
- EVILABRA. Reporte de La Estrategia Para La Vigilancia de Laboratorio de La RAM (EVILABRA); 2018–2022; INCIENSA: San José, Costa Rica, 2023; p. 29. [Google Scholar]
- Korzeniewska, E.; Harnisz, M. Extended-Spectrum Beta-Lactamase (ESBL)-Positive Enterobacteriaceae in Municipal Sewage and Their Emission to the Environment. J. Environ. Manag. 2013, 128, 904–911. [Google Scholar] [CrossRef]
- Amos, G.C.A.; Hawkey, P.M.; Gaze, W.H.; Wellington, E.M. Waste Water Effluent Contributes to the Dissemination of CTX-M-15 in the Natural Environment. J. Antimicrob. Chemother. 2014, 69, 1785–1791. [Google Scholar] [CrossRef]
- Cho, S.; Jackson, C.R.; Frye, J.G. Freshwater Environment as a Reservoir of Extended-Spectrum β-Lactamase-Producing Enterobacteriaceae. J. Appl. Microbiol. 2023, 134, lxad034. [Google Scholar] [CrossRef]
- Szczepanowski, R.; Linke, B.; Krahn, I.; Gartemann, K.-H.; Gützkow, T.; Eichler, W.; Pühler, A.; Schlüter, A. Detection of 140 Clinically Relevant Antibiotic-Resistance Genes in the Plasmid Metagenome of Wastewater Treatment Plant Bacteria Showing Reduced Susceptibility to Selected Antibiotics. Microbiology 2009, 155, 2306–2319. [Google Scholar] [CrossRef]
- Tennstedt, T.; Szczepanowski, R.; Braun, S.; Pühler, A.; Schlüter, A. Occurrence of Integron-Associated Resistance Gene Cassettes Located on Antibiotic Resistance Plasmids Isolated from a Wastewater Treatment Plant. FEMS Microbiol. Ecol. 2003, 45, 239–252. [Google Scholar] [CrossRef]
- Ramsay, J.P.; Firth, N. Diverse Mobilization Strategies Facilitate Transfer of Non-Conjugative Mobile Genetic Elements. Curr. Opin. Microbiol. 2017, 38, 1–9. [Google Scholar] [CrossRef]
- Paganini, J.A.; Plantinga, N.L.; Arredondo-Alonso, S.; Willems, R.J.L.; Schürch, A.C. Recovering Escherichia coli Plasmids in the Absence of Long-Read Sequencing Data. Microorganisms 2021, 9, 1613. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.; Zdarska, V.; Kolar, M.; Mlynarcik, P. Analysis of BlaEC Family Class C Beta-Lactamase. FEMS Microbiol. Lett. 2023, 370, fnad097. [Google Scholar] [CrossRef]
- Mammeri, H.; Poirel, L.; Fortineau, N.; Nordmann, P. Naturally Occurring Extended-Spectrum Cephalosporinases in Escherichia coli. Antimicrob. Agents Chemother. 2006, 50, 2573. [Google Scholar] [CrossRef]
- Tomás, M.; Doumith, M.; Warner, M.; Turton, J.F.; Beceiro, A.; Bou, G.; Livermore, D.M.; Woodford, N. Efflux Pumps, OprD Porin, AmpC β-Lactamase, and Multiresistance in Pseudomonas Aeruginosa Isolates from Cystic Fibrosis Patients. Antimicrob. Agents Chemother. 2010, 54, 2219. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, M.M.; Literacka, E.; Zioga, A.; Giani, T.; Baraniak, A.; Fiett, J.; Sadowy, E.; Tassios, P.T.; Rossolini, G.M.; Gniadkowski, M.; et al. Evolution and Spread of a Multidrug-Resistant Proteus Mirabilis Clone with Chromosomal AmpC-Type Cephalosporinases in Europe. Antimicrob. Agents Chemother. 2011, 55, 2735–2742. [Google Scholar] [CrossRef] [PubMed]
- Aogáin, M.M.; Rogers, T.R.; Crowley, B. Identification of Emergent blaCMY-2-Carrying Proteus Mirabilis Lineages by Whole-Genome Sequencing. New Microbes New Infect. 2015, 9, 58. [Google Scholar] [CrossRef]
- SINIGIRH—Sistema Nacional de Información Para La Gestión Integrada Del Recurso Hídrico. Available online: https://mapas.da.go.cr/mapnew.php (accessed on 10 March 2024).
- Guo, J.; Li, J.; Chen, H.; Bond, P.L.; Yuan, Z. Metagenomic Analysis Reveals Wastewater Treatment Plants as Hotspots of Antibiotic Resistance Genes and Mobile Genetic Elements. Water Res. 2017, 123, 468–478. [Google Scholar] [CrossRef]
- Thongsamer, T.; Neamchan, R.; Blackburn, A.; Acharya, K.; Sutheeworapong, S.; Tirachulee, B.; Pattanachan, P.; Vinitnantharat, S.; Zhou, X.-Y.; Su, J.-Q.; et al. Environmental Antimicrobial Resistance Is Associated with Faecal Pollution in Central Thailand’s Coastal Aquaculture Region. J. Hazard. Mater. 2021, 416, 125718. [Google Scholar] [CrossRef]
- Agramont, J.; Gutiérrez-Cortez, S.; Joffré, E.; Sjöling, Å.; Calderon Toledo, C. Fecal Pollution Drives Antibiotic Resistance and Class 1 Integron Abundance in Aquatic Environments of the Bolivian Andes Impacted by Mining and Wastewater. Microorganisms 2020, 8, 1122. [Google Scholar] [CrossRef]
- Mahaney, A.P.; Franklin, R.B. Persistence of Wastewater-Associated Antibiotic Resistant Bacteria in River Microcosms. Sci. Total Environ. 2022, 819, 153099. [Google Scholar] [CrossRef]
- Jutkina, J.; Marathe, N.P.; Flach, C.-F.; Larsson, D.G.J. Antibiotics and Common Antibacterial Biocides Stimulate Horizontal Transfer of Resistance at Low Concentrations. Sci. Total Environ. 2018, 616–617, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Parsons, T.R.; Maita, Y.; Lalli, C.M. A Manual of Chemical and Biological Methods for Seawater Analysis; Pergamon Press: Oxford, UK, 1984; ISBN 978-0-08-030288-1. [Google Scholar]
- Strickland, J.D.H.; Parsons, T.R. A Practical Handbook of Seawater Analysis, 2nd ed.; Fisheries Research Board of Canada: Ottawa, ON, Canada, 1972. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 10 March 2024).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Chklovski, A.; Parks, D.H.; Woodcroft, B.J.; Tyson, G.W. CheckM2: A Rapid, Scalable and Accurate Tool for Assessing Microbial Genome Quality Using Machine Learning. Nat. Methods 2023, 20, 1203–1212. [Google Scholar] [CrossRef]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A Toolkit to Classify Genomes with the Genome Taxonomy Database. Bioinformatics 2020, 36, 1925–1927. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Hahnke, R.L.; Petersen, J.; Scheuner, C.; Michael, V.; Fiebig, A.; Rohde, C.; Rohde, M.; Fartmann, B.; Goodwin, L.A.; et al. Complete Genome Sequence of DSM 30083T, the Type Strain (U5/41T) of Escherichia coli, and a Proposal for Delineating Subspecies in Microbial Taxonomy. Stand. Genomic Sci. 2014, 9, 2. [Google Scholar] [CrossRef]
- Sahl, J.W.; Morris, C.R.; Emberger, J.; Fraser, C.M.; Ochieng, J.B.; Juma, J.; Fields, B.; Breiman, R.F.; Gilmour, M.; Nataro, J.P.; et al. Defining the Phylogenomics of Shigella Species: A Pathway to Diagnostics. J. Clin. Microbiol. 2015, 53, 951–960. [Google Scholar] [CrossRef]
- Shen, Z.; Koh, X.P.; Yu, Y.; Woo, C.F.; Tong, Y.; Lau, S.C.K. Draft Genome Sequences of 16 Strains of Escherichia Cryptic Clade II Isolated from Intertidal Sediment in Hong Kong. Microbiol. Resour. Announc. 2019, 8, e00416-19. [Google Scholar] [CrossRef]
- Mire, M.; Kim, C.; Baffaut, C.; Liu, F.; Wuliji, T.; Zheng, G. Escherichia Cryptic Clade II through Clade VIII: Rapid Detection and Prevalence in Feces and Surface Water. Sci. Total Environ. 2022, 848, 157741. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Enright, A.J.; Van Dongen, S.; Ouzounis, C.A. An Efficient Algorithm for Large-Scale Detection of Protein Families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing Large Minimum Evolution Trees with Profiles Instead of a Distance Matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.; Nash, J.H.E. MOB-Suite: Software Tools for Clustering, Reconstruction and Typing of Plasmids from Draft Assemblies. Microb. Genom. 2018, 4, e000206. [Google Scholar] [CrossRef]
- Néron, B.; Littner, E.; Haudiquet, M.; Perrin, A.; Cury, J.; Rocha, E.P.C. IntegronFinder 2.0: Identification and Analysis of Integrons across Bacteria, with a Focus on Antibiotic Resistance in Klebsiella. Microorganisms 2022, 10, 700. [Google Scholar] [CrossRef]
- Li, Y.; Feng, X.; Chen, X.; Yang, S.; Zhao, Z.; Chen, Y.; Li, S.C. PlasmidScope: A Comprehensive Plasmid Database with Rich Annotations and Online Analytical Tools. Nucleic Acids Res. 2024, gkae930. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-Mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Tseemann/Abricate 2024.
- Feldgarden, M.; Brover, V.; Gonzalez-Escalona, N.; Frye, J.G.; Haendiges, J.; Haft, D.H.; Hoffmann, M.; Pettengill, J.B.; Prasad, A.B.; Tillman, G.E.; et al. AMRFinderPlus and the Reference Gene Catalog Facilitate Examination of the Genomic Links among Antimicrobial Resistance, Stress Response, and Virulence. Sci. Rep. 2021, 11, 12728. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Pankhurst, L.; Hubbard, A.; Votintseva, A.; Stoesser, N.; Sheppard, A.E.; Mathers, A.; Norris, R.; Navickaite, I.; Eaton, C.; et al. Resolving Plasmid Structures in Enterobacteriaceae Using the MinION Nanopore Sequencer: Assessment of MinION and MinION/Illumina Hybrid Data Assembly Approaches. Microb. Genom. 2017, 3, e000118. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG Database: An Updated Version Includes Eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The Protein Families Database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Yu, M.K.; Fogarty, E.C.; Eren, A.M. Diverse Plasmid Systems and Their Ecology across Human Gut Metagenomes Revealed by PlasX and MobMess. Nat. Microbiol. 2024, 9, 830–847. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate (Site) | Sequencing Method | Contigs | Size (Mb) | N50 | L50 | %GC | Completeness | Contamination | Taxonomy According to TYGS (dDDH in %) |
|---|---|---|---|---|---|---|---|---|---|
| A222-7 (1) | Illumina | 73 | 4.90 | 130,684 | 11 | 50.61 | 100 | 0.05 | Escherichia coli (73.8) |
| A229-5 (1) | Illumina | 88 | 4.86 | 111,924 | 14 | 50.76 | 100 | 0.04 | Escherichia coli (73.8) |
| A229-12 (1) | Illumina | 99 | 4.75 | 103,479 | 15 | 50.78 | 100 | 0.09 | Escherichia coli (74.7) |
| A236-7 (1) | Illumina | 64 | 4.80 | 167,951 | 10 | 50.7 | 100 | 0.15 | Escherichia coli (74) |
| A236-12 (1) | Illumina | 73 | 4.7 | 147,946 | 8 | 50.59 | 100 | 0.19 | Escherichia coli (75.1) |
| A223-7 (2) | Illumina | 45 | 4.97 | 413,561 | 6 | 52.48 | 100 | 0.05 | Citrobacter sp. (42.5) * |
| A230-5 (2) | Illumina | 40 | 5.41 | 296,086 | 6 | 57.74 | 100 | 0.09 | Klebsiella quasipneumoniae (71.4) |
| A230-7 (2) | Illumina | 50 | 5.48 | 349,092 | 6 | 57.22 | 100 | 0.23 | Klebsiella pneumoniae (93.4) |
| A230-8 (2) | Illumina | 159 | 4.96 | 77,565 | 23 | 50.62 | 100 | 0.12 | Escherichia coli (74.1) |
| A237-6 (2) | Illumina | 67 | 5.22 | 188,161 | 10 | 57.56 | 100 | 0.85 | Klebsiella pneumoniae (93.4) |
| A224-7 (3) | Illumina | 84 | 5.60 | 141,929 | 14 | 57.21 | 100 | 0.13 | Klebsiella variicola (92.2) |
| A231-5 (3) | Illumina | 51 | 5.47 | 215,170 | 9 | 57.27 | 100 | 0.26 | Klebsiella pneumoniae (93.5) |
| A238-6 (3) | Illumina | 75 | 5.54 | 191,124 | 10 | 57.19 | 100 | 0.19 | Klebsiella pneumoniae (93) |
| A224-8 (3) | Illumina and PacBio | 5 | 4.70 | 4,712,205 | 1 | 50.73 | 100 | 0.04 | Escherichia coli (75.2) |
| A231-12 (3) | Illumina and PacBio | 4 | 5.08 | 4,757,699 | 1 | 50.61 | 100 | 0.02 | Escherichia coli (73.8) |
| Site | Statistic | ARGs | Plasmids |
|---|---|---|---|
| 1 | Minimum | 1 | 1 |
| Median | 1 | 2 | |
| Maximum | 1 | 4 | |
| Abundance | 4 | 11 | |
| Richness | 3 | 11 | |
| 2 | Minimum | 1 | 1 |
| Median | 4 | 2 | |
| Maximum | 7 | 6 | |
| Abundance | 20 | 16 | |
| Richness | 19 | 16 | |
| 3 | Minimum | 4 | 2 |
| Median | 7 | 4 | |
| Maximum | 13 | 7 | |
| Abundance | 41 | 23 | |
| Richness | 29 | 23 |
| Isolate (Site) | Phenotype | nfsA Mutations | nfsB Mutations | oqx Genes |
|---|---|---|---|---|
| K. pneumoniae A238-6 (3) | Resistant | E29A, G125W, G204S | oqxB, oqxA | |
| K. pneumoniae A231-5 (3) | Resistant | Q195L | oqxB11, oqxA5 | |
| K. pneumoniae A237-6 (2) | Resistant | K222R | oqxB25, oqxA6 | |
| K. pneumoniae A230-7 (2) | Resistant | E29A, G125W, G204S | oqxA, oqxB | |
| K. quasipneumoniae A230-5 (2) | Resistant | E29A, Q94E, Q147K, E191G, E194D | Q181K | oqxA10, oqxB11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendoza-Guido, B.; Barrantes, K.; Rodríguez, C.; Rojas-Jimenez, K.; Arias-Andres, M. The Impact of Urban Pollution on Plasmid-Mediated Resistance Acquisition in Enterobacteria from a Tropical River. Antibiotics 2024, 13, 1089. https://doi.org/10.3390/antibiotics13111089
Mendoza-Guido B, Barrantes K, Rodríguez C, Rojas-Jimenez K, Arias-Andres M. The Impact of Urban Pollution on Plasmid-Mediated Resistance Acquisition in Enterobacteria from a Tropical River. Antibiotics. 2024; 13(11):1089. https://doi.org/10.3390/antibiotics13111089
Chicago/Turabian StyleMendoza-Guido, Bradd, Kenia Barrantes, César Rodríguez, Keilor Rojas-Jimenez, and Maria Arias-Andres. 2024. "The Impact of Urban Pollution on Plasmid-Mediated Resistance Acquisition in Enterobacteria from a Tropical River" Antibiotics 13, no. 11: 1089. https://doi.org/10.3390/antibiotics13111089
APA StyleMendoza-Guido, B., Barrantes, K., Rodríguez, C., Rojas-Jimenez, K., & Arias-Andres, M. (2024). The Impact of Urban Pollution on Plasmid-Mediated Resistance Acquisition in Enterobacteria from a Tropical River. Antibiotics, 13(11), 1089. https://doi.org/10.3390/antibiotics13111089