
A Cysteine-Reloading Process Initiating the Biosynthesis of the Bicyclic Scaffold of Dithiolopyrrolones



Abstract
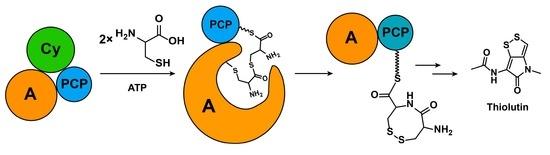
{kind=link}
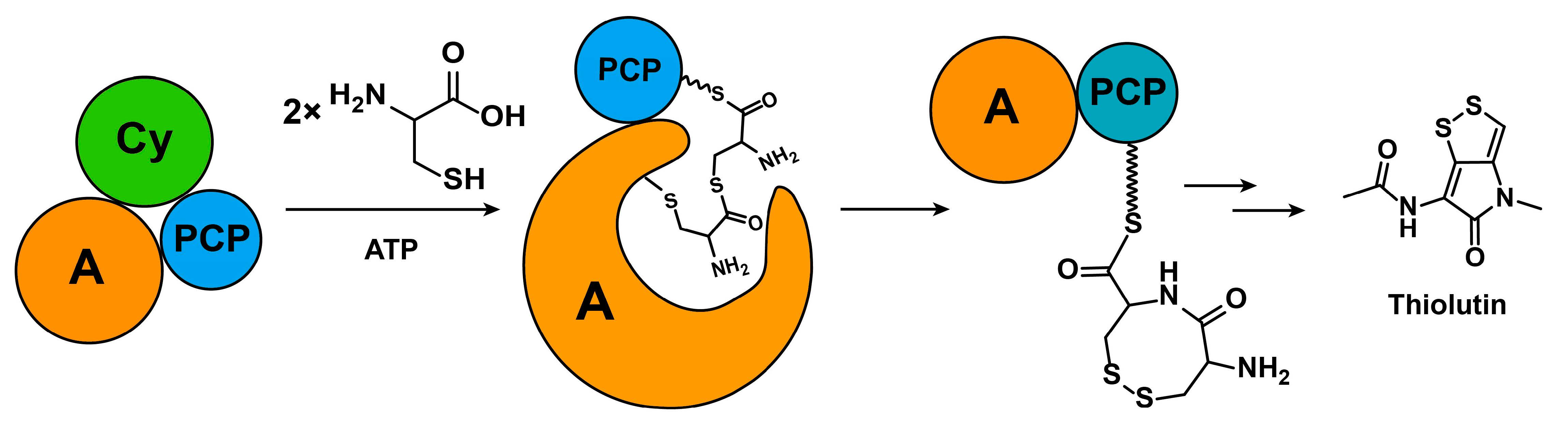
{kind=link}
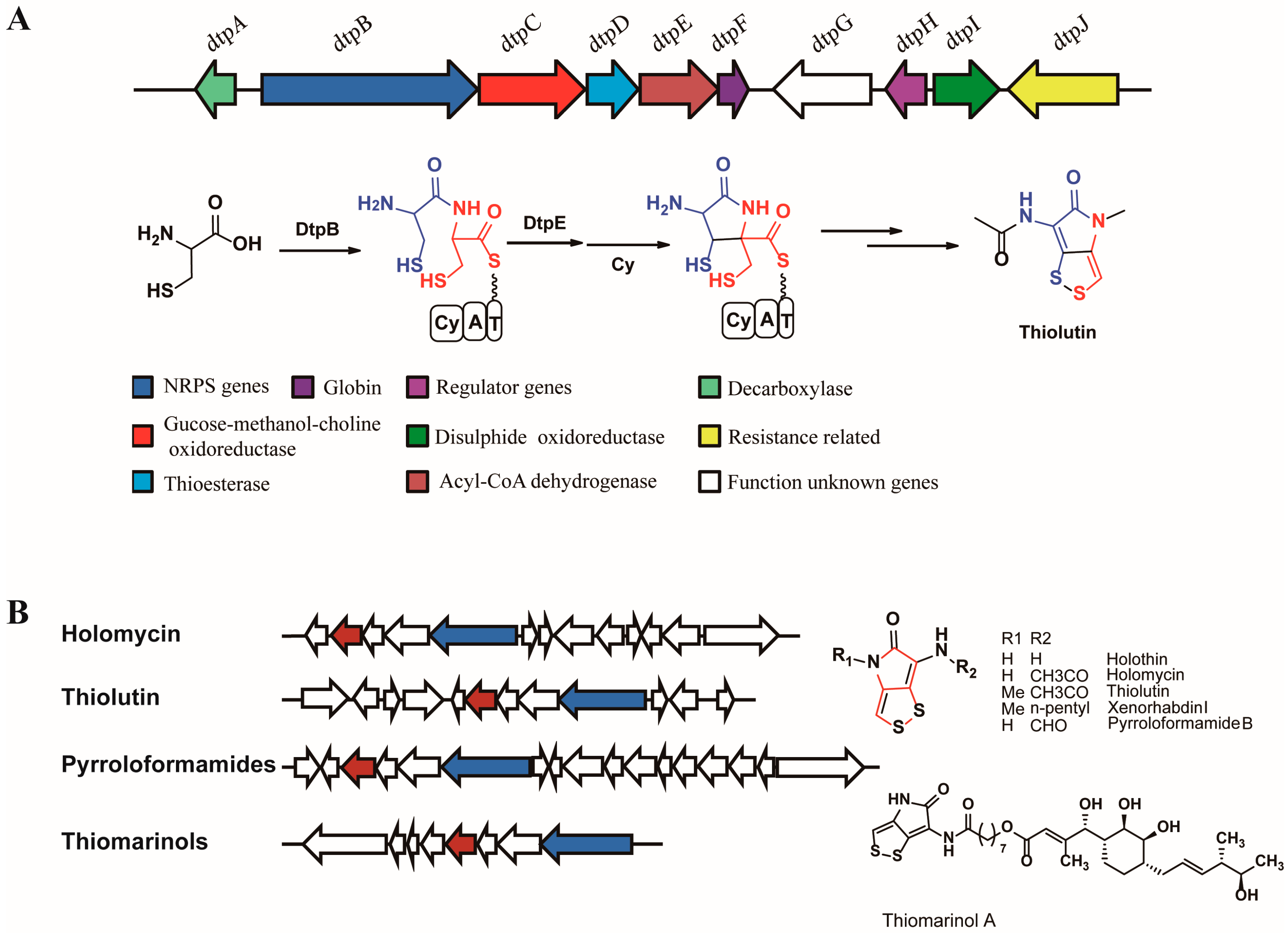
{kind=link}
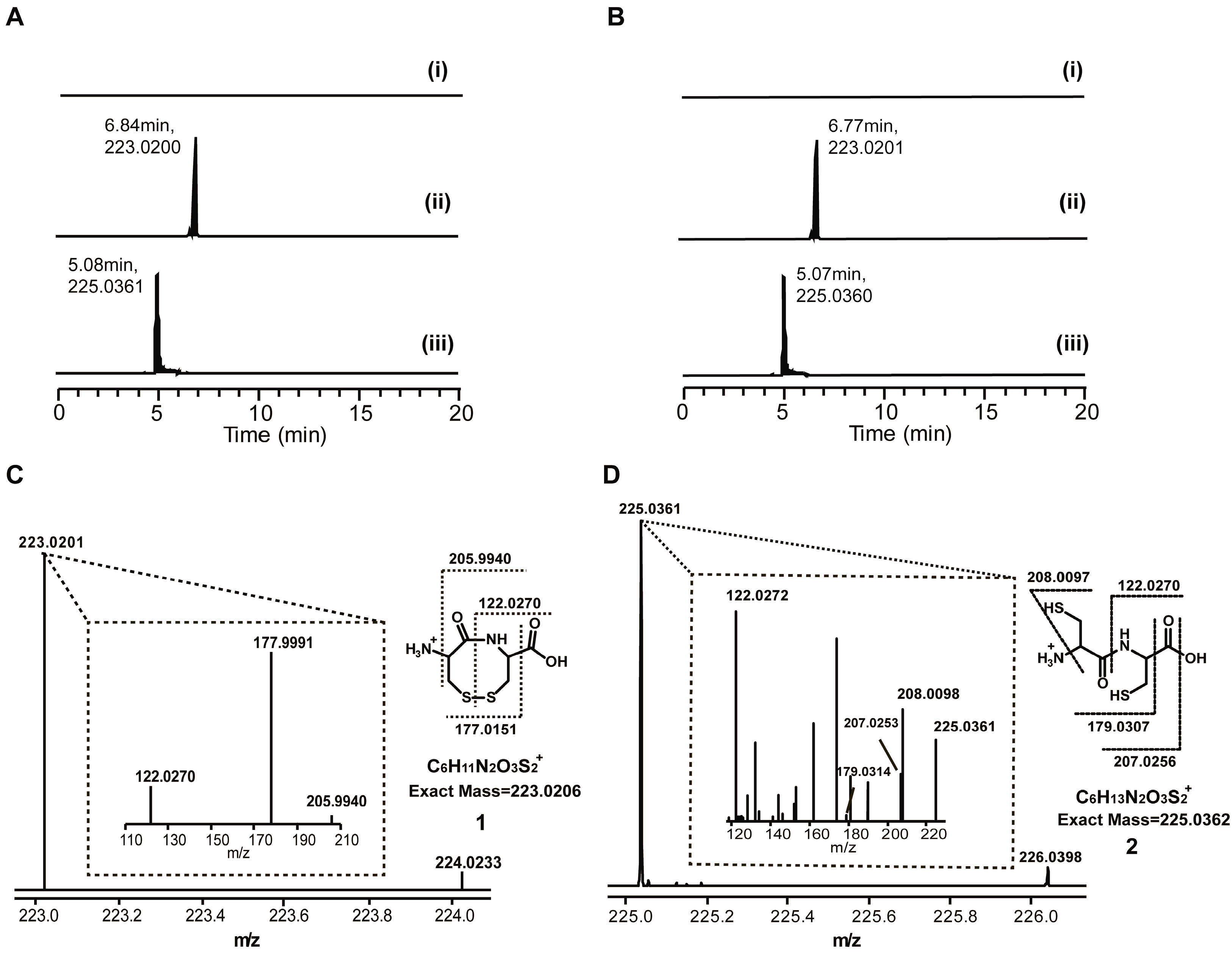
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
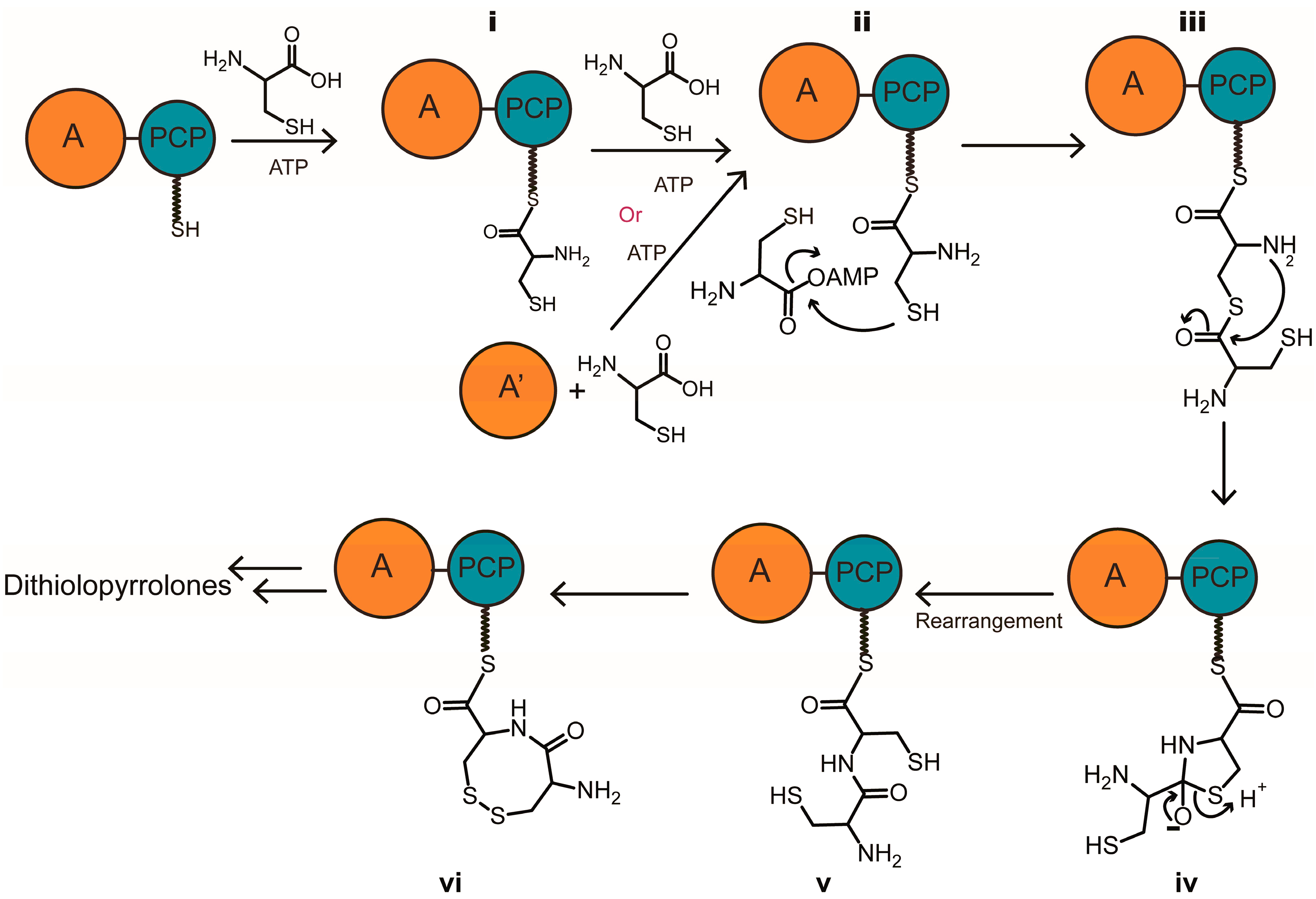
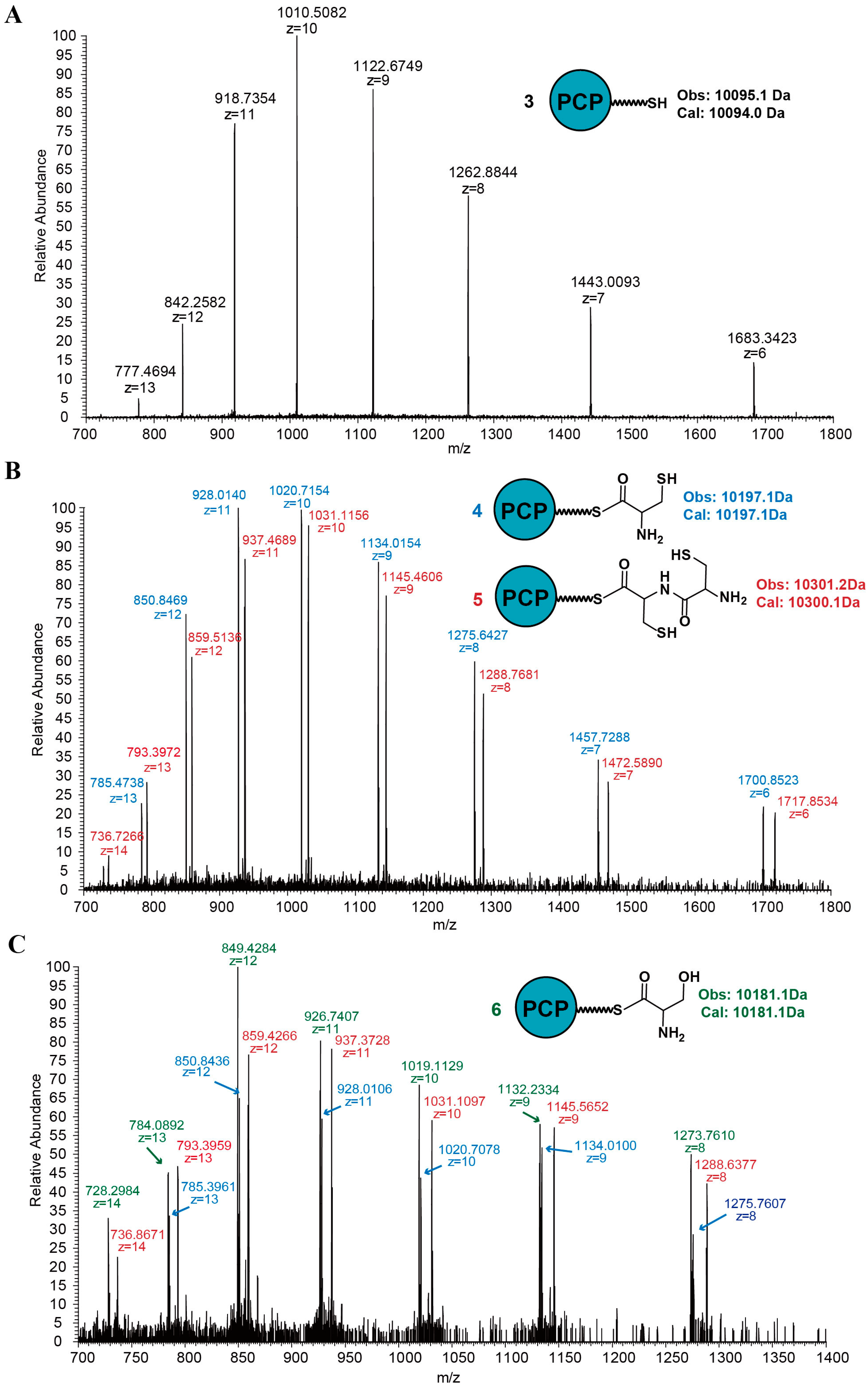
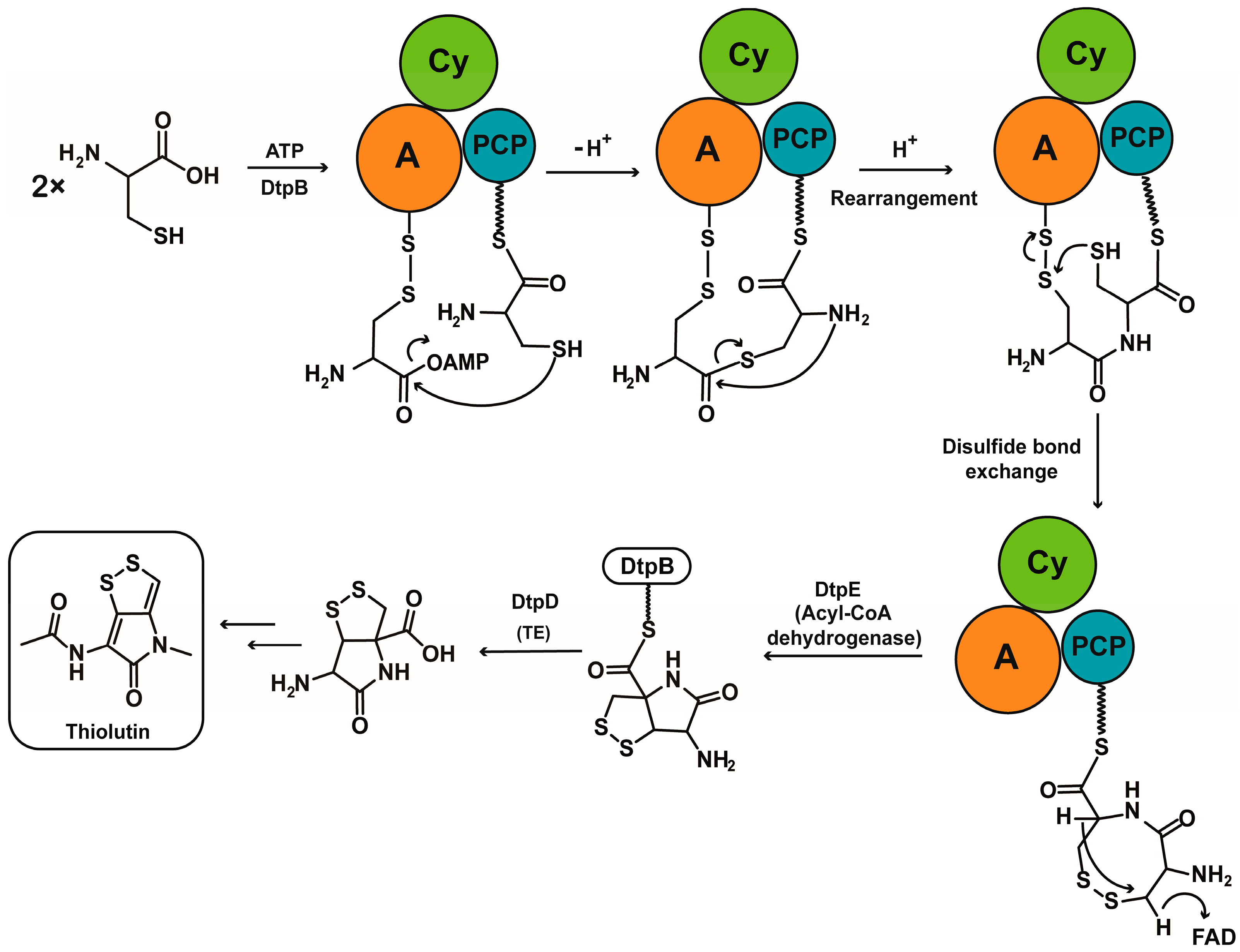
2.1. Discovery of a Novel Intermediate Derived from Cysteine Led to a Proposed Mechanism for the Cysteine Reloading Process
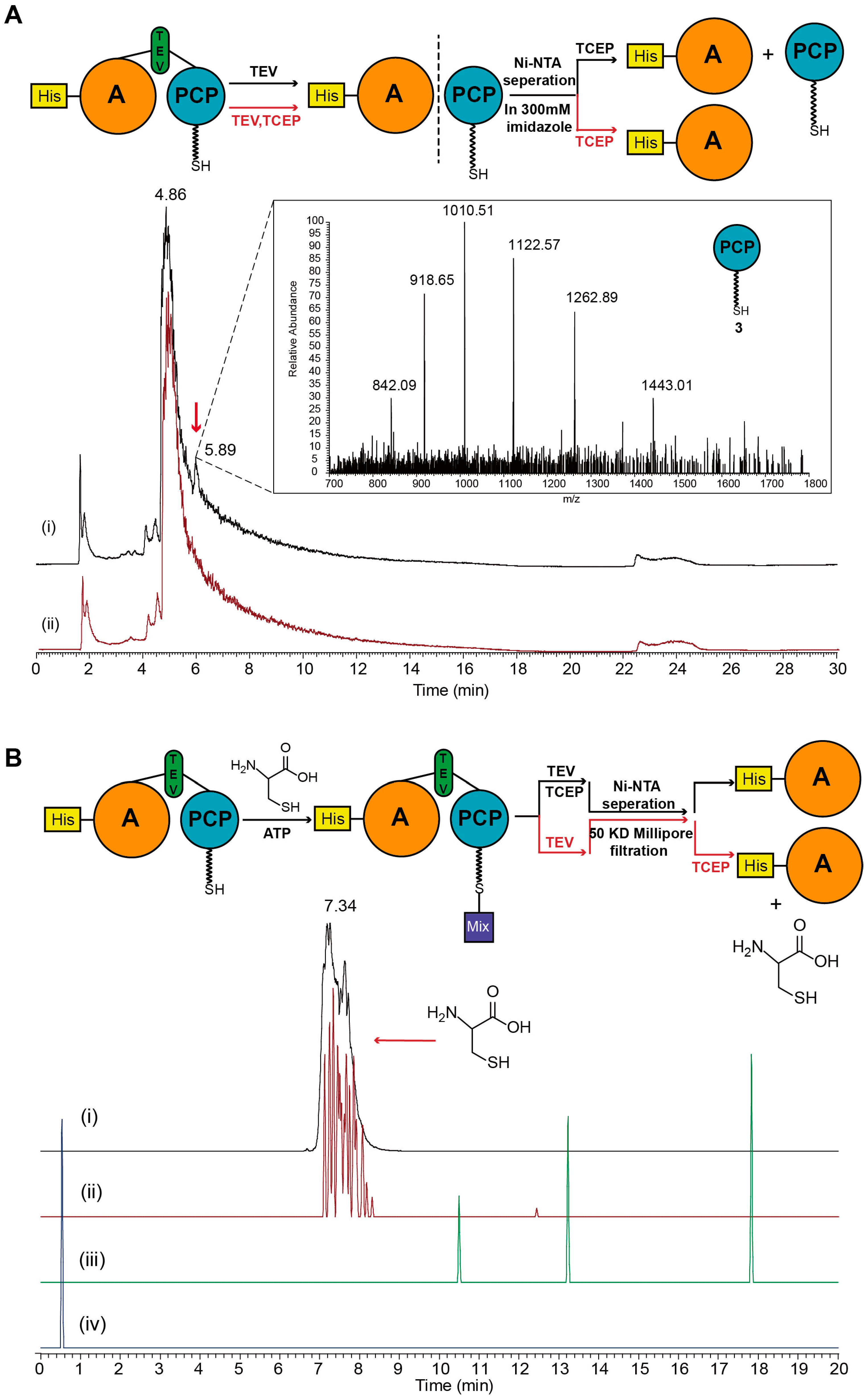
2.2. Experiments to Investigate and Correct the Proposed Mechanism
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Cloning and Expression of DtpB and DtpB-APCP
5.2. Cysteine Loading Assays with DtpB or DtpB-APCP
5.3. Cysteine Loading Assays with DtpB or DtpB-APCP
5.4. Cys Loading Assays with PCP Domain and A Domain in Intermolecular Stage
5.5. Cys Loading Assays Conjugated with TEV Digestion
5.6. Amino Acid Loading Assays with Mixed Substrates
5.7. Site-Directed Mutagenesis on DtpB-TEV and DtpB-APCP-TEV
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kenig, M.; Reading, C. Holomycin and an antibiotic (MM 19290) related to tunicamycin, metabolites of Streptomyces clavuligerus. J. Antibiot. 1979, 32, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Okamura, K.; Soga, K.; Shimauchi, Y.; Ishikura, T.; Lein, J. Holomycin and N-propionylholothin, antibiotics produced by a cephamycin C producer. J. Antibiot. 1977, 30, 334–336. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.E.; Fried, J.H.; Harrison, I.T.; Rapp, E.; Ross, C.H. Synthesis of holomycin and derivatives. J. Org. Chem. 1977, 42, 2891–2893. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Huang, S.; Yu, Y.; Deng, H. Dithiolopyrrolone Natural Products: Isolation, Synthesis and Biosynthesis. Mar. Drugs 2013, 11, 3970–3997. [Google Scholar] [CrossRef]
- McInerney, B.V.; Gregson, R.P.; Lacey, M.J.; Akhurst, R.J.; Lyons, G.R.; Rhodes, S.H.; Smith, D.R.; Engelhardt, L.M.; White, A.H. Biologically active metabolites from Xenorhabdus spp., Part 1. Dithiolopyrrolone derivatives with antibiotic activity. J. Nat. Prod. 1991, 54, 774–784. [Google Scholar] [CrossRef]
- Zhang, S.D.; Isbrandt, T.; Lindqvist, L.L.; Larsen, T.O.; Gram, L. Holomycin, an Antibiotic Secondary Metabolite, Is Required for Biofilm Formation by the Native Producer Photobacterium galatheae S2753. Appl. Environ. Microbiol. 2021, 87, e00169-21. [Google Scholar] [CrossRef]
- Chan, A.N.; Shiver, A.L.; Wever, W.J.; Razvi, S.Z.; Traxler, M.F.; Li, B. Role for dithiolopyrrolones in disrupting bacterial metal homeostasis. Proc. Natl. Acad. Sci. USA 2017, 114, 2717–2722. [Google Scholar] [CrossRef]
- Li, B.; Walsh, C.T. Identification of the gene cluster for the dithiolopyrrolone antibiotic holomycin in Streptomyces clavuligerus. Proc. Natl. Acad. Sci. USA 2010, 107, 19731–19735. [Google Scholar] [CrossRef]
- Huang, S.; Zhao, Y.; Qin, Z.; Wang, X.; Onega, M.; Chen, L.; He, J.; Yu, Y.; Deng, H. Identification and heterologous expression of the biosynthetic gene cluster for holomycin produced by Streptomyces clavuligerus. Process Biochem. 2011, 46, 811–816. [Google Scholar] [CrossRef]
- Ding, H.; Wang, J.N.; Zhang, D.S.; Ma, Z.J. Derivatives of Holomycin and Cyclopropaneacetic Acid from Streptomyces sp. DT-A37. Chem. Biodivers. 2017, 14, e1700140. [Google Scholar] [CrossRef]
- Huang, S.; Tong, M.H.; Qin, Z.; Deng, Z.; Deng, H.; Yu, Y. Identification and characterization of the biosynthetic gene cluster of thiolutin, a tumor angiogenesis inhibitor, in Saccharothrix algeriensis NRRL B-24137. Anticancer Agents Med. Chem. 2015, 15, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Dunn, Z.D.; Wever, W.J.; Economou, N.J.; Bowers, A.A.; Li, B. Enzymatic Basis of “Hybridity” in Thiomarinol Biosynthesis. Angew. Chem. Int. Ed. 2015, 54, 5137–5141. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Liang, H.; Qin, X.; Cao, D.; Zhu, X.; Ju, J.; Shen, B.; Duan, Y.; Huang, Y. The Isolation of Pyrroloformamide Congeners and Characterization of Their Biosynthetic Gene Cluster. J. Nat. Prod. 2020, 83, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Bai, S.; Liu, J.; Yang, L.; Han, L.; Huang, X.; He, J. Identification of an unusual type II thioesterase in the dithio lopyrrolone antibiotics biosynthetic pathway. Biochem. Biophys. Res. Commun. 2016, 473, 329–335. [Google Scholar] [CrossRef]
- Abe, T.; Hashimoto, Y.; Zhuang, Y.; Ge, Y.; Kumano, T.; Kobayashi, M. Peptide Bond Synthesis by a Mechanism Involving an Enzymatic Reaction and a Subsequent Chemical Reaction. J. Biol. Chem. 2016, 291, 1735–1750. [Google Scholar] [CrossRef]
- Abe, T.; Kobayashi, K.; Kawamura, S.; Sakaguchi, T.; Shiiba, K.; Kobayashi, M. Dipeptide synthesis by internal adenylation domains of a multidomain enzyme involved in nonribosomal peptide synthesis. J. Gen. Appl. Microbiol. 2019, 65, 1–10. [Google Scholar] [CrossRef]
- May, J.J.; Wendrich, T.M.; Marahiel, M.A. The dhb Operon of Bacillus subtilisEncodes the Biosynthetic Template for the Catecholic Siderophore 2,3-Dihydroxybenzoate-Glycine-Threonine Trimeric Ester Bacillibactin. J. Biol. Chem. 2001, 276, 7209–7217. [Google Scholar] [CrossRef]
- Gehring, A.M.; Bradley, K.A.; Walsh, C.T. Enterobactin biosynthesis in Escherichia coli: Isochorismate lyase (EntB) is a bifunctional enzyme that is phosphopantetheinylated by EntD and then acylated by EntE using ATP and 2,3-dihydroxybenzoate. Biochemistry 1997, 36, 8495–8503. [Google Scholar] [CrossRef]
- Shen, B.; Du, L.; Sanchez, C.; Edwards, D.J.; Chen, M.; Murrell, J.M. Cloning and Characterization of the Bleomycin Biosynthetic Gene Cluster from Streptomyces verticillus ATCC15003. J. Nat. Prod. 2002, 65, 422–431. [Google Scholar] [CrossRef]
- Bučević-Popović, V.; Šprung, M.; Soldo, B.; Pavela-Vrančič, M. The A9 Core Sequence from NRPS Adenylation Domain Is Relevant for Thioester Formation. ChemBioChem 2012, 13, 1913–1920. [Google Scholar] [CrossRef]
- Drake, E.J.; Miller, B.R.; Shi, C.; Tarrasch, J.T.; Sundlov, J.A.; Leigh Allen, C.; Skiniotis, G.; Aldrich, C.C.; Gulick, A.M. Structures of two distinct conformations of holo-non-ribosomal peptide synthetases. Nature 2016, 529, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Gulick, A.M. Conformational Dynamics in the Acyl-CoA Synthetases, Adenylation Domains of Non-ribosomal Peptide Synthetases, and Firefly Luciferase. ACS Chem. Biol. 2009, 4, 811–827. [Google Scholar] [CrossRef] [PubMed]
- Labby, K.J.; Watsula, S.G.; Garneau-Tsodikova, S. Interrupted adenylation domains: Unique bifunctional enzymes involved in nonribosomal peptide biosynthesis. Nat. Prod. Rep. 2015, 32, 641–653. [Google Scholar] [CrossRef]
- Li, B.; Walsh, C.T. Streptomyces clavuligerus HlmI Is an Intramolecular Disulfide-Forming Dithiol Oxi dase in Holomycin Biosynthesis. Biochemistry 2011, 50, 4615–4622. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Tu, Y.; Pan, T.; Deng, Z.; Duan, L. A Cysteine-Reloading Process Initiating the Biosynthesis of the Bicyclic Scaffold of Dithiolopyrrolones. Antibiotics 2023, 12, 787. https://doi.org/10.3390/antibiotics12040787
Chen Y, Tu Y, Pan T, Deng Z, Duan L. A Cysteine-Reloading Process Initiating the Biosynthesis of the Bicyclic Scaffold of Dithiolopyrrolones. Antibiotics. 2023; 12(4):787. https://doi.org/10.3390/antibiotics12040787
Chicago/Turabian StyleChen, Yan, Yanqin Tu, Tingyu Pan, Zixin Deng, and Lian Duan. 2023. "A Cysteine-Reloading Process Initiating the Biosynthesis of the Bicyclic Scaffold of Dithiolopyrrolones" Antibiotics 12, no. 4: 787. https://doi.org/10.3390/antibiotics12040787
APA StyleChen, Y., Tu, Y., Pan, T., Deng, Z., & Duan, L. (2023). A Cysteine-Reloading Process Initiating the Biosynthesis of the Bicyclic Scaffold of Dithiolopyrrolones. Antibiotics, 12(4), 787. https://doi.org/10.3390/antibiotics12040787