


Computationally Designed AMPs with Antibacterial and Antibiofilm Activity against MDR Acinetobacter baumannii

 ,
, 
Abstract
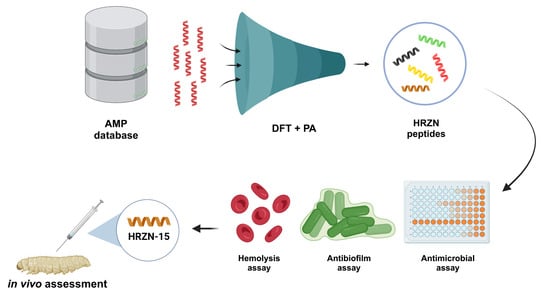
1. Introduction
2. Results
2.1. Design of Peptides
2.2. Antimicrobial Activity Prediction
2.3. Antimicrobial Susceptibility Testing
2.4. Time-Kill Kinetics
2.5. Antibiofilm Assays
2.5.1. Minimum Biofilm Inhibitory Concentration (MBIC)
2.5.2. Minimum Biofilm Eradication Concentration (MBEC)
2.6. Mechanism of Action, Scanning Electron Microscopy, and Resistance Induction
2.7. Toxicity Assessment
3. Discussion
4. Materials and Methods
4.1. DFT plus PA Computational Approach to Design New Peptides
4.1.1. HRZN-13 and -14
4.1.2. HRZN-15
4.1.3. HRZN-16 and -17
4.2. Peptide Synthesis
4.3. Bacterial Strains
4.4. Screening Peptides for Antibacterial and Antibiofilm Activities
4.5. Minimum Inhibitory Concentration (MIC)
4.6. Time-Kill Kinetics
4.7. Minimum Biofilm Inhibitory Concentration (MBIC)
4.8. Minimum Biofilm Eradication Concentration (MBEC)
4.9. Resistance Induction
4.10. Membrane Permeabilization Assay
4.11. Membrane Depolarization Assay
4.12. Scanning Electron Microscopy
4.13. Hemolysis
4.14. Waxworm Toxicity Testing
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Prioritization of Pathogens to Guide Discovery, Research and Development of New Antibiotics for Drug-Resistant Bacterial Infections, Including Tuberculosis; World Health Organization: Geneva, Switzerland, 2023; pp. 41–79. [Google Scholar]
- Heitkamp, R.A.; Li, P.; Mende, K.; Demons, S.T.; Tribble, D.R.; Tyner, S.D. Association of Enterococcus spp. with Severe Combat Extremity Injury, Intensive Care, and Polymicrobial Wound Infection. Surg. Infect. 2018, 19, 95–103. [Google Scholar] [CrossRef]
- Calhoun, J.H.; Murray, C.K.; Manring, M.M. Multidrug-resistant organisms in military wounds from Iraq and Afghanistan. Clin. Orthop. Relat. Res. 2008, 466, 1356–1362. [Google Scholar] [CrossRef]
- Yun, H.C.; Murray, C.K. Infection Prevention in the Deployed Environment. US Army Med. Dep. J. 2016, 114–118. [Google Scholar]
- Peters, B.M.; Jabra-Rizk, M.A.; O’May, G.A.; Costerton, J.W.; Shirtliff, M.E. Polymicrobial interactions: Impact on pathogenesis and human disease. Clin. Microbiol. Rev. 2012, 25, 193–213. [Google Scholar] [CrossRef] [PubMed]
- Duplantier, A.J.; van Hoek, M.L. The Human Cathelicidin Antimicrobial Peptide LL-37 as a Potential Treatment for Polymicrobial Infected Wounds. Front. Immunol. 2013, 4, 143. [Google Scholar] [CrossRef]
- Penesyan, A.; Nagy, S.S.; Kjelleberg, S.; Gillings, M.R.; Paulsen, I.T. Rapid microevolution of biofilm cells in response to antibiotics. NPJ Biofilms Microbiomes 2019, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.; Riddle, M.S.; Danko, J.R.; Blazes, D.L.; Hayden, R.; Tasker, S.A.; Dunne, J.R. Trauma-related infections in battlefield casualties from Iraq. Ann. Surg. 2007, 245, 803–811. [Google Scholar] [CrossRef]
- Sheppard, F.R.; Keiser, P.; Craft, D.W.; Gage, F.; Robson, M.; Brown, T.S.; Petersen, K.; Sincock, S.; Kasper, M.; Hawksworth, J.; et al. The majority of US combat casualty soft-tissue wounds are not infected or colonized upon arrival or during treatment at a continental US military medical facility. Am. J. Surg. 2010, 200, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Ressner, R.A.; Murray, C.K.; Griffith, M.E.; Rasnake, M.S.; Hospenthal, D.R.; Wolf, S.E. Outcomes of bacteremia in burn patients involved in combat operations overseas. J. Am. Coll. Surg. 2008, 206, 439–444. [Google Scholar] [CrossRef]
- Murray, C.K.; Obremskey, W.T.; Hsu, J.R.; Andersen, R.C.; Calhoun, J.H.; Clasper, J.C.; Whitman, T.J.; Curry, T.K.; Fleming, M.E.; Wenke, J.C.; et al. Prevention of infections associated with combat-related extremity injuries. J. Trauma 2011, 71, S235–S257. [Google Scholar] [CrossRef]
- Sensenig, R.A.; Murray, C.K.; Mende, K.; Wolf, S.E.; Chung, K.K.; Hospenthal, D.R.; Yun, H.C. Longitudinal characterization of Acinetobacter baumannii-calcoaceticus complex, Klebsiella pneumoniae, and methicillin-resistant Staphylococcus aureus colonizing and infecting combat casualties. Am J. Infect. Control 2012, 40, 183–185. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, P.; Baker, S. Developing new therapeutic approaches for treating infections caused by multi-drug resistant Acinetobacter baumannii: Acinetobacter baumannii therapeutics. J. Infect. 2020, 81, 857–861. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.M.C.; Dean, S.N.; Propst, C.N.; Bishop, B.M.; van Hoek, M.L. Komodo dragon-inspired synthetic peptide DRGN-1 promotes wound-healing of a mixed-biofilm infected wound. NPJ Biofilms Microbiomes 2017, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Dean, S.N.; Bishop, B.M.; van Hoek, M.L. Natural and synthetic cathelicidin peptides with anti-microbial and anti-biofilm activity against Staphylococcus aureus. BMC Microbiol. 2011, 11, 114. [Google Scholar] [CrossRef]
- Barksdale, S.M.; Hrifko, E.J.; Chung, E.M.; van Hoek, M.L. Peptides from American alligator plasma are antimicrobial against multi-drug resistant bacterial pathogens including Acinetobacter baumannii. BMC Microbiol. 2016, 16, 189. [Google Scholar] [CrossRef]
- Barksdale, S.M.; Hrifko, E.J.; van Hoek, M.L. Cathelicidin antimicrobial peptide from Alligator mississippiensis has antibacterial activity against multi-drug resistant Acinetobacter baumanii and Klebsiella pneumoniae. Dev. Comp. Immunol. 2017, 70, 135–144. [Google Scholar] [CrossRef]
- Dean, S.N.; Bishop, B.M.; van Hoek, M.L. Susceptibility of Pseudomonas aeruginosa Biofilm to Alpha-Helical Peptides: D-enantiomer of LL-37. Front. Microbiol. 2011, 2, 128. [Google Scholar] [CrossRef]
- Amer, L.S.; Bishop, B.M.; van Hoek, M.L. Antimicrobial and antibiofilm activity of cathelicidins and short, synthetic peptides against Francisella. Biochem. Biophys. Res. Commun. 2010, 396, 246–251. [Google Scholar] [CrossRef]
- Chung, M.C.; Dean, S.N.; van Hoek, M.L. Acyl carrier protein is a bacterial cytoplasmic target of cationic antimicrobial peptide LL-37. Biochem. J. 2015, 470, 243–253. [Google Scholar] [CrossRef]
- Han, S.; Bishop, B.M.; van Hoek, M.L. Antimicrobial activity of human beta-defensins and induction by Francisella. Biochem. Biophys. Res. Commun. 2008, 371, 670–674. [Google Scholar] [CrossRef]
- Blower, R.J.; Barksdale, S.M.; van Hoek, M.L. Snake Cathelicidin NA-CATH and Smaller Helical Antimicrobial Peptides Are Effective against Burkholderia thailandensis. PLoS Negl. Trop. Dis. 2015, 9, e0003862. [Google Scholar] [CrossRef]
- Blower, R.J.; Popov, S.G.; van Hoek, M.L. Cathelicidin peptide rescues G. mellonella infected with B. anthracis. Virulence 2018, 9, 287–293. [Google Scholar] [CrossRef] [PubMed]
- de Latour, F.A.; Amer, L.S.; Papanstasiou, E.A.; Bishop, B.M.; van Hoek, M.L. Antimicrobial activity of the Naja atra cathelicidin and related small peptides. Biochem. Biophys. Res. Commun. 2010, 396, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Dean, S.N.; Milton, M.E.; Cavanagh, J.; van Hoek, M.L. Francisella novicida Two-Component System Response Regulator BfpR Modulates iglC Gene Expression, Antimicrobial Peptide Resistance, and Biofilm Production. Front. Cell. Infect. Microbiol. 2020, 10, 82. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Singh, S.; van Hoek, M.L. Short, Synthetic Cationic Peptides Have Antibacterial Activity against Mycobacterium smegmatis by Forming Pores in Membrane and Synergizing with Antibiotics. Antibiotics 2015, 4, 358–378. [Google Scholar] [CrossRef]
- Hitt, S.J.; Bishop, B.M.; van Hoek, M.L. Komodo-dragon cathelicidin-inspired peptides are antibacterial against carbapenem-resistant Klebsiella pneumoniae. J. Med. Microbiol. 2020, 69, 1262–1272. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, A.; Gupta, K.; van Hoek, M.L. Characterization of Cimex lectularius (bedbug) defensin peptide and its antimicrobial activity against human skin microflora. Biochem. Biophys. Res. Commun. 2016, 470, 955–960. [Google Scholar] [CrossRef][Green Version]
- Kaushal, A.; Gupta, K.; Shah, R.; van Hoek, M.L. Antimicrobial activity of mosquito cecropin peptides against Francisella. Dev. Comp. Immunol. 2016, 63, 171–180. [Google Scholar] [CrossRef]
- van Hoek, M.L. Antimicrobial peptides in reptiles. Pharmaceuticals 2014, 7, 723–753. [Google Scholar] [CrossRef]
- Rajasekaran, G.; Kim, E.Y.; Shin, S.Y. LL-37-derived membrane-active FK-13 analogs possessing cell selectivity, anti-biofilm activity and synergy with chloramphenicol and anti-inflammatory activity. Biochim. Biophys. Acta (BBA)-Biomembr. 2017, 1859, 722–733. [Google Scholar] [CrossRef]
- Feng, X.; Sambanthamoorthy, K.; Palys, T.; Paranavitana, C. The human antimicrobial peptide LL-37 and its fragments possess both antimicrobial and antibiofilm activities against multidrug-resistant Acinetobacter baumannii. Peptides 2013, 49, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Deslouches, B.; Steckbeck, J.D.; Craigo, J.K.; Doi, Y.; Mietzner, T.A.; Montelaro, R.C. Rational Design of Engineered Cationic Antimicrobial Peptides Consisting Exclusively of Arginine and Tryptophan, and Their Activity against Multidrug-Resistant Pathogens. Antimicrob. Agents Chemother. 2013, 57, 2511–2521. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Dobbins, D.; Ghahramani, P.; Friedland, I.; Steckbeck, J. A Phase 1 Study of the Safety, Tolerability, and Pharmacokinetics of Single Ascending Doses of a First-in-Human Engineered Cationic Peptide, PLG0206, Intravenously Administered in Healthy Subjects. Antimicrob. Agents Chemother. 2022, 66, e01441-21. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.K.; Seo, C.H.; Luchian, T.; Park, Y. Pse-T2, an Antimicrobial Peptide with High-Level, Broad-Spectrum Antimicrobial Potency and Skin Biocompatibility against Multidrug-Resistant Pseudomonas aeruginosa Infection. Antimicrob. Agents Chemother. 2018, 62, 10-1182. [Google Scholar] [CrossRef]
- Flamm, R.K.; Rhomberg, P.R.; Simpson, K.M.; Farrell, D.J.; Sader, H.S.; Jones, R.N. In Vitro Spectrum of Pexiganan Activity When Tested against Pathogens from Diabetic Foot Infections and with Selected Resistance Mechanisms. Antimicrob. Agents Chemother. 2015, 59, 1751–1754. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2015, 44, D1087–D1093. [Google Scholar] [CrossRef]
- Cardoso, M.H.; Orozco, R.Q.; Rezende, S.B.; Rodrigues, G.; Oshiro, K.G.N.; Cândido, E.S.; Franco, O.L. Computer-Aided Design of Antimicrobial Peptides: Are We Generating Effective Drug Candidates? Front. Microbiol. 2020, 10, 3097. [Google Scholar] [CrossRef]
- Wang, G.; Vaisman, I.I.; van Hoek, M.L. Machine Learning Prediction of Antimicrobial Peptides. In Computational Peptide Science: Methods and Protocols; Simonson, T., Ed.; Springer: New York, NY, USA, 2022; pp. 1–37. [Google Scholar]
- Bobde, S.S.; Alsaab, F.M.; Wang, G.; Van Hoek, M.L. Ab initio Designed Antimicrobial Peptides Against Gram-Negative Bacteria. Front. Microbiol. 2021, 12, 3460. [Google Scholar] [CrossRef]
- Kyriakidis, I.; Vasileiou, E.; Pana, Z.D.; Tragiannidis, A. Acinetobacter baumannii Antibiotic Resistance Mechanisms. Pathogens 2021, 10, 373. [Google Scholar] [CrossRef]
- Lee, C.R.; Lee, J.H.; Park, M.; Park, K.S.; Bae, I.K.; Kim, Y.B.; Cha, C.J.; Jeong, B.C.; Lee, S.H. Biology of Acinetobacter baumannii: Pathogenesis, Antibiotic Resistance Mechanisms, and Prospective Treatment Options. Front. Cell. Infect. Microbiol. 2017, 7, 55. [Google Scholar] [CrossRef]
- Witten, J.; Witten, Z. Deep learning regression model for antimicrobial peptide design. bioRxiv 2019. [CrossRef]
- Novković, M.; Simunić, J.; Bojović, V.; Tossi, A.; Juretić, D. DADP: The database of anuran defense peptides. Bioinformatics 2012, 28, 1406–1407. [Google Scholar] [CrossRef] [PubMed]
- Pirtskhalava, M.; Gabrielian, A.; Cruz, P.; Griggs, H.L.; Squires, R.B.; Hurt, D.E.; Grigolava, M.; Chubinidze, M.; Gogoladze, G.; Vishnepolsky, B.; et al. DBAASP v.2: An enhanced database of structure and antimicrobial/cytotoxic activity of natural and synthetic peptides. Nucleic Acids Res. 2015, 44, D1104–D1112. [Google Scholar] [CrossRef]
- Fan, L.; Sun, J.; Zhou, M.; Zhou, J.; Lao, X.; Zheng, H.; Xu, H. DRAMP: A comprehensive data repository of antimicrobial peptides. Sci. Rep. 2016, 6, 24482. [Google Scholar] [CrossRef] [PubMed]
- Piotto, S.P.; Sessa, L.; Concilio, S.; Iannelli, P. YADAMP: Yet another database of antimicrobial peptides. Int. J. Antimicrob. Agents 2012, 39, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Wang, G. Ab initio design of potent anti-MRSA peptides based on database filtering technology. J. Am. Chem. Soc. 2012, 134, 12426–12429. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Mourtada, R.; Herce, H.D.; Yin, D.J.; Moroco, J.A.; Wales, T.E.; Engen, J.R.; Walensky, L.D. Design of stapled antimicrobial peptides that are stable, nontoxic and kill antibiotic-resistant bacteria in mice. Nat. Biotechnol. 2019, 37, 1186–1197. [Google Scholar] [CrossRef]
- Lee, E.Y.; Fulan, B.M.; Wong, G.C.; Ferguson, A.L. Mapping membrane activity in undiscovered peptide sequence space using machine learning. Proc. Natl. Acad. Sci. USA 2016, 113, 13588–13593. [Google Scholar] [CrossRef]
- Waghu, F.H.; Gopi, L.; Barai, R.S.; Ramteke, P.; Nizami, B.; Idicula-Thomas, S. CAMP: Collection of sequences and structures of antimicrobial peptides. Nucleic Acids Res. 2014, 42, D1154–D1158. [Google Scholar] [CrossRef]
- Waghu, F.H.; Barai, R.S.; Gurung, P.; Idicula-Thomas, S. CAMPR3: A database on sequences, structures and signatures of antimicrobial peptides. Nucleic Acids Res. 2016, 44, D1094–D1097. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.; Karnik, S.; Nilawe, P.; Jayaraman, V.K.; Idicula-Thomas, S. ClassAMP: A Prediction Tool for Classification of Antimicrobial Peptides. IEEE/ACM Trans. Comput. Biol. Bioinform. 2012, 9, 1535–1538. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Karnik, S.; Barai, R.S.; Jayaraman, V.K.; Idicula-Thomas, S. CAMP: A useful resource for research on antimicrobial peptides. Nucleic Acids Res. 2009, 38, D774–D780. [Google Scholar] [CrossRef] [PubMed]
- Vishnepolsky, B.; Pirtskhalava, M. Prediction of Linear Cationic Antimicrobial Peptides Based on Characteristics Responsible for Their Interaction with the Membranes. J. Chem. Inf. Model. 2014, 54, 1512–1523. [Google Scholar] [CrossRef] [PubMed]
- Dean, S.N.; Alvarez, J.A.E.; Zabetakis, D.; Walper, S.A.; Malanoski, A.P. PepVAE: Variational Autoencoder Framework for Antimicrobial Peptide Generation and Activity Prediction. Front. Microbiol. 2021, 12, 725727. [Google Scholar] [CrossRef]
- Thompson, M.G.; Black, C.C.; Pavlicek, R.L.; Honnold, C.L.; Wise, M.C.; Alamneh, Y.A.; Moon, J.K.; Kessler, J.L.; Si, Y.; Williams, R.; et al. Validation of a novel murine wound model of Acinetobacter baumannii infection. Antimicrob. Agents Chemother. 2014, 58, 1332–1342. [Google Scholar] [CrossRef]
- Zurawski, D.V.; Black, C.C.; Alamneh, Y.A.; Biggemann, L.; Banerjee, J.; Thompson, M.G.; Wise, M.C.; Honnold, C.L.; Kim, R.K.; Paranavitana, C.; et al. A Porcine Wound Model of Acinetobacter baumannii Infection. Adv. Wound Care 2019, 8, 14–27. [Google Scholar] [CrossRef]
- Zurawski, D.V.; Thompson, M.G.; McQueary, C.N.; Matalka, M.N.; Sahl, J.W.; Craft, D.W.; Rasko, D.A. Genome sequences of four divergent multidrug-resistant Acinetobacter baumannii strains isolated from patients with sepsis or osteomyelitis. J. Bacteriol. 2012, 194, 1619–1620. [Google Scholar] [CrossRef]
- Jacobs, A.C.; Thompson, M.G.; Black, C.C.; Kessler, J.L.; Clark, L.P.; McQueary, C.N.; Gancz, H.Y.; Corey, B.W.; Moon, J.K.; Si, Y.; et al. AB5075, a Highly Virulent Isolate of Acinetobacter baumannii, as a Model Strain for the Evaluation of Pathogenesis and Antimicrobial Treatments. mBio 2014, 5, e01076-14. [Google Scholar] [CrossRef]
- Wiegand, I.; Hilpert, K.; Hancock, R.E.W. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef]
- Zhao, J.; Han, M.-L.; Zhu, Y.; Lin, Y.-W.; Wang, Y.-W.; Lu, J.; Hu, Y.; Tony Zhou, Q.; Velkov, T.; Li, J. Comparative metabolomics reveals key pathways associated with the synergistic activity of polymyxin B and rifampicin combination against multidrug-resistant Acinetobacter baumannii. Biochem. Pharmacol. 2021, 184, 114400. [Google Scholar] [CrossRef]
- Mwangi, J.; Yin, Y.; Wang, G.; Yang, M.; Li, Y.; Zhang, Z.; Lai, R. The antimicrobial peptide ZY4 combats multidrug-resistant Pseudomonas aeruginosa and Acinetobacter baumannii infection. Proc. Natl. Acad. Sci. USA 2019, 116, 26516–26522. [Google Scholar] [CrossRef]
- Akbari, R.; Hakemi Vala, M.; Sabatier, J.-M.; Pooshang Bagheri, K. Fast killing kinetics, significant therapeutic index, and high stability of melittin-derived antimicrobial peptide. Amino Acids 2022, 54, 1275–1285. [Google Scholar] [CrossRef]
- de la Fuente-Núñez, C.; Reffuveille, F.; Haney, E.F.; Straus, S.K.; Hancock, R.E.W. Broad-Spectrum Anti-biofilm Peptide That Targets a Cellular Stress Response. PLoS Pathog. 2014, 10, e1004152. [Google Scholar] [CrossRef] [PubMed]
- Overhage, J.; Campisano, A.; Bains, M.; Torfs, E.C.; Rehm, B.H.; Hancock, R.E. Human host defense peptide LL-37 prevents bacterial biofilm formation. Infect. Immun. 2008, 76, 4176–4182. [Google Scholar] [CrossRef]
- Beganovic, M.; Luther, M.K.; Daffinee, K.E.; LaPlante, K.L. Biofilm prevention concentrations (BPC) of minocycline compared to polymyxin B, meropenem, and amikacin against Acinetobacter baumannii. Diagn. Microbiol. Infect. Dis. 2019, 94, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, A.M.; van Hoek, M.L. Development of a Defibrinated Human Blood Hemolysis Assay for Rapid Testing of Hemolytic Activity of Antimicrobial Peptides Compared to Computational Prediction with LL-37; George Mason Univeristy: Fairfax, VA, USA, 2023; manuscript in preparation; to be submitted. [Google Scholar]
- Oren, Z.; Lerman, J.C.; Gudmundsson, G.H.; Agerberth, B.; Shai, Y. Structure and organization of the human antimicrobial peptide LL-37 in phospholipid membranes: Relevance to the molecular basis for its non-cell-selective activity. Biochem. J. 1999, 341 Pt 3, 501–513. [Google Scholar] [CrossRef]
- Zurawski, D.V.; Banerjee, J.; Alamneh, Y.A.; Shearer, J.P.; Demons, S.T. Skin and Soft Tissue Models for Acinetobacter baumannii Infection. Methods Mol. Biol. 2019, 1946, 271–287. [Google Scholar] [CrossRef]
- Yang, H.; Lv, N.; Hu, L.; Liu, Y.; Cheng, J.; Ye, Y.; Li, J. In vivo activity of vancomycin combined with colistin against multidrug-resistant strains of Acinetobacter baumannii in a Galleria mellonella model. Infect. Dis. 2016, 48, 189–194. [Google Scholar] [CrossRef]
- Yang, H.; Chen, G.; Hu, L.; Liu, Y.; Cheng, J.; Li, H.; Ye, Y.; Li, J. In vivo activity of daptomycin/colistin combination therapy in a Galleria mellonella model of Acinetobacter baumannii infection. Int. J. Antimicrob. Agents 2015, 45, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Yang, H.; Hu, L.; Ye, Y.; Li, J. Activity of levofloxacin in combination with colistin against Acinetobacter baumannii: In vitro and in a Galleria mellonella model. J. Microbiol. Immunol. Infect. 2017, 50, 821–830. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wand, M.E.; Bock, L.J.; Turton, J.F.; Nugent, P.G.; Sutton, J.M. Acinetobacter baumannii virulence is enhanced in Galleria mellonella following biofilm adaptation. J. Med. Microbiol. 2012, 61, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Ten, K.E.; Muzahid, N.H.; Rahman, S.; Tan, H.S. Use of the waxworm Galleria mellonella larvae as an infection model to study Acinetobacter baumannii. PLoS ONE 2023, 18, e0283960. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Duma, L.; Rossez, Y. Galleria mellonella as a Good Model to Study Acinetobacter baumannii Pathogenesis. Pathogens 2021, 10, 1483. [Google Scholar] [CrossRef]
- Peleg, A.Y.; Jara, S.; Monga, D.; Eliopoulos, G.M.; Moellering, R.C., Jr.; Mylonakis, E. Galleria mellonella as a model system to study Acinetobacter baumannii pathogenesis and therapeutics. Antimicrob. Agents Chemother. 2009, 53, 2605–2609. [Google Scholar] [CrossRef]
- Hornsey, M.; Wareham, D.W. In vivo efficacy of glycopeptide-colistin combination therapies in a Galleria mellonella model of Acinetobacter baumannii infection. Antimicrob. Agents Chemother. 2011, 55, 3534–3537. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Håkansson, J.; Ringstad, L.; Björn, C. Antimicrobial Peptides: An Emerging Category of Therapeutic Agents. Front. Cell. Infect. Microbiol. 2016, 6, 194. [Google Scholar] [CrossRef]
- Jiang, Z.; Vasil, A.I.; Gera, L.; Vasil, M.L.; Hodges, R.S. Rational design of alpha-helical antimicrobial peptides to target Gram-negative pathogens, Acinetobacter baumannii and Pseudomonas aeruginosa: Utilization of charge, ‘specificity determinants,’ total hydrophobicity, hydrophobe type and location as design parameters to improve the therapeutic ratio. Chem. Biol. Drug Des. 2011, 77, 225–240. [Google Scholar] [CrossRef]
- Dennison, S.R.; Phoenix, D.A. Influence of C-Terminal Amidation on the Efficacy of Modelin-5. Biochemistry 2011, 50, 1514–1523. [Google Scholar] [CrossRef]
- Andreu, D.; Rivas, L. Animal antimicrobial peptides: An overview. Pept. Sci. 1998, 47, 415–433. [Google Scholar] [CrossRef]
- Shai, Y. Mode of action of membrane active antimicrobial peptides. Pept. Sci. 2002, 66, 236–248. [Google Scholar] [CrossRef]
- Xhindoli, D.; Pacor, S.; Benincasa, M.; Scocchi, M.; Gennaro, R.; Tossi, A. The human cathelicidin LL-37—A pore-forming antibacterial peptide and host-cell modulator. Biochim. Biophys. Acta (BBA)-Biomembr. 2016, 1858, 546–566. [Google Scholar] [CrossRef] [PubMed]
- Ludtke, S.J.; He, K.; Heller, W.T.; Harroun, T.A.; Yang, L.; Huang, H.W. Membrane Pores Induced by Magainin. Biochemistry 1996, 35, 13723–13728. [Google Scholar] [CrossRef] [PubMed]
- Browne, K.; Chakraborty, S.; Chen, R.; Willcox, M.D.; Black, D.S.; Walsh, W.R.; Kumar, N. A New Era of Antibiotics: The Clinical Potential of Antimicrobial Peptides. Int. J. Mol. Sci. 2020, 21, 7047. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, F.C.; Cardoso, M.H.; Gil-Ley, A.; Luchi, L.V.; da Silva, M.G.L.; Macedo, M.L.R.; de la Fuente-Nunez, C.; Franco, O.L. Geometric deep learning as a potential tool for antimicrobial peptide prediction. Front. Bioinform. 2023, 3, 1216362. [Google Scholar] [CrossRef] [PubMed]
- Cesaro, A.; Torres, M.T.; de la Fuente-Nunez, C. Methods for the design and characterization of peptide antibiotics. Methods Enzymol. 2022, 663, 303–326. [Google Scholar] [CrossRef]
- Porto, W.F.; Irazazabal, L.; Alves, E.S.F.; Ribeiro, S.M.; Matos, C.O.; Pires, A.S.; Fensterseifer, I.C.M.; Miranda, V.J.; Haney, E.F.; Humblot, V.; et al. In silico optimization of a guava antimicrobial peptide enables combinatorial exploration for peptide design. Nat. Commun. 2018, 9, 1490. [Google Scholar] [CrossRef]
- Torres, M.D.T.; Cao, J.; Franco, O.L.; Lu, T.K.; de la Fuente-Nunez, C. Synthetic Biology and Computer-Based Frameworks for Antimicrobial Peptide Discovery. ACS Nano 2021, 15, 2143–2164. [Google Scholar] [CrossRef]
- Torres, M.D.T.; Sothiselvam, S.; Lu, T.K.; de la Fuente-Nunez, C. Peptide Design Principles for Antimicrobial Applications. J. Mol. Biol. 2019, 431, 3547–3567. [Google Scholar] [CrossRef]
- Wan, F.; Kontogiorgos-Heintz, D.; de la Fuente-Nunez, C. Deep generative models for peptide design. Digit. Discov. 2022, 1, 195–208. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Chapple, D.S. Peptide Antibiotics. Antimicrob. Agents Chemother. 1999, 43, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Mant, C.T.; Farmer, S.W.; Hancock, R.E.; Vasil, M.L.; Hodges, R.S. Rational design of alpha-helical antimicrobial peptides with enhanced activities and specificity/therapeutic index. J. Biol. Chem. 2005, 280, 12316–12329. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Benz, R.; Hancock, R.E. Influence of proline residues on the antibacterial and synergistic activities of alpha-helical peptides. Biochemistry 1999, 38, 8102–8111. [Google Scholar] [CrossRef] [PubMed]
- Boyle, A.L. Chapter 3-Applications of de novo designed peptides. In Peptide Applications in Biomedicine, Biotechnology and Bioengineering; Koutsopoulos, S., Ed.; Woodhead Publishing: Sawston, UK, 2018; pp. 51–86. [Google Scholar] [CrossRef]
- Jacobs, A.C.; Zurawski, D.V. Laboratory Maintenance of Acinetobacter baumannii. Curr. Protoc. Microbiol. 2014, 35, 6G.1.1–6G.1.6. [Google Scholar] [CrossRef]
- Clinical and Laboratory Standards Institute (CLSI). Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard-Tenth Edition; CLSI Document M07-A10; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2015; ISBN 1-56238-987-4/1-56238-988-2. [Google Scholar]
- Sato, Y.; Unno, Y.; Ubagai, T.; Ono, Y. Sub-minimum inhibitory concentrations of colistin and polymyxin B promote Acinetobacter baumannii biofilm formation. PLoS ONE 2018, 13, e0194556. [Google Scholar] [CrossRef]
- Wang, J.; Woo, M.; Yan, C. Spot plating assay for the determination of survival and plating efficiency of Escherichia coli in sub-MIC levels of antibiotics. JEMI Methods 2017, 1, 26–29. [Google Scholar]
- Meng, Q.; Lin, F.; Ling, B. In Vitro Activity of Peptide Antibiotics in Combination With Other Antimicrobials on Extensively Drug-Resistant Acinetobacter baumannii in the Planktonic and Biofilm Cell. Front. Pharmacol. 2022, 13, 890955. [Google Scholar] [CrossRef]
- Innovotech. MBEC Assay® For High-Throughput Antimicrobial Susceptibility Testing of Biofilms PROCEDURAL MANUAL Version 2.1. 2019. Available online: https://www.innovotech.ca/mbec-procedural-manual-v2-2 (accessed on 25 January 2023).
- Ajish, C.; Yang, S.; Kumar, S.D.; Kim, E.Y.; Min, H.J.; Lee, C.W.; Shin, S.-H.; Shin, S.Y. A novel hybrid peptide composed of LfcinB6 and KR-12-a4 with enhanced antimicrobial, anti-inflammatory and anti-biofilm activities. Sci. Rep. 2022, 12, 4365. [Google Scholar] [CrossRef]
- Wu, M.; Maier, E.; Benz, R.; Hancock, R.E. Mechanism of interaction of different classes of cationic antimicrobial peptides with planar bilayers and with the cytoplasmic membrane of Escherichia coli. Biochemistry 1999, 38, 7235–7242. [Google Scholar] [CrossRef]
- Juba, M.L.; Porter, D.K.; Williams, E.H.; Rodriguez, C.A.; Barksdale, S.M.; Bishop, B.M. Helical cationic antimicrobial peptide length and its impact on membrane disruption. Biochim. Et Biophys. Acta (BBA)-Biomembr. 2015, 1848, 1081–1091. [Google Scholar] [CrossRef]
- Kalab, M.; Yang, A.-F.; Chabot, D. Conventional Scanning Electron Microscopy of Bacteria. Infocus Mag. 2008, 2008, 42–61. [Google Scholar] [CrossRef]
- Venkatesh Babu, G.; Perumal, P.; Muthu, S.; Pichai, S.; Sankar Narayan, K.; Malairaj, S. Enhanced method for High Spatial Resolution surface imaging and analysis of fungal spores using Scanning Electron Microscopy. Sci. Rep. 2018, 8, 16278. [Google Scholar] [CrossRef] [PubMed]
- Propst, C.N.; Pylypko, S.L.; Blower, R.J.; Ahmad, S.; Mansoor, M.; van Hoek, M.L. Francisella philomiragia Infection and Lethality in Mammalian Tissue Culture Cell Models, Galleria mellonella, and BALB/c Mice. Front. Microbiol. 2016, 7, 696. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence | Charge | % Hydrophobicity (Hydrophobic Moment) | Predicted Structure AlphaFold2 | Helical Wheel |
|---|---|---|---|---|---|
| HRZN-13 | FLWRISKFLGKKL-NH2 | +5 | 54% (0.537) |  |  |
| HRZN-14 | FLWRISKFLGRKL-NH2 | +5 | 54% (0.539) |  |  |
| HRZN-15 | FLPWISKFLGKIL-NH2 | +3 | 62% (0.659) |  |  |
| HRZN-16 | FLKKIWKLLGKLL-NH2 | +5 | 62% (0.877) |  |  |
| HRZN-17 | KLWKLLKKLGRLL-NH2 | +6 | 54% (0.804) |  |  |
| IDR-1018 | VRLIVAVRIWRR-NH2 | +5 | 67% (0.271) |  |  |
| LL-37 | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES | +6 | 35% (0.521) |  |  |
| Name | Ferguson | CAMPR3 | ClassAMP | DBAASP | PepVAE3 | ||||
|---|---|---|---|---|---|---|---|---|---|
| SVM | SVM | RF | ANN | DA | SVM | RF | |||
| HRZN-13 | 1 | 1.00 | 1.00 | AMP | 0.99 | 0.99 | 1.00 | AMP | AMP |
| HRZN-14 | 1 | 0.99 | 0.99 | AMP | 0.99 | 0.99 | 1.00 | AMP | AMP |
| HRZN-15 | 0.98 | 0.96 | 0.99 | AMP | 1.00 | 0.99 | 1.00 | AMP | AMP |
| HRZN-16 | 1 | 1.00 | 0.98 | AMP | 1.00 | 0.96 | 0.99 | AMP | AMP |
| HRZN-17 | 1 | 0.93 | 0.60 | AMP | 0.99 | 0.94 | 0.96 | AMP | AMP |
| IDR-1018 | 0.15 | 0.99 | 0.97 | AMP | 0.99 | 0.97 | 0.97 | Non-AMP | AMP |
| LL-37 | 1 | 0.76 | 0.75 | AMP | 0.77 | 0.97 | 0.95 | AMP | AMP |
| Peptide/Organism | A. baumannii AB5075 (MRSN959) | A. baumannii BAA-1710 | A. baumannii BAA-1794 | A. baumannii BAA-1800 |
|---|---|---|---|---|
| HRZN-13 | 32 | 32 | 64 | 64 |
| HRZN-14 | 16 | 32 | 64 | 32 |
| HRZN-15 | 4–8 | 4 | 4 | 4 |
| HRZN-16 | 16 | 16 | 16 | 16 |
| HRZN-17 | 32 | 32 | 32 | 32 |
| LL-37 | 8 | 32 | 16–32 | 8 |
| Polymyxin B | 0.25–0.5 | 0.5 | 0.5 | 0.5 |
| Peptide | MBIC100 (µg/mL) | MBEC (µg/mL) |
|---|---|---|
| HRZN-15 | 8 | 16 |
| LL-37 | 64 | ~32 |
| IDR-1018 | >64 * | >32 |
| Polymyxin B | 1 | * 2 |
| Strain | Source | Source Information |
|---|---|---|
| AB5075 (MRSN 959) | BEI Resources | Human tibia/osteomyelitis |
| BAA-1710 | ATCC | Human blood |
| BAA-1794 | ATCC | Human sputum |
| BAA-1800 | ATCC | Human deep trachea |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alsaab, F.M.; Dean, S.N.; Bobde, S.; Ascoli, G.G.; van Hoek, M.L. Computationally Designed AMPs with Antibacterial and Antibiofilm Activity against MDR Acinetobacter baumannii. Antibiotics 2023, 12, 1396. https://doi.org/10.3390/antibiotics12091396
Alsaab FM, Dean SN, Bobde S, Ascoli GG, van Hoek ML. Computationally Designed AMPs with Antibacterial and Antibiofilm Activity against MDR Acinetobacter baumannii. Antibiotics. 2023; 12(9):1396. https://doi.org/10.3390/antibiotics12091396
Chicago/Turabian StyleAlsaab, Fahad M., Scott N. Dean, Shravani Bobde, Gabriel G. Ascoli, and Monique L. van Hoek. 2023. "Computationally Designed AMPs with Antibacterial and Antibiofilm Activity against MDR Acinetobacter baumannii" Antibiotics 12, no. 9: 1396. https://doi.org/10.3390/antibiotics12091396
APA StyleAlsaab, F. M., Dean, S. N., Bobde, S., Ascoli, G. G., & van Hoek, M. L. (2023). Computationally Designed AMPs with Antibacterial and Antibiofilm Activity against MDR Acinetobacter baumannii. Antibiotics, 12(9), 1396. https://doi.org/10.3390/antibiotics12091396