
Investigation of the Impact of Antibiotic Administration on the Preterm Infants’ Gut Microbiome Using Next-Generation Sequencing—Based 16S rRNA Gene Analysis


Abstract
1. Introduction
2. Results
2.1. Evaluation of Infants’ Microbiota
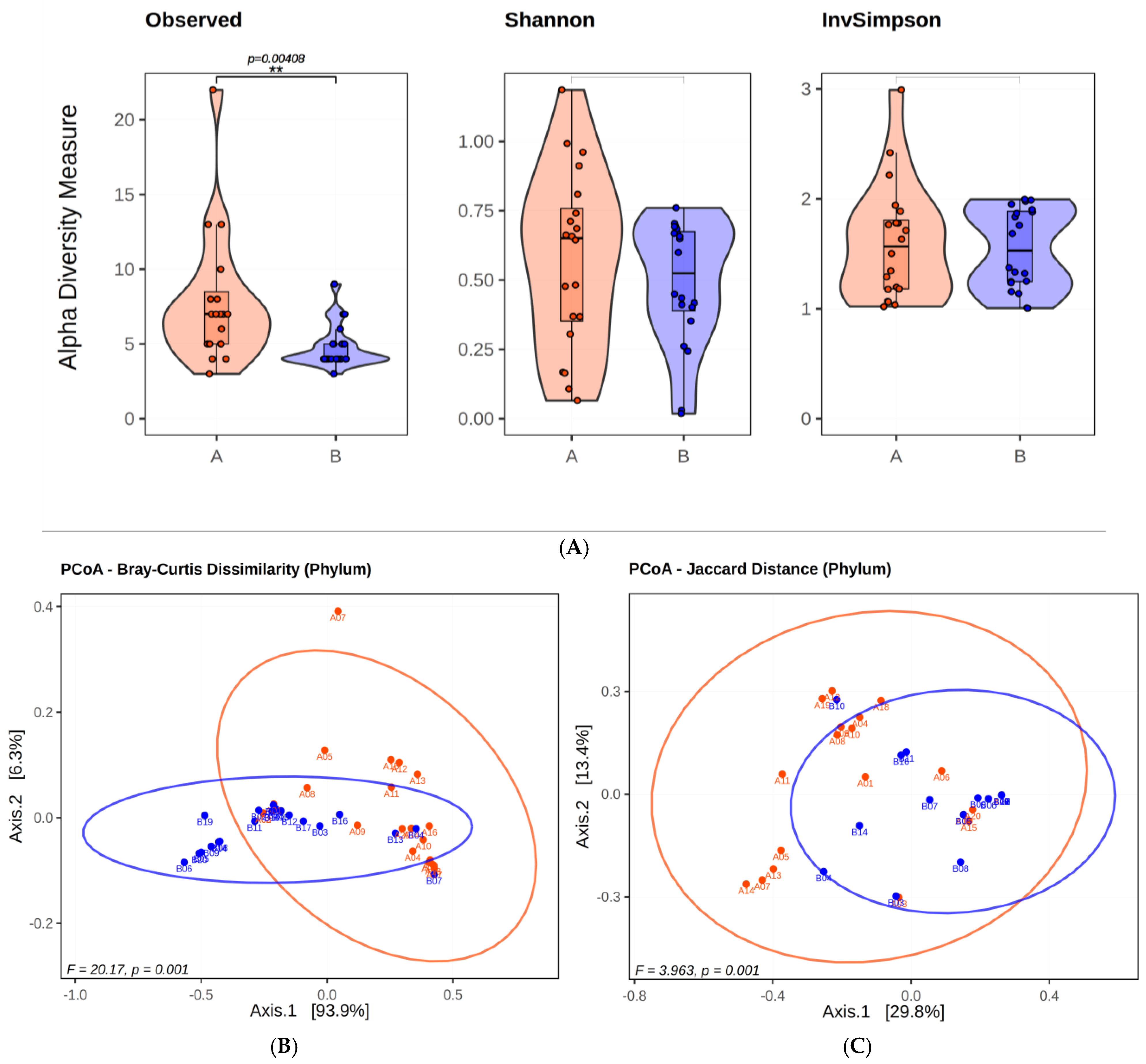
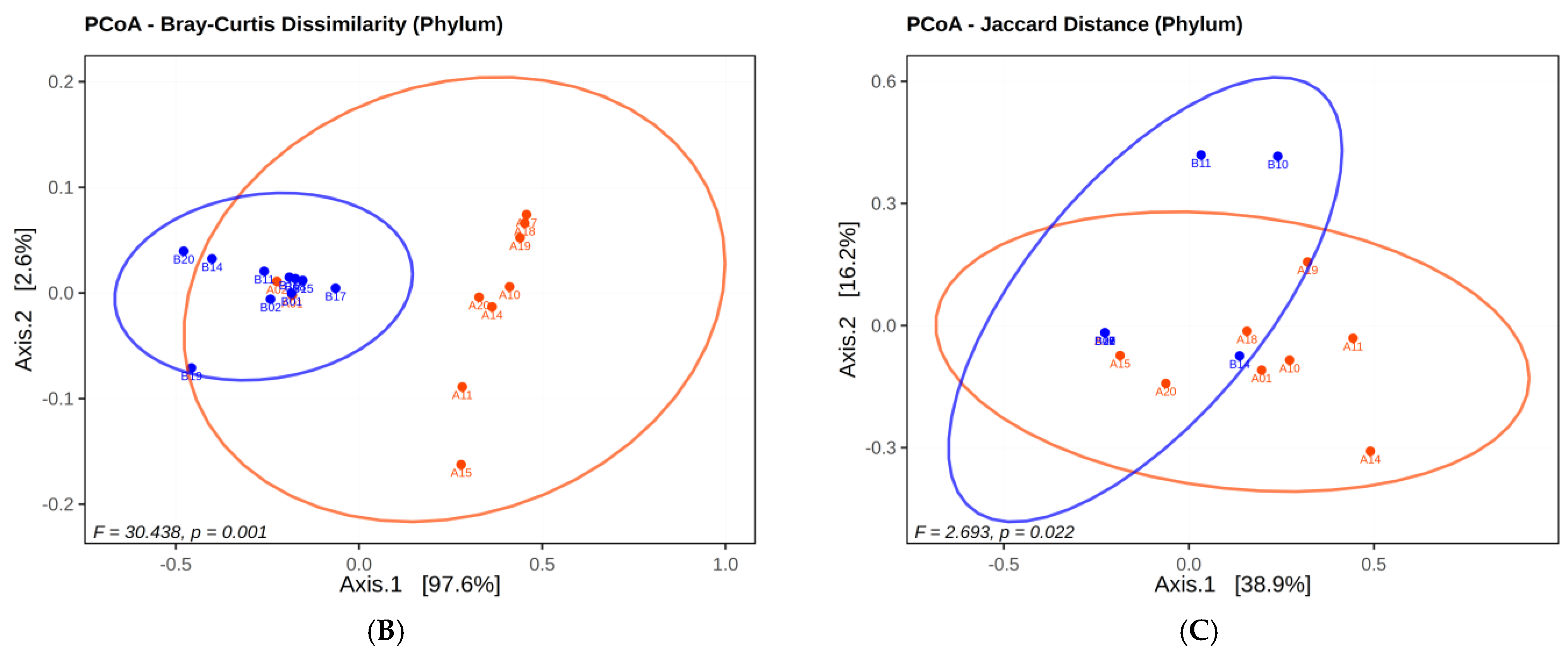
2.1.1. Microbial Diversty Analysis
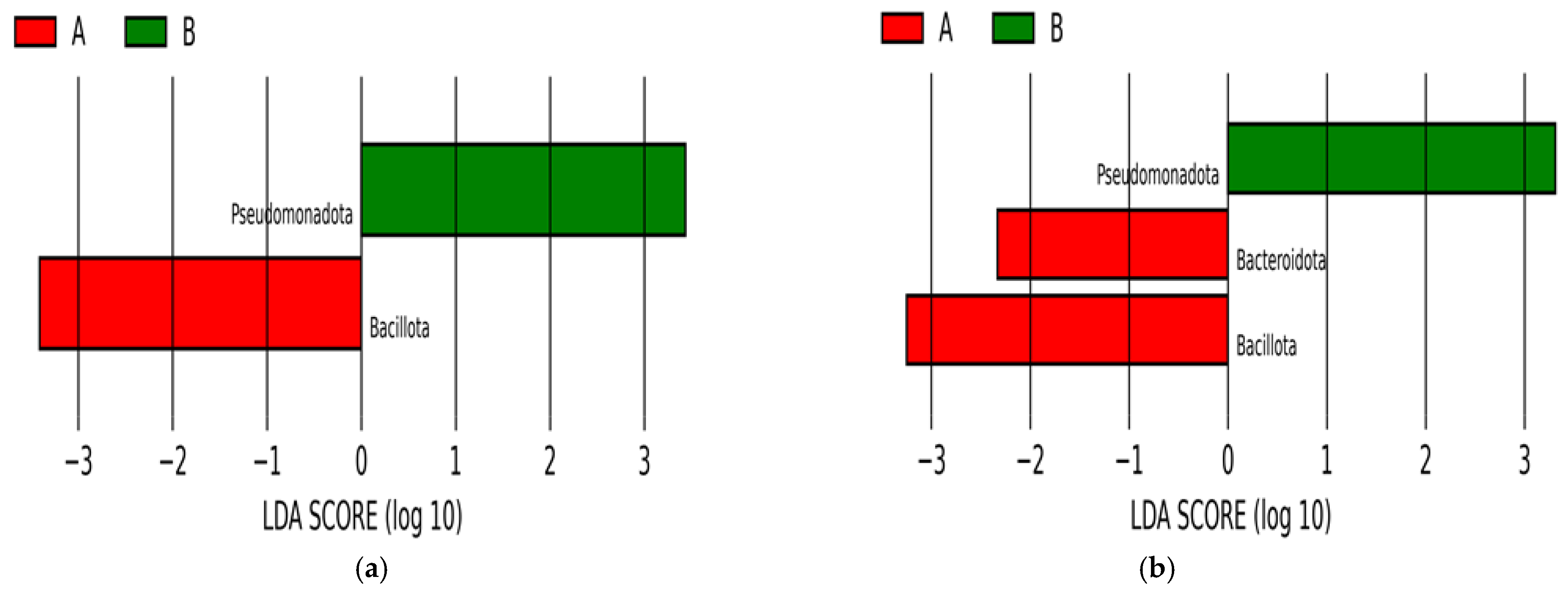
2.1.2. Analysis of Infants’ Microbiota at Phyla Level
- 1st Group. Seven phyla were within the antibiotic spectrum and known to cause human disease.
- 2nd Group. Seven phyla were within the antibiotic spectrum but did not cause human disease.
- 3rd Group. Eleven phyla were not causing human disease nor in the spectrum of antibiotics.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phyla | Number of Samples | Average Incidence Percentage (%) | p-Value (<0.05) | Comment | ||
|---|---|---|---|---|---|---|
| A | B | A | B | |||
| 1st Group: Phyla related with human disease and within antibiotic efficiency spectrum | ||||||
| Bacillota | 20 | 20 | 71.851 | 36.9886 | 0.0009319 | Significant decrease |
| Pseudomonadota | 20 | 20 | 21.7138 | 62.3941 | 0.00006 | Significant increase |
| Bacteroidota | 19 | 17 | 5.157 | 0.565 | 0.006066 | Significant decrease |
| Actinomycetota | 19 | 19 | 1.0071 | 0.0423 | 0.00005 | Significant decrease |
| Fusobacteriota | 4 | 1 | 0.0285 | 0.00001 | 0.1056454 | not-significant |
| Campylobacterota | 5 | 4 | 0.0067 | 0.0041 | 0.3525421 | not-significant |
| Spirochaetota | 5 | 1 | 0.0033 | 0.0001 | 0.0590582 | not-significant |
| 2nd Group: Phyla not related with human disease but within antibiotic efficiency spectrum | ||||||
| Cyanobacteriota | 13 | 3 | 0.0655 | 0.0005 | 0.001511 | Significant decrease |
| Verrucomicrobiota | 7 | 3 | 0.0246 | 0.0006 | 0.0243902 | Significant decrease |
| Myxococcota | 2 | 0 | 0.019 | 0 | 0.3710934 | not-significant |
| Deinococcota | 4 | 0 | 0.0053 | 0 | 0.1003482 | not-significant |
| Chloroflexota | 2 | 2 | 0.0009 | 0.0004 | 0.4226781 | not-significant |
| Gemmatimonadota | 1 | 0 | 0.0002 | 0 | 1 | not-significant |
| Aquificota | 3 | 0 | 0.0042 | 0 | 0.1814492 | not-significant |
| 3rd Group: Phyla not related with human disease and not within antibiotic efficiency spectrum | ||||||
| Thermotogota | 5 | 0 | 0.0402 | 0 | 0.0590582 | not-significant |
| Thermodesulfobacteriota | 9 | 3 | 0.0277 | 0.0034 | 0.0366713 | Significant decrease |
| Candidatus Melainabacteria | 7 | 0 | 0.0173 | 0 | 0.0222541 | Significant decrease |
| Thermomicrobiota | 1 | 0 | 0.0041 | 0 | 1 | not-significant |
| Bdellovibrionota | 2 | 0 | 0.0021 | 0 | 0.3710934 | not-significant |
| Chrysiogenota | 2 | 0 | 0.0008 | 0 | 0.3710934 | not-significant |
| Rhodothermota | 2 | 1 | 0.0004 | 0.0002 | 1 | not-significant |
| Nitrospirota | 1 | 1 | 0.0003 | 0 | 1 | not-significant |
| Candidatus Thermoplasmatota | 1 | 0 | 0.0002 | 0 | 1 | not-significant |
| Nitrospinota | 1 | 0 | 0.0002 | 0 | 1 | not-significant |
| Lentisphaerota | 1 | 2 | 0.0001 | 0.0005 | 0.5862137 | not-significant |
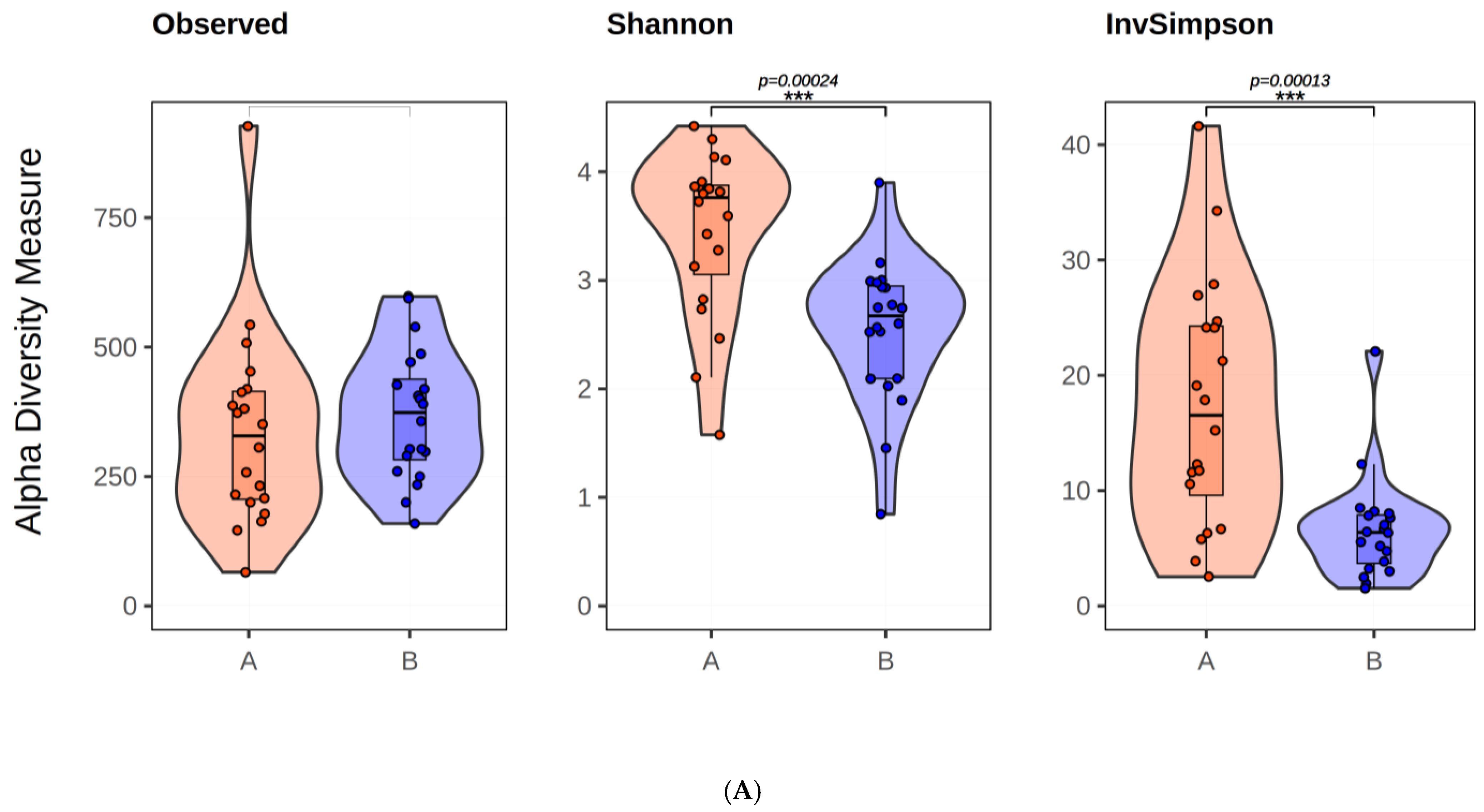
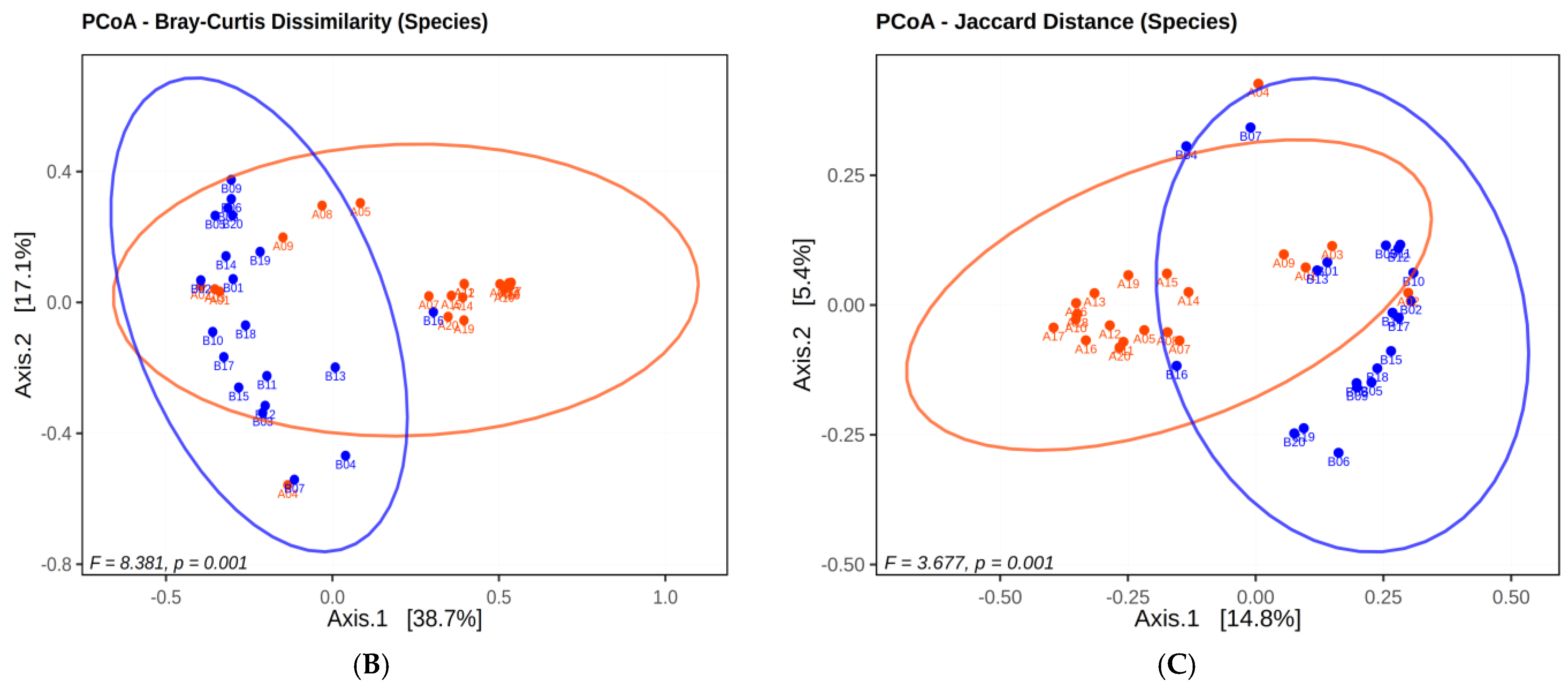
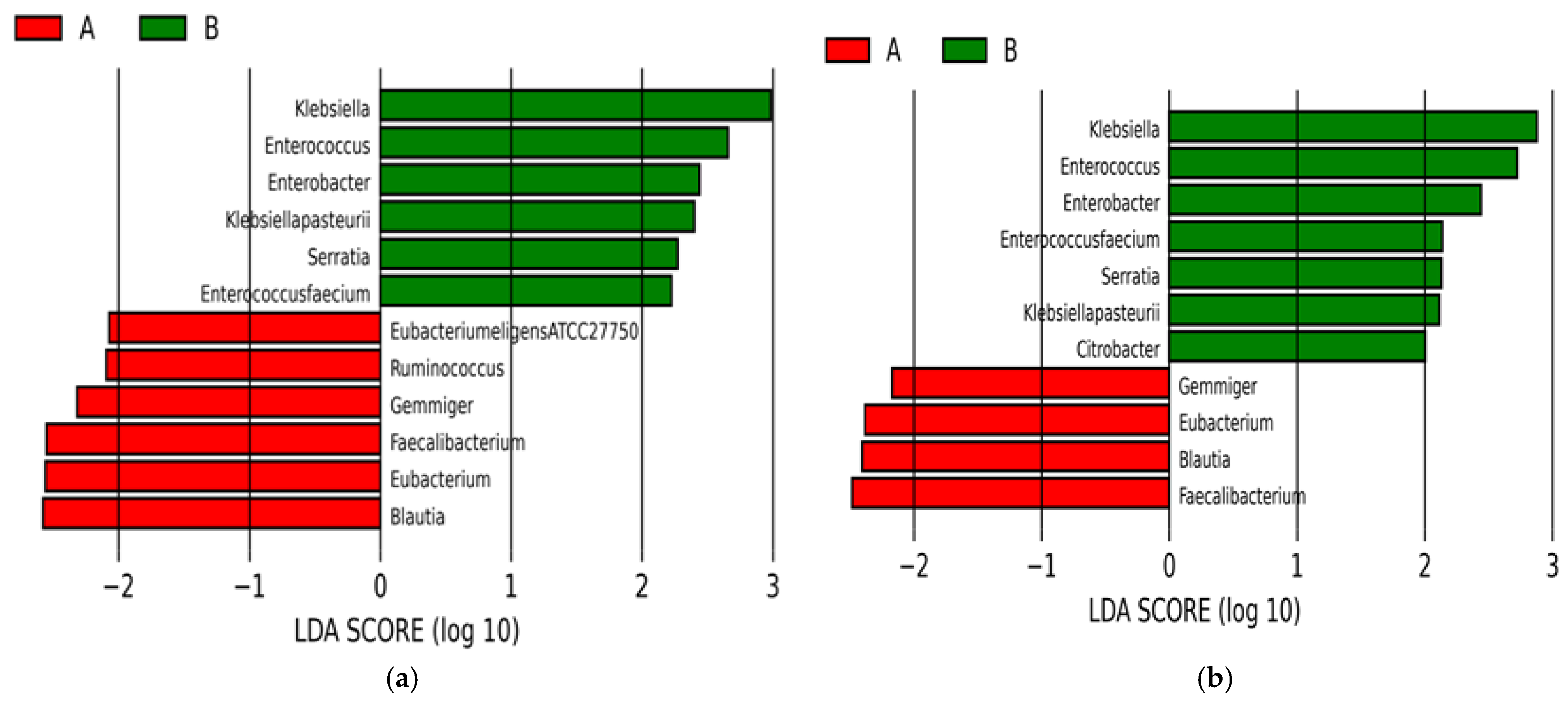
2.1.3. Microbial Diversity Analysis at Species Level
2.2. Evaluation of Twin Infants
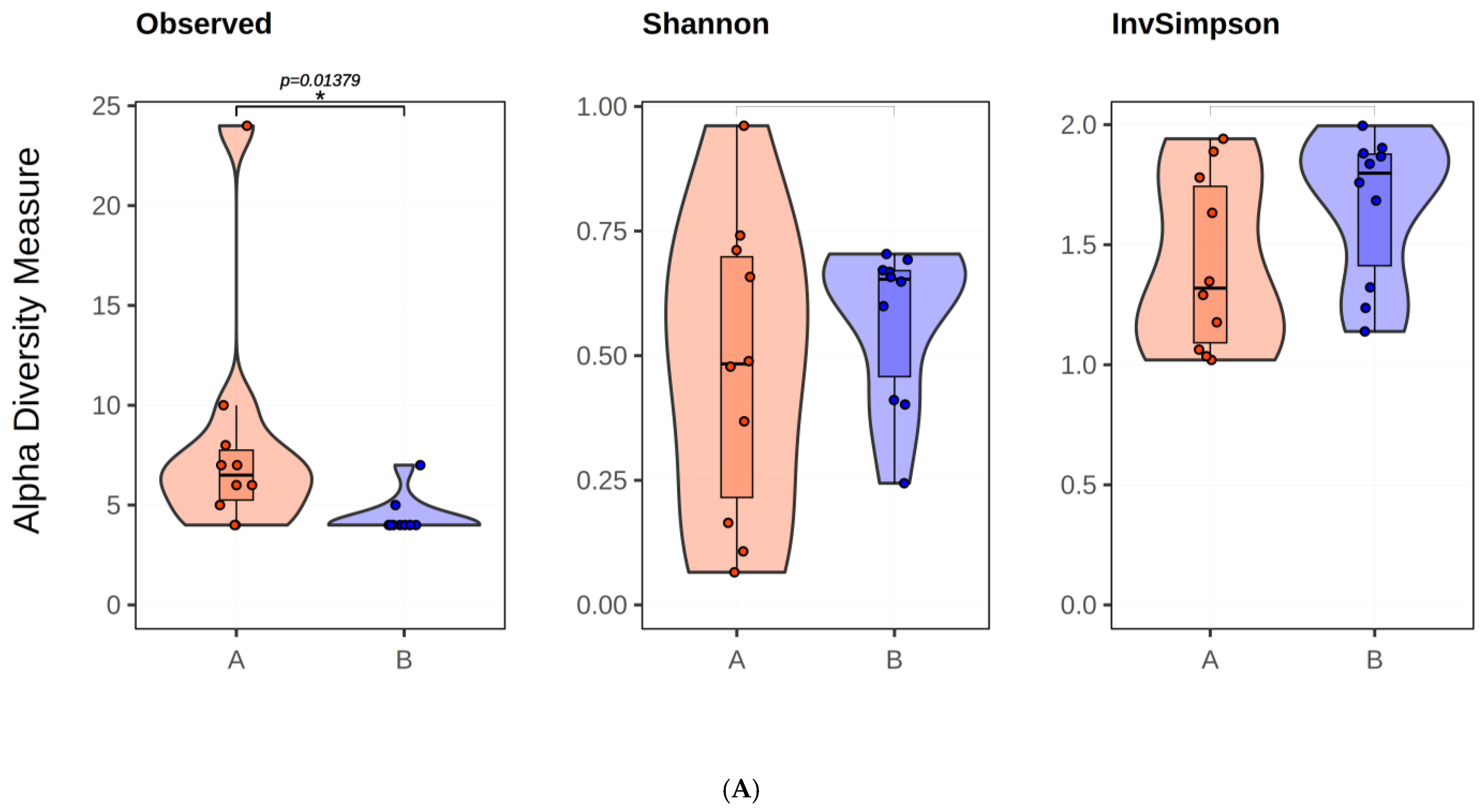
2.2.1. Microbial Diversity Analysis in Twins
2.2.2. Analysis of Twin Infants’ Microbiota at Phyla Level
2.2.3. LEfSe Analysis Results
3. Discussion
4. Material and Methods
4.1. Determination of Patient Population and Isolation of VRE and CRKP from Rectal Samples
4.2. gDNA Extraction and 16S rRNA Sequencing
4.3. Bioinformatics and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Bengmark, S. Ecological control of the gastrointestinal tract. The role of probiotic flora. Gut 1998, 42, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Patangia, D.V.; Anthony Ryan, C.; Dempsey, E.; Paul Ross, R.; Stanton, C. Impact of antibiotics on the human microbiome and consequences for host health. Microbiologyopen 2022, 11, e1260. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Nageshwar Reddy, D. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8787–8803. [Google Scholar] [CrossRef]
- Ellis, J.L.; Karl, J.P.; Oliverio, A.M.; Fu, X.; Soares, J.W.; Wolfe, B.E.; Hernandez, C.J.; Mason, J.B.; Booth, S.L. Dietary vitamin K is remodeled by gut microbiota and influences community composition. Gut Microbes 2021, 13, 1887721. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef]
- Li, J.; Jia, H.; Cai, X.; Zhong, H.; Feng, Q.; Sunagawa, S.; Arumugam, M.; Kultima, J.R.; Prifti, E.; Nielsen, T.; et al. An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 2014, 32, 834–841. [Google Scholar] [CrossRef]
- Derrien, M.; Alvarez, A.S.; de Vos, W.M. The Gut Microbiota in the First Decade of Life. Trends Microbiol. 2019, 27, 997–1010. [Google Scholar] [CrossRef]
- Agostoni, C.; Axelsson, I.; Braegger, C.; Goulet, O.; Koletzko, B.; Michaelsen, K.F.; Rigo, J.; Shamir, R.; Szajewska, H.; Turck, D.; et al. ESPGHAN Committee on Nutrition. Probiotic bacteria in dietetic products for infants: A commentary by the ESPGHAN Committee on Nutrition. J. Pediatr. Gastroenterol. Nutr. 2004, 38, 365–374. [Google Scholar] [CrossRef]
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The impact of the gut microbiota on human health: An integrative view. Cell 2012, 148, 1258–1270. [Google Scholar] [CrossRef]
- DiGiulio, D.B.; Romero, R.; Amogan, H.P.; Kusanovic, J.P.; Bik, E.M.; Gotsch, F.; Kim, C.J.; Erez, O.; Edwin, S.; Relman, D.A. Microbial prevalence, diversity and abundance in amniotic fluid during preterm labor: A molecular and culture-based investigation. PLoS ONE 2008, 26, e3056. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.L.; Versalovic, J. The human microbiome and its potential importance to pediatrics. Pediatrics 2012, 129, 950–960. [Google Scholar] [CrossRef]
- Arboleya, S.; Sánchez, B.; Solís, G.; Fernández, N.; Suárez, M.; Hernández-Barranco, A.M.; Milani, C.; Margolles, A.; de Los Reyes-Gavilán, C.G.; Ventura, M.; et al. Impact of Prematurity and Perinatal Antibiotics on the Developing Intestinal Microbiota: A Functional Inference Study. Int. J. Mol. Sci. 2016, 17, 649. [Google Scholar] [CrossRef] [PubMed]
- Woese, C.R.; Kandler, O.; Wheelis, M.L. Towards a natural system of organisms: Proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl. Acad. Sci. USA 1990, 87, 4576–4579. [Google Scholar] [CrossRef] [PubMed]
- Branton, D.; Deamer, D.W.; Marziali, A.; Bayley, H.; Benner, S.A.; Butler, T.; Di Ventra, M.; Garaj, S.; Hibbs, A.; Huang, X.; et al. The potential and challenges of nanopore sequencing. Nat. Biotechnol. 2008, 26, 1146–1153. [Google Scholar] [CrossRef]
- Alcon-Giner, C.; Caim, S.; Mitra, S.; Ketskemety, J.; Wegmann, U.; Wain, J.; Belteki, G.; Clarke, P.; Hall, L.J. Optimisation of 16S rRNA gut microbiota profiling of extremely low birth weight infants. BMC Genom. 2017, 18, 841. [Google Scholar] [CrossRef]
- Jethwani, P.; Grover, K. Gut microbiota in health and diseases. Int. J. Curr. Microbiol. Appl. Sci. 2019, 8, 1586–1599. [Google Scholar] [CrossRef]
- Bäckhed, F.; Roswall, J.; Peng, Y.; Feng, Q.; Jia, H.; Kovatcheva-Datchary, P.; Li, Y.; Xia, Y.; Xie, H.; Zhong, H.; et al. Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life. Cell Host Microbe 2015, 17, 690–703, Erratum in Cell Host Microbe. 2015, 17, 852. [Google Scholar] [CrossRef]
- Lawn, J.E.; Kerber, K.; Enweronu-Laryea, C.; Cousens, S. 3.6 million neonatal deaths—What is progressing and what is not? Semin. Perinatol. 2010, 34, 371–386. [Google Scholar] [CrossRef]
- Perez-Palacios, P.; Girlich, D.; Soraa, N.; Lamrani, A.; Maoulainine, F.M.R.; Bennaoui, F.; Amri, H.; El Idrissi, N.S.; Bouskraoui, M.; Birer, A.; et al. Multidrug-resistant Enterobacterales responsible for septicaemia in a neonatal intensive care unit in Morocco. J. Glob. Antimicrob. Resist. 2023, 33, 208–217. [Google Scholar] [CrossRef]
- CDC-Center of Disease Control and Prevention. Management of Multidrug-Resistant Organisms in Healthcare Settings, 2006. Available online: https://www.cdc.gov/infection-control/media/pdfs/Guideline-MDRO-H.pdf (accessed on 19 September 2024).
- Demir, H.K.; Nakipoglu, Y. Investigation of the prevalence of carbapenem resistance genes in faecal carriage of carbapenem resistant Klebsiella spp. isolates by multiplex real-time PCR method. J. Infect. Dev. Ctries 2023, 17, 1606–1612. [Google Scholar] [CrossRef] [PubMed]
- Pammi, M.; Haque, K.N. Pentoxifylline for treatment of sepsis and necrotizing enterocolitis in neonates. Cochrane Database Syst. Rev. 2015, 3, CD004205, Erratum in Cochrane Database Syst Rev. 2023, 6, CD004205. [Google Scholar] [CrossRef]
- Gasparrini, A.J.; Wang, B.; Sun, X.; Kennedy, E.A.; Hernandez-Leyva, A.; Ndao, I.M.; Tarr, P.I.; Warner, B.B.; Dantas, G. Persistent metagenomic signatures of early-life hospitalization and antibiotic treatment in the infant gut microbiota and resistome. Nat. Microbiol. 2019, 4, 2285–2297. [Google Scholar] [CrossRef] [PubMed]
- Fouhy, F.; Guinane, C.M.; Hussey, S.; Wall, R.; Ryan, C.A.; Dempsey, E.M.; Murphy, B.; Ross, R.P.; Fitzgerald, G.F.; Stanton, C.; et al. High-throughput sequencing reveals the incomplete, short-term recovery of infant gut microbiota following parenteral antibiotic treatment with ampicillin and gentamicin. Antimicrob. Agents Chemother. 2012, 56, 5811–5820. [Google Scholar] [CrossRef]
- Dardas, M.; Gill, S.R.; Grier, A.; Pryhuber, G.S.; Gill, A.L.; Lee, Y.H.; Guillet, R. The impact of postnatal antibiotics on the preterm intestinal microbiome. Pediatr. Res. 2014, 76, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Zwittink, R.D.; van Zoeren-Grobben, D.; Renes, I.B.; van Lingen, R.A.; Norbruis, O.F.; Martin, R.; Groot Jebbink, L.J.; Knol, J.; Belzer, C. Dynamics of the bacterial gut microbiota in preterm and term infants after intravenous amoxicillin/ceftazidime treatment. BMC Pediatr. 2020, 20, 195. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Cobas, A.E.; Gosalbes, M.J.; Friedrichs, A.; Knecht, H.; Artacho, A.; Eismann, K.; Otto, W.; Rojo, D.; Bargiela, R.; von Bergen, M.; et al. Gut microbiota disturbance during antibiotic therapy: A multi-omic approach. Gut 2013, 62, 1591–1601. [Google Scholar] [CrossRef]
- Wandro, S.; Osborne, S.; Enriquez, C.; Bixby, C.; Arrieta, A.; Whiteson, K. The Microbiome and Metabolome of Preterm Infant Stool Are Personalized and Not Driven by Health Outcomes, Including Necrotizing Enterocolitis and Late-Onset Sepsis. mSphere 2018, 3, e00104-18. [Google Scholar] [CrossRef]
- Yokoyama, S.; Suzuki, T. Isolation and characterization of a novel equol-producing bacterium from human feces. Biosci. Biotechnol. Biochem. 2008, 72, 2660–2666. [Google Scholar] [CrossRef]
- Lu, S.; Huang, Q.; Wei, B.; Chen, Y. Effects of β-Lactam Antibiotics on Gut Microbiota Colonization and Metabolites in Late Preterm Infants. Curr. Microbiol. 2020, 77, 3888–3896. [Google Scholar] [CrossRef]
- Korpela, K.; Blakstad, E.W.; Moltu, S.J.; Strømmen, K.; Nakstad, B.; Rønnestad, A.E.; Brække, K.; Iversen, P.O.; Drevon, C.A.; de Vos, W. Intestinal microbiota development and gestational age in preterm neonates. Sci. Rep. 2018, 8, 2453. [Google Scholar] [CrossRef] [PubMed]
- Drell, T.; Lutsar, I.; Stšepetova, J.; Parm, U.; Metsvaht, T.; Ilmoja, M.L.; Simm, J.; Sepp, E. The development of gut microbiota in critically ill extremely low birth weight infants assessed with 16S rRNA gene-based sequencing. Gut Microbes 2014, 5, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Procop, G.W.; Church, D.L.; Hall, G.S.; Koneman, E.W. Konemans’ Color Atlas and Textbook of Diagnostic Microbiology, 7th ed.; Lippincott Williams and Wilkins: New York, NY, USA, 2017. [Google Scholar]
- EUCAST—European Committee on Antimicrobial Susceptibility Testing. Disk Diffusion Method for Antimicrobial Susceptibility Testing, Version 12.0. Available online: https://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Disk_test_documents/2024_manuals/Manual_v_12.0_EUCAST_Disk_Test_2024.pdf (accessed on 19 September 2024).
- Dos Santos, H.R.M.; Argolo, C.S.; Argôlo-Filho, R.C.; Loguercio, L.L. A 16S rDNA PCR-based theoretical to actual delta approach on culturable mock communities revealed severe losses of diversity information. BMC Microbiol. 2019, 19, 74. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]

| Infant No ** | Birth Age (Weeks + Days) | Birth Weight (g) | Antibiotics Used in Treatment * (Days) | Colonization of the First Mixed VRE + CRKP (Days) |
|---|---|---|---|---|
| 1 | 31 + 1/7 | 1470 | GEN + AMP (3), TEC + CTX (4) | 25 |
| 2 | 31 + 1/7 | 1410 | GEN + AMP (3), TEC + CTX (4) | 25 |
| 3 | 29 + 2/7 | 855 | GEN + AMP (3), TEC + CTX (4) | 69 |
| 4 | 25 + 2/7 | 450 | GEN + AMP (3), TEC + CTX (2), V + MEM (7), V + MEM + AK (9), LEV + SXT (7) | 96 |
| 5 | 31 + 4/7 | 1385 | GEN + AMP (3), TEC + CTX (4) | 26 |
| 6 | 29 + 3/7 | 1200 | GEN + P (3), TEC + GEN (6) | 15 |
| 7 | 28 + 6/7 | 1440 | GEN + SAM (3), TEC + TZP (4) | 22 |
| 8 | 27 + 5/7 | 920 | AMP + CTX (4), V + TZP (5) | 40 |
| 9 | 27 + 4/7 | 640 | GEN + AMP (3), TEC + CTX (4) | 32 |
| 10 | 29 + 4/7 | 1045 | GEN + AMP (3), TEC + CTX (4), V + MEM (5) | 35 |
| 11 | 29 + 4/7 | 1355 | GEN + AMP (3), TEC + CTX (4), TEC + MEM (5) | 45 |
| 12 | 31 + 1/7 | 1270 | GEN + AMP (3), TEC + CTX (3) | 55 |
| 13 | 31 | 1425 | GEN + AMP (4), TEC + CTX (3), V + TZP (3) | 36 |
| 14 | 29 + 5/7 | 1070 | GEN + AMP (3), TEC + TZP (2) | 74 |
| 15 | 29 + 5/7 | 1005 | GEN + AMP (3), TEC + TZP (2) | 60 |
| 16 | 26 + 5/7 | 840 | GEN + AMP (3), TEC + CTX (3) | 33 |
| 17 | 26 + 1/7 | 920 | TEC + CTX (2), V + MEM (4) | 60 |
| 18 | 26 + 1/7 | 930 | TEC + CTX (2), V + MEM (4) | 68 |
| 19 | 29 + 3/7 | 1350 | GEN + AMP (3), TEC + CTX + MEM (7) | 29 |
| 20 | 29 + 3/7 | 880 | GEN + AMP (3), TEC + CTX+MEM (7) | 30 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aktaş, A.; Ekren, B.Y.; Yaşa, B.; Sezerman, O.U.; Nakipoğlu, Y. Investigation of the Impact of Antibiotic Administration on the Preterm Infants’ Gut Microbiome Using Next-Generation Sequencing—Based 16S rRNA Gene Analysis. Antibiotics 2024, 13, 977. https://doi.org/10.3390/antibiotics13100977
Aktaş A, Ekren BY, Yaşa B, Sezerman OU, Nakipoğlu Y. Investigation of the Impact of Antibiotic Administration on the Preterm Infants’ Gut Microbiome Using Next-Generation Sequencing—Based 16S rRNA Gene Analysis. Antibiotics. 2024; 13(10):977. https://doi.org/10.3390/antibiotics13100977
Chicago/Turabian StyleAktaş, Ahmet, Berkay Yekta Ekren, Beril Yaşa, Osman Uğur Sezerman, and Yaşar Nakipoğlu. 2024. "Investigation of the Impact of Antibiotic Administration on the Preterm Infants’ Gut Microbiome Using Next-Generation Sequencing—Based 16S rRNA Gene Analysis" Antibiotics 13, no. 10: 977. https://doi.org/10.3390/antibiotics13100977
APA StyleAktaş, A., Ekren, B. Y., Yaşa, B., Sezerman, O. U., & Nakipoğlu, Y. (2024). Investigation of the Impact of Antibiotic Administration on the Preterm Infants’ Gut Microbiome Using Next-Generation Sequencing—Based 16S rRNA Gene Analysis. Antibiotics, 13(10), 977. https://doi.org/10.3390/antibiotics13100977