


Peptide Stapling Applied to Antimicrobial Peptides
 , ,
, ,  and
and
Abstract
:
1. Introduction
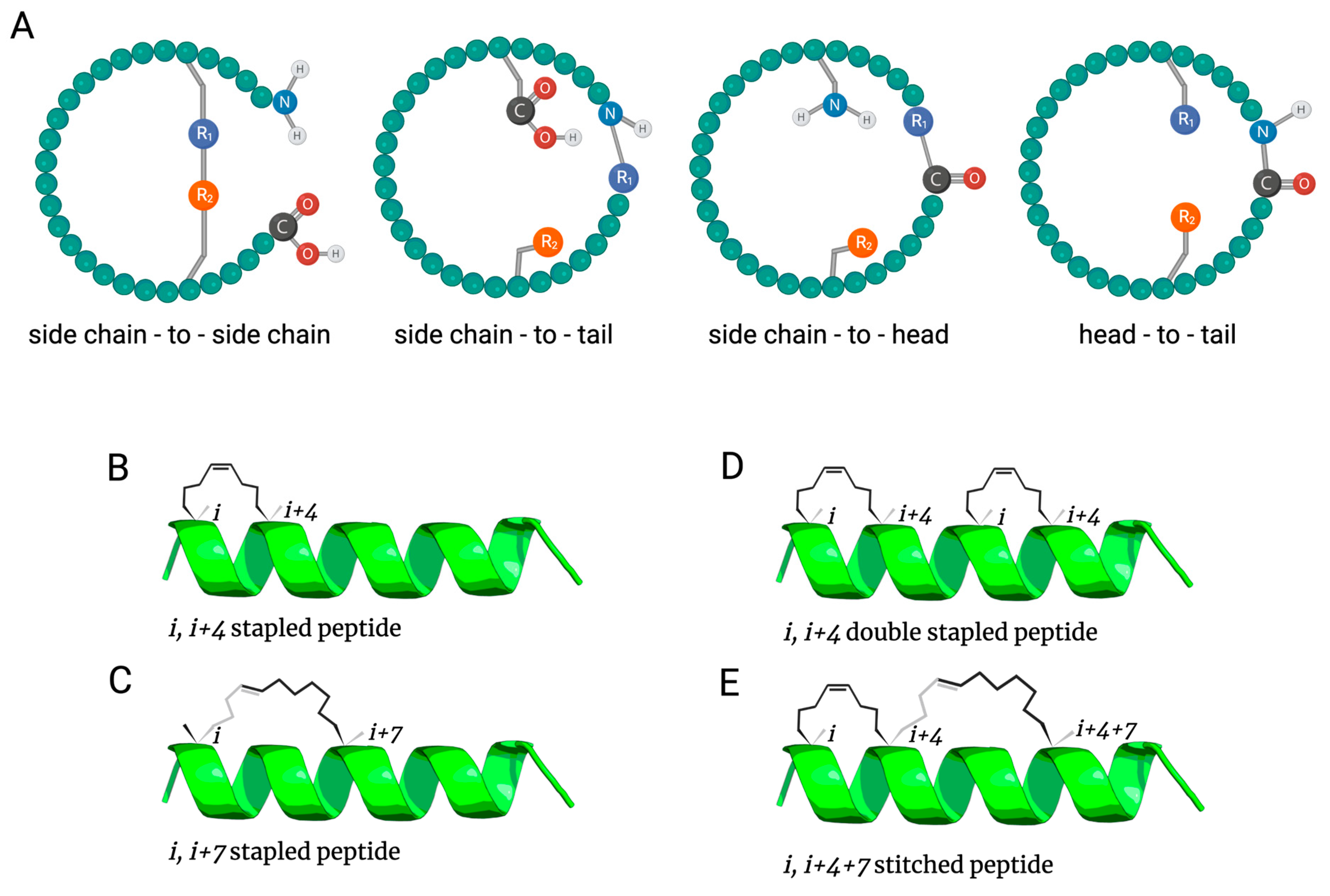
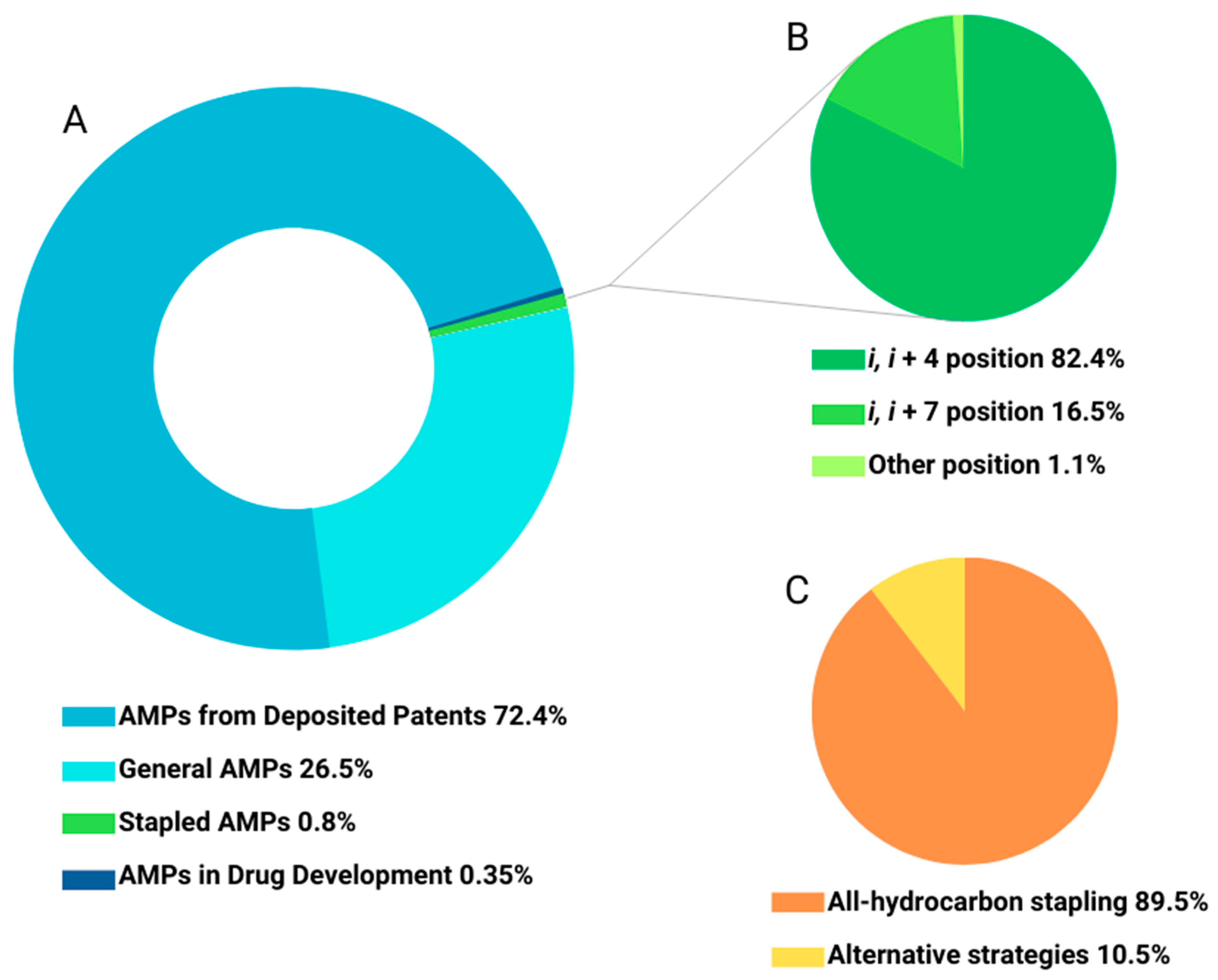
2. Antimicrobial Peptide Stapling
2.1. All-Hydrocarbon Stapling

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Origin | Stapling Style | Amino Acid Substitution | Antimicrobial Activity | Cytotoxicity | Helix Content | Proteolysis Susceptibility | References |
|---|---|---|---|---|---|---|---|---|
| P9 | CXCL10 chemokine | All-hydrocarbon (i, i + 4); position 8 and 12 | No | Improved | Increased | Increased | NA | [48] |
| MPIS | Polybia-MPI | All-hydrocarbon (i, i + 4); position 6 and 10 | No | Improved: Gram-positive Indifferent: Gram-negative | Increased | Increased | Decreased | [47] |
| MPIS-D8N | Polybia-MPI | All-hydrocarbon (i, i + 4); position 6 and 10 | Position 8 D substituted for N | Improved: Gram-positive Indifferent: Gram-negative | Increased | Increased | Decreased | [47] |
| MPIS-Q12K | Polybia-MPI | All-hydrocarbon (i, i + 4); position 6 and 10 | Position 12 Q substituted for K | Improved: Gram-positive Indifferent: Gram-negative | Increased | Increased | Decreased | [47] |
| Ac-DS-14W | Non-natural alanine/lysine-based | Double all-hydrocarbon in tandem (i, i + 4); positions 2 and 6/9 and 13 | No | Improved: Gram-positive Indifferent: Gram-negative | Increased | Increased | Decreased | [50] |
| Ac-DS-5W- | Non-natural alanine/lysine-based (Ac-DS-14W analog) | Double all-hydrocarbon in tandem (i, i + 4); positions 2 and 6/9 and 13 | W insertion at position 5 | Improved | Indifferent | Increased | NA | [50] |
| S-6K-F17 | Synthetic peptide 6K-F17 | All-hydrocarbon (i, i + 4); position 10 and 14 | No | Improved | Increased | Increased | NA | [40] |
| S-6K-F17-2G | Synthetic peptide 6K-F17 | All-hydrocarbon (i, i + 4); position 10 and 14 | G insertion at positions 8 and 16 | Indifferent | Decreased | Indifferent | NA | [40] |
| S-6K-F17-3G | Synthetic peptide 6K-F17 | All-hydrocarbon (i, i + 4); position 10 and 14 | G insertion at positions 8, 13 and 16 | Indifferent | Decreased | Indifferent | NA | [40] |
| S-6K-F17-3GN | Synthetic peptide 6K-F17 | All-hydrocarbon (i, i + 4); position 10 and 14 | G insertion at positions 8, 13 and 16 N insertion at position 7 | Indifferent | Decreased | Indifferent | NA | [40] |
| Sau-2 | Aurein1.2 | All-hydrocarbon (i, i + 4); position 2 and 6 | No | Improved | NA | Increased | Decreased | [46] |
| Peptide 2 | Peptide 1, Mag 2 derivative | All-hydrocarbon (i, i + 4); position 1 and 5 | No | Improved | Decreased | Increased | NA | [26] |
| Peptide 8 | Peptide 1, Mag 2 derivative | All-hydrocarbon (i, i + 7); position 8 and 15 | No | Decreased | Increased | Increased | NA | [26] |
| Mag (i + 4) 1,15 (A9K) | Mag 2 | Double all-hydrocarbon (i, i + 4); positions 2 and 6/16 and 20 | Position 9 A substituted for K | Improved | Decreased | Increased | Decreased | [51] |
| C-MPI-1 | Polybia-MPI | Triazole stapling (i, i + 4); position 8 and 12 | No | Decreased | Increased | Increased | Decreased | [52] |
| C-MPI-2 | Polybia-MPI | Triazole stapling (i, i + 6); position 2 and 8 | No | Non-active | NA | Indifferent | NA | [52] |
| Peptide 12 (OH-CM6) | OH-CATH30 | Lysine N-alkylation (i, i + 4); position 12 and 16 | No | Improved | Increased, but good therapeutic index | Increased | Decreased | [53] |
| V26-SP-8 | VapC26 α454–65 | All-hydrocarbon (i, i + 7); position 1 and 8 | No | Improved | NA | Increased | NA | [54] |
| S-TM4 (88–100) | TM4 (88–100) | All-hydrocarbon (i, i + 4); position 4 and 8 | No | Improved | Decreased | Increased | Decreased | [55] |
2.2. Alternative Stapling Strategies
3. Stapling for Protein–Protein Interaction Targets
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO Global Action Plan on Antimicrobial Resistance. Microbe Mag. 2015, 10, 354–355. [CrossRef]
- IACG No Time to Wait: Securing the Future from Drug-Resistant Infections. Rep. Secr.-Gen. United Nations 2019, 54, 113–114.
- Browne, K.; Chakraborty, S.; Chen, R.; Willcox, M.D.P.; Black, D.S.; Walsh, W.R.; Kumar, N. A New Era of Antibiotics: The Clinical Potential of Antimicrobial Peptides. Int. J. Mol. Sci. 2020, 21, 7047. [Google Scholar] [CrossRef]
- Haney, E.F.; Straus, S.K.; Hancock, R.E.W. Reassessing the Host Defense Peptide Landscape. Front. Chem. 2019, 7, 43. [Google Scholar] [CrossRef]
- de Souza, C.M.; da Silva, Á.P.; Júnior, N.G.O.; Martínez, O.F.; Franco, O.L. Peptides as a Therapeutic Strategy against Klebsiella Pneumoniae. Trends Pharmacol. Sci. 2022, 43, 335–348. [Google Scholar] [CrossRef]
- Marr, A.K.; Gooderham, W.J.; Hancock, R.E. Antibacterial Peptides for Therapeutic Use: Obstacles and Realistic Outlook. Curr. Opin. Pharmacol. 2006, 6, 468–472. [Google Scholar] [CrossRef]
- Kumar, P.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility in Vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L.; et al. The Value of Antimicrobial Peptides in the Age of Resistance. Lancet Infect. Dis. 2020, 20, e216–e230. [Google Scholar] [CrossRef]
- Migoń, D.; Neubauer, D.; Kamysz, W. Hydrocarbon Stapled Antimicrobial Peptides. Protein J. 2018, 37, 2–12. [Google Scholar] [CrossRef]
- Tan, P.; Fu, H.; Ma, X. Design, Optimization, and Nanotechnology of Antimicrobial Peptides: From Exploration to Applications. Nano Today 2021, 39, 101229. [Google Scholar] [CrossRef]
- Li, W.; Separovic, F.; O’Brien-Simpson, N.M.; Wade, J.D. Chemically Modified and Conjugated Antimicrobial Peptides against Superbugs. Chem. Soc. Rev. 2021, 50, 4932–4973. [Google Scholar] [CrossRef]
- Han, Y.; Zhang, M.; Lai, R.; Zhang, Z. Chemical Modifications to Increase the Therapeutic Potential of Antimicrobial Peptides. Peptides 2021, 146, 170666. [Google Scholar] [CrossRef]
- Kapil, S.; Sharma, V. d-Amino Acids in Antimicrobial Peptides: A Potential Approach to Treat and Combat Antimicrobial Resistance. Can. J. Microbiol. 2021, 67, 119–137. [Google Scholar] [CrossRef] [PubMed]
- Manteghi, R.; Pallagi, E.; Olajos, G.; Csóka, I. Pegylation and Formulation Strategy of Anti-Microbial Peptide (AMP) According to the Quality by Design Approach. Eur. J. Pharm. Sci. 2020, 144, 105197. [Google Scholar] [CrossRef]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic Peptides: Current Applications and Future Directions. Signal Transduct. Target Ther. 2022, 7, 48. [Google Scholar] [CrossRef]
- Andreev, K.; Martynowycz, M.W.; Ivankin, A.; Huang, M.L.; Kuzmenko, I.; Meron, M.; Lin, B.; Kirshenbaum, K.; Gidalevitz, D. Cyclization Improves Membrane Permeation by Antimicrobial Peptoids. Langmuir 2016, 32, 12905–12913. [Google Scholar] [CrossRef] [PubMed]
- Gan, B.H.; Gaynord, J.; Rowe, S.M.; Deingruber, T.; Spring, D.R. The Multifaceted Nature of Antimicrobial Peptides: Current Synthetic Chemistry Approaches and Future Directions. Chem. Soc. Rev. 2021, 50, 7820–7880. [Google Scholar] [CrossRef] [PubMed]
- Khatri, B.; Nuthakki, V.R.; Chatterjee, J. Strategies to Enhance Metabolic Stabilities. Methods Mol. Biol. 2019, 2001, 17–40. [Google Scholar] [CrossRef] [PubMed]
- Skowron, K.J.; Speltz, T.E.; Moore, T.W. Recent Structural Advances in Constrained Helical Peptides. Med. Res. Rev. 2019, 39, 749–770. [Google Scholar] [CrossRef]
- Selvarajan, V.; Tram, N.D.T.; Xu, J.; Ngen, S.T.Y.; Koh, J.-J.; Teo, J.W.P.; Yuen, T.-Y.; Ee, P.L.R. Stapled β-Hairpin Antimicrobial Peptides with Improved Stability and Activity against Drug-Resistant Gram-Negative Bacteria. J. Med. Chem. 2023, 66, 8498–8509. [Google Scholar] [CrossRef]
- Walensky, L.D.; Bird, G.H. Hydrocarbon-Stapled Peptides: Principles, Practice, and Progress. J. Med. Chem. 2014, 57, 6275–6288. [Google Scholar] [CrossRef] [PubMed]
- Schafmeister, C.E.; Po, J.; Verdine, G.L. An All-Hydrocarbon Cross-Linking System for Enhancing the Helicity and Metabolic Stability of Peptides. J. Am. Chem. Soc. 2000, 122, 5891–5892. [Google Scholar] [CrossRef]
- Kim, Y.W.; Kutchukian, P.S.; Verdine, G.L. Introduction of All-Hydrocarbon i, i + 3 Staples into α-Helices via Ring-Closing Olefin Metathesis. Org. Lett. 2010, 12, 3046–3049. [Google Scholar] [CrossRef]
- Li, X.; Chen, S.; Zhang, W.-D.; Hu, H.-G. Stapled Helical Peptides Bearing Different Anchoring Residues. Chem. Rev. 2020, 120, 10079–10144. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-L.; Hsueh, J.-Y.; Yip, B.-S.; Chih, Y.-H.; Peng, K.-L.; Cheng, J.-W. Antimicrobial Peptides Display Strong Synergy with Vancomycin Against Vancomycin-Resistant E. faecium, S. Aureus, and Wild-Type E. coli. Int. J. Mol. Sci. 2020, 21, 4578. [Google Scholar] [CrossRef]
- Hirano, M.; Saito, C.; Yokoo, H.; Goto, C.; Kawano, R.; Misawa, T.; Demizu, Y. Development of Antimicrobial Stapled Peptides Based on Magainin 2 Sequence. Molecules 2021, 26, 444. [Google Scholar] [CrossRef]
- Lau, Y.H.; De Andrade, P.; Wu, Y.; Spring, D.R. Peptide Stapling Techniques Based on Different Macrocyclisation Chemistries. Chem. Soc. Rev. 2015, 44, 91–102. [Google Scholar] [CrossRef]
- Guarracino, D.A.; Riordan, J.A.; Barreto, G.M.; Oldfield, A.L.; Kouba, C.M.; Agrinsoni, D. Macrocyclic Control in Helix Mimetics. Chem. Rev. 2019, 119, 9915–9949. [Google Scholar] [CrossRef]
- Klein, M. Stabilized Helical Peptides: Overview of the Technologies and Its Impact on Drug Discovery. Expert Opin. Drug Discov. 2017, 12, 1117–1125. [Google Scholar] [CrossRef]
- Ali, A.M.; Atmaj, J.; Van Oosterwijk, N.; Groves, M.R.; Dömling, A. Stapled Peptides Inhibitors: A New Window for Target Drug Discovery. Comput. Struct. Biotechnol. J. 2019, 17, 263–281. [Google Scholar] [CrossRef]
- Wang, N.; Xie, G.; Liu, C.; Cong, W.; He, S.; Li, Y.; Fan, L.; Hu, H.G. Design, Synthesis, and Antitumor Activities Study of Stapled A4K14-Citropin 1.1 Peptides. Front. Chem. 2020, 8, 616147. [Google Scholar] [CrossRef]
- Bluntzer, M.T.J.; O’Connell, J.; Baker, T.S.; Michel, J.; Hulme, A.N. Designing Stapled Peptides to Inhibit Protein-Protein Interactions: An Analysis of Successes in a Rapidly Changing Field. Pept. Sci. 2021, 113, e24191. [Google Scholar] [CrossRef]
- Wang, C.; Xia, S.; Zhang, P.; Zhang, T.; Wang, W.; Tian, Y.; Meng, G.; Jiang, S.; Liu, K. Discovery of Hydrocarbon-Stapled Short α-Helical Peptides as Promising Middle East Respiratory Syndrome Coronavirus (MERS-CoV) Fusion Inhibitors. J. Med. Chem. 2018, 61, 2018–2026. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhao, N.; An, L.; Dai, Z.; Chen, X.; Yang, F.; You, Q.; Di, B.; Hu, C.; Xu, L. Apoptosis-Inducing Activity of Synthetic Hydrocarbon-Stapled Peptides in H358 Cancer Cells Expressing KRASG12C. Acta Pharm. Sin. B 2021, 11, 2670–2684. [Google Scholar] [CrossRef]
- Yu, J.J.; Zhou, D.D.; Cui, B.; Zhang, C.; Tan, F.W.; Chang, S.; Li, K.; Lv, X.X.; Zhang, X.W.; Shang, S.; et al. Disruption of the EGFR-SQSTM1 Interaction by a Stapled Peptide Suppresses Lung Cancer via Activating Autophagy and Inhibiting EGFR Signaling. Cancer Lett. 2020, 474, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.I.; Pham, T.K.; Kim, D.; Park, M.; Kim, B.O.; Cho, Y.H.; Kim, Y.W.; Lee, C. Identification of Brevinin-1EMa-Derived Stapled Peptides as Broad-Spectrum Virus Entry Blockers. Virology 2021, 561, 6–16. [Google Scholar] [CrossRef]
- Curreli, F.; Victor, S.M.B.; Ahmed, S.; Drelich, A.; Tong, X.; Tseng, C.-T.K.; Hillyer, C.D.; Debnath, A.K. Stapled Peptides Based on Human Angiotensin-Converting Enzyme 2 (ACE2) Potently Inhibit SARS-CoV-2 Infection In Vitro. mBio 2020, 11, e02451-20. [Google Scholar] [CrossRef]
- Maas, M.N.; Hintzen, J.C.J.; Löffler, P.M.G.; Mecinović, J. Targeting SARS-CoV-2 Spike Protein by Stapled HACE2 Peptides. Chem. Commun. 2021, 57, 3283–3286. [Google Scholar] [CrossRef]
- de Campos, L.J.; Palermo, N.Y.; Conda-Sheridan, M. Targeting SARS-CoV-2 Receptor Binding Domain with Stapled Peptides: AnIn SilicoStudy. J. Phys. Chem. B 2021, 125, 6572–6586. [Google Scholar] [CrossRef]
- Stone, T.A.; Cole, G.B.; Nguyen, H.Q.; Sharpe, S.; Deber, C.M. Influence of Hydrocarbon-Stapling on Membrane Interactions of Synthetic Antimicrobial Peptides. Bioorg. Med. Chem. 2018, 26, 1189–1196. [Google Scholar] [CrossRef]
- Shi, G.; Kang, X.; Dong, F.; Liu, Y.; Zhu, N.; Hu, Y.; Xu, H.; Lao, X.; Zheng, H. DRAMP 3.0: An Enhanced Comprehensive Data Repository of Antimicrobial Peptides. Nucleic Acids Res. 2021, 50, D488–D496. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-W.; Grossmann, T.N.; Verdine, G.L. Synthesis of All-Hydrocarbon Stapled α-Helical Peptides by Ring-Closing Olefin Metathesis. Nat. Protoc. 2011, 6, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.S.; Lane, D.P.; Verma, C.S. Stapled Peptide Design: Principles and Roles of Computation. Drug Discov. Today 2016, 21, 1642–1653. [Google Scholar] [CrossRef] [PubMed]
- Bird, G.H.; Madani, N.; Perry, A.F.; Princiotto, A.M.; Supko, J.G.; He, X.; Gavathiotis, E.; Sodroski, J.G.; Walensky, L.D. Hydrocarbon Double-Stapling Remedies the Proteolytic Instability of a Lengthy Peptide Therapeutic. Proc. Natl. Acad. Sci. USA 2010, 107, 14093–14098. [Google Scholar] [CrossRef]
- Hilinski, G.J.; Kim, Y.W.; Hong, J.; Kutchukian, P.S.; Crenshaw, C.M.; Berkovitch, S.S.; Chang, A.; Ham, S.; Verdine, G.L. Stitched α-Helical Peptides via Bis Ring-Closing Metathesis. J. Am. Chem. Soc. 2014, 136, 12314–12322. [Google Scholar] [CrossRef]
- Zheng, M.; Wang, R.; Chen, S.; Zou, Y.; Yan, L.; Zhao, L.; Li, X. Design, Synthesis and Antifungal Activity of Stapled Aurein1.2 Peptides. Antibiotics 2021, 10, 956. [Google Scholar] [CrossRef]
- Luong, H.X.; Kim, D.H.; Lee, B.J.; Kim, Y.W. Antimicrobial Activity and Stability of Stapled Helices of Polybia-MP1. Arch. Pharm. Res. 2017, 40, 1414–1419. [Google Scholar] [CrossRef]
- Crawford, M.A.; Ward, A.E.; Gray, V.; Bailer, P.; Fisher, D.J.; Kubicka, E.; Cui, Z.; Luo, Q.; Gray, M.C.; Criss, A.K.; et al. Disparate Regions of the Human Chemokine CXCL10 Exhibit Broad-Spectrum Antimicrobial Activity against Biodefense and Antibiotic-Resistant Bacterial Pathogens. ACS Infect. Dis. 2023, 9, 122–139. [Google Scholar] [CrossRef]
- Souza, B.M.; Mendes, M.A.; Santos, L.D.; Marques, M.R.; César, L.M.M.; Almeida, R.N.A.; Pagnocca, F.C.; Konno, K.; Palma, M.S. Structural and Functional Characterization of Two Novel Peptide Toxins Isolated from the Venom of the Social Wasp Polybia Paulista. Peptides 2005, 26, 2157–2164. [Google Scholar] [CrossRef]
- Dinh, T.T.T.; Kim, D.H.; Luong, H.X.; Lee, B.J.; Kim, Y.W. Antimicrobial Activity of Doubly-Stapled Alanine/Lysine-Based Peptides. Bioorg. Med. Chem. Lett. 2015, 25, 4016–4019. [Google Scholar] [CrossRef]
- Mourtada, R.; Herce, H.D.; Yin, D.J.; Moroco, J.A.; Wales, T.E.; Engen, J.R.; Walensky, L.D. Design of Stapled Antimicrobial Peptides That Are Stable, Nontoxic and Kill Antibiotic-Resistant Bacteria in Mice. Nat. Biotechnol. 2019, 37, 1186–1197. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, W.; Gou, S.; Huang, H.; Yao, J.; Yang, Z.; Liu, H.; Zhong, C.; Liu, B.; Ni, J.; et al. Intramolecular Cyclization of the Antimicrobial Peptide Polybia-MPI with Triazole Stapling: Influence on Stability and Bioactivity. J. Pept. Sci. 2017, 23, 824–832. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Hu, Y.; Pu, Q.; He, T.; Zhang, Q.; Wu, W.; Xia, X.; Zhang, J. Novel Stapling by Lysine Tethering Provides Stable and Low Hemolytic Cationic Antimicrobial Peptides. J. Med. Chem. 2020, 63, 4081–4089. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.-M.; Moon, H.; Han, S.-W.; Kim, B.W.; Kim, D.-H.; Kim, B.M.; Lee, B.-J. Toxin-Activating Stapled Peptides Discovered by Structural Analysis Were Identified as New Therapeutic Candidates That Trigger Antibacterial Activity against Mycobacterium Tuberculosis in the Mycobacterium Smegmatis Model. Microorganisms 2021, 9, 568. [Google Scholar] [CrossRef]
- Bellmann-Sickert, K.; Stone, T.A.; Poulsen, B.E.; Deber, C.M. Efflux by Small Multidrug Resistance Proteins Is Inhibited by Membrane-Interactive Helix-Stapled Peptides. J. Biol. Chem. 2015, 290, 1752–1759. [Google Scholar] [CrossRef]
- Stark, M.; Liu, L.-P.; Deber, C.M. Cationic Hydrophobic Peptides with Antimicrobial Activity. Antimicrob. Agents Chemother. 2002, 46, 3585–3590. [Google Scholar] [CrossRef]
- Rozek, T.; Wegener, K.L.; Bowie, J.H.; Olver, I.N.; Carver, J.A.; Wallace, J.C.; Tyler, M.J. The Antibiotic and Anticancer Active Aurein Peptides from the Australian Bell Frogs Litoria Aurea and Litoria Raniformis. Eur. J. Biochem. 2000, 267, 5330–5341. [Google Scholar] [CrossRef]
- Jamieson, A.; Robertson, N. Regulation of Protein–Protein Interactions Using Stapled Peptides. Rep. Org. Chem. 2015, 65, 65–74. [Google Scholar] [CrossRef]
- Xie, X.; Gao, L.; Shull, A.Y.; Teng, Y. Stapled Peptides: Providing the Best of Both Worlds in Drug Development. Future Med. Chem. 2016, 8, 1969–1980. [Google Scholar] [CrossRef]
- Cromm, P.M.; Spiegel, J.; Grossmann, T.N. Hydrocarbon Stapled Peptides as Modulators of Biological Function. ACS Chem. Biol. 2015, 10, 1362–1375. [Google Scholar] [CrossRef]
- Walensky, L.D.; Kung, A.L.; Escher, I.; Malia, T.J.; Barbuto, S.; Wright, R.D.; Wagner, G.; Verdine, G.L.; Korsmeyer, S.J. Activation of Apoptosis in Vivo by a Hydrocarbon-Stapled BH3 Helix. Science 2004, 305, 1466–1470. [Google Scholar] [CrossRef] [PubMed]
- Glas, A.; Bier, D.; Hahne, G.; Rademacher, C.; Ottmann, C.; Grossmann, T.N. Constrained Peptides with Target-Adapted Cross-Links as Inhibitors of a Pathogenic Protein-Protein Interaction. Angew. Chem. Int. Ed. 2014, 53, 2489–2493. [Google Scholar] [CrossRef] [PubMed]
- Modell, A.E.; Blosser, S.L.; Arora, P.S. Systematic Targeting of Protein–Protein Interactions. Trends Pharmacol. Sci. 2016, 37, 702–713. [Google Scholar] [CrossRef] [PubMed]
- Pelay-Gimeno, M.; Glas, A.; Koch, O.; Grossmann, T.N. Structure-Based Design of Inhibitors of Protein-Protein Interactions: Mimicking Peptide Binding Epitopes. Angew. Chem. Int. Ed. 2015, 54, 8896–8927. [Google Scholar] [CrossRef]

| Modification Type | Advantages |
|---|---|
| INSERTION OF D-AMINO ACIDS | ↑ proteolytic stability ↑ membrane insertation |
| PEGYLATION | ↑ proteolytic stability ↓ toxicity ↑ biocompatibility ↑ plasma half-live |
| ACETYLATION | ↑ proteolytic stability |
| DIMERIZATION | ↑ antimicrobial activity ↑ membrane interaction ↑ membrane permeability |
| LIPIDATION | ↑ antimicrobial activity ↑ proteolytic stability ↑ membrane permeability ↑ bioavailability |
| CYCLIZATION | ↑ antimicrobial activity ↑ proteolytic stability |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lourenço, A.L.P.; Rios, T.B.; da Silva, Á.P.; Franco, O.L.; Ramada, M.H.S. Peptide Stapling Applied to Antimicrobial Peptides. Antibiotics 2023, 12, 1400. https://doi.org/10.3390/antibiotics12091400
Lourenço ALP, Rios TB, da Silva ÁP, Franco OL, Ramada MHS. Peptide Stapling Applied to Antimicrobial Peptides. Antibiotics. 2023; 12(9):1400. https://doi.org/10.3390/antibiotics12091400
Chicago/Turabian StyleLourenço, Ana Laura Pereira, Thuanny Borba Rios, Állan Pires da Silva, Octávio Luiz Franco, and Marcelo Henrique Soller Ramada. 2023. "Peptide Stapling Applied to Antimicrobial Peptides" Antibiotics 12, no. 9: 1400. https://doi.org/10.3390/antibiotics12091400
APA StyleLourenço, A. L. P., Rios, T. B., da Silva, Á. P., Franco, O. L., & Ramada, M. H. S. (2023). Peptide Stapling Applied to Antimicrobial Peptides. Antibiotics, 12(9), 1400. https://doi.org/10.3390/antibiotics12091400