
Design and Synthesis of Novel Antimicrobial Agents



Abstract
1. Introduction
2. Antibiotic Classification

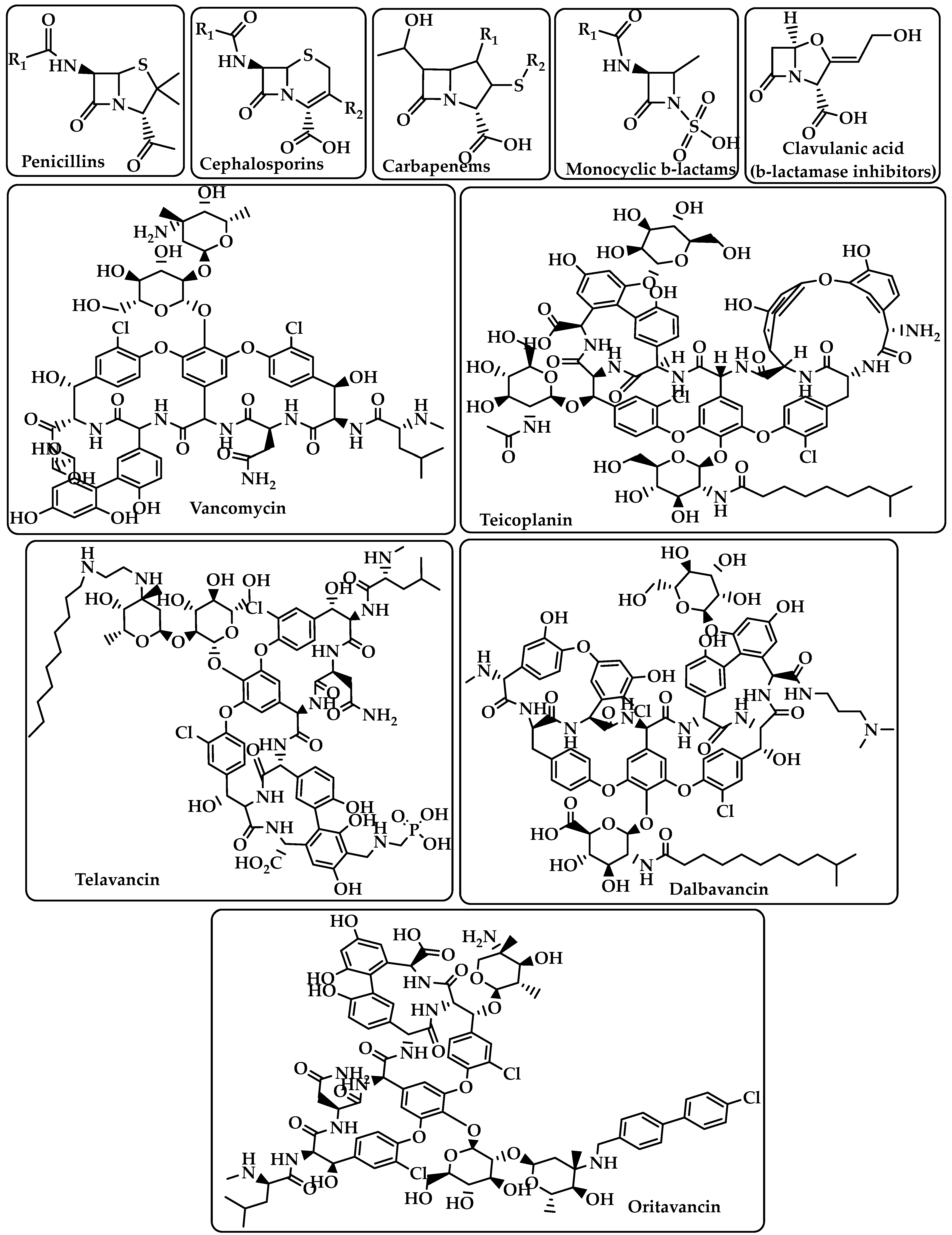{kind=link}
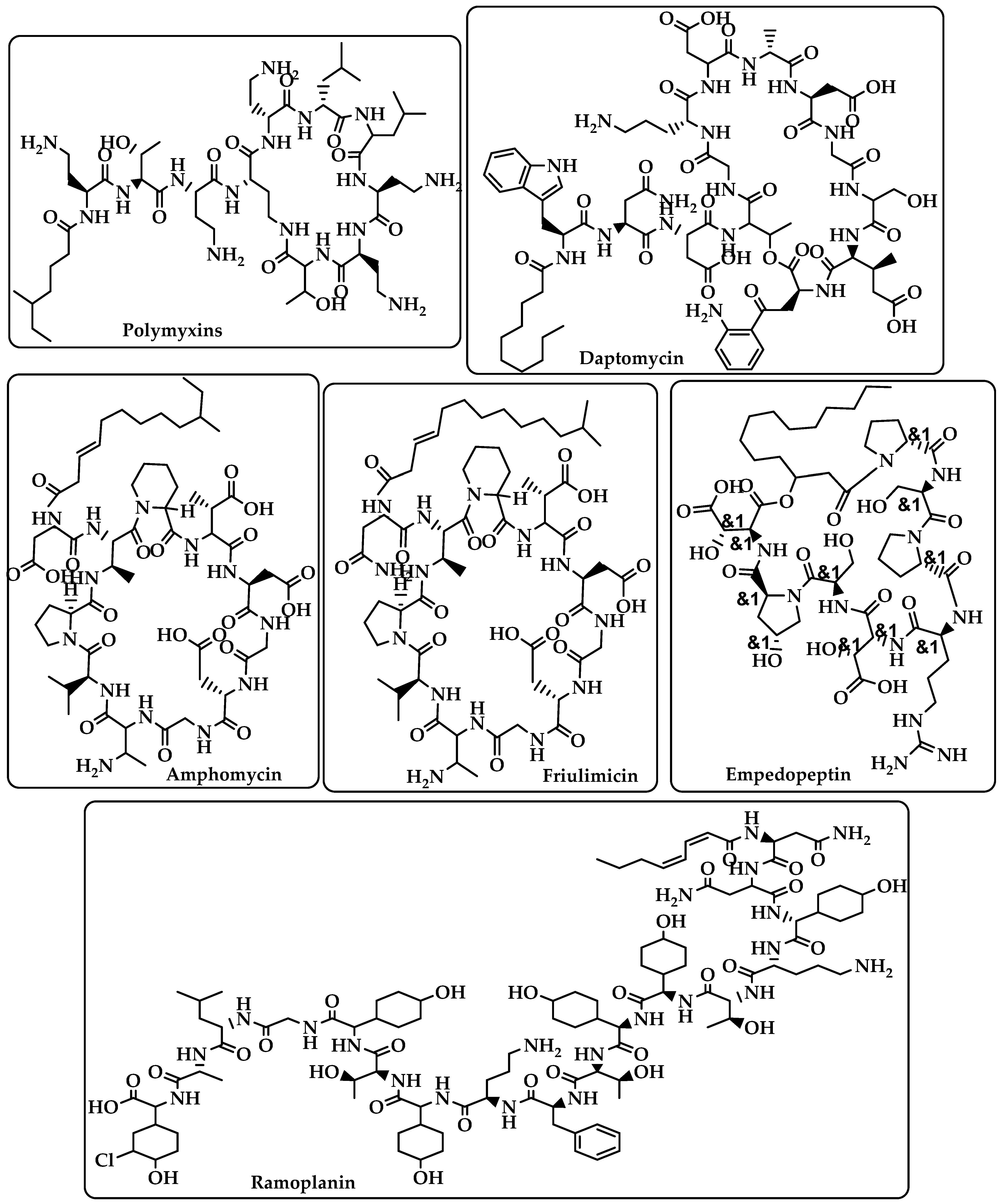{kind=link}
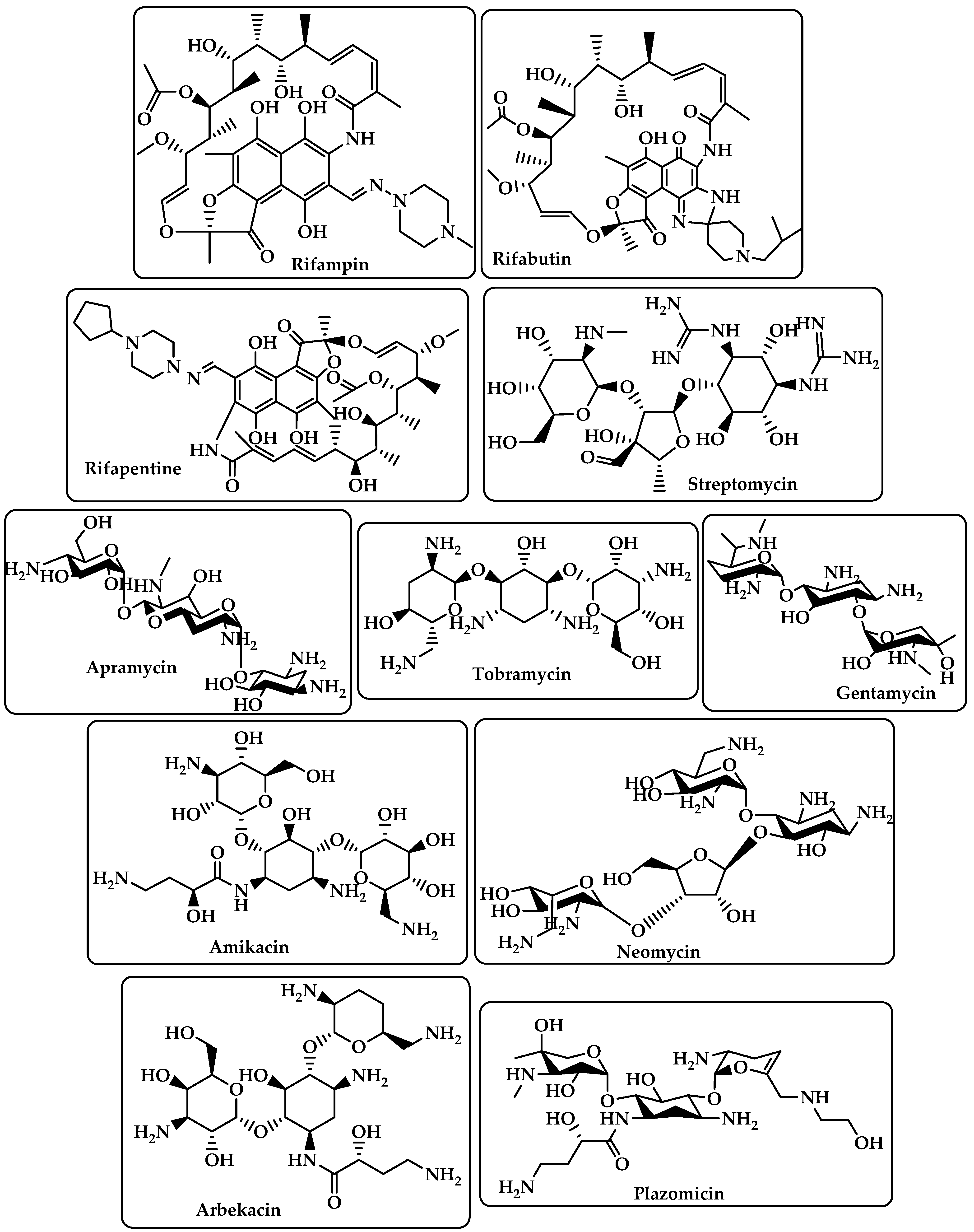{kind=link}
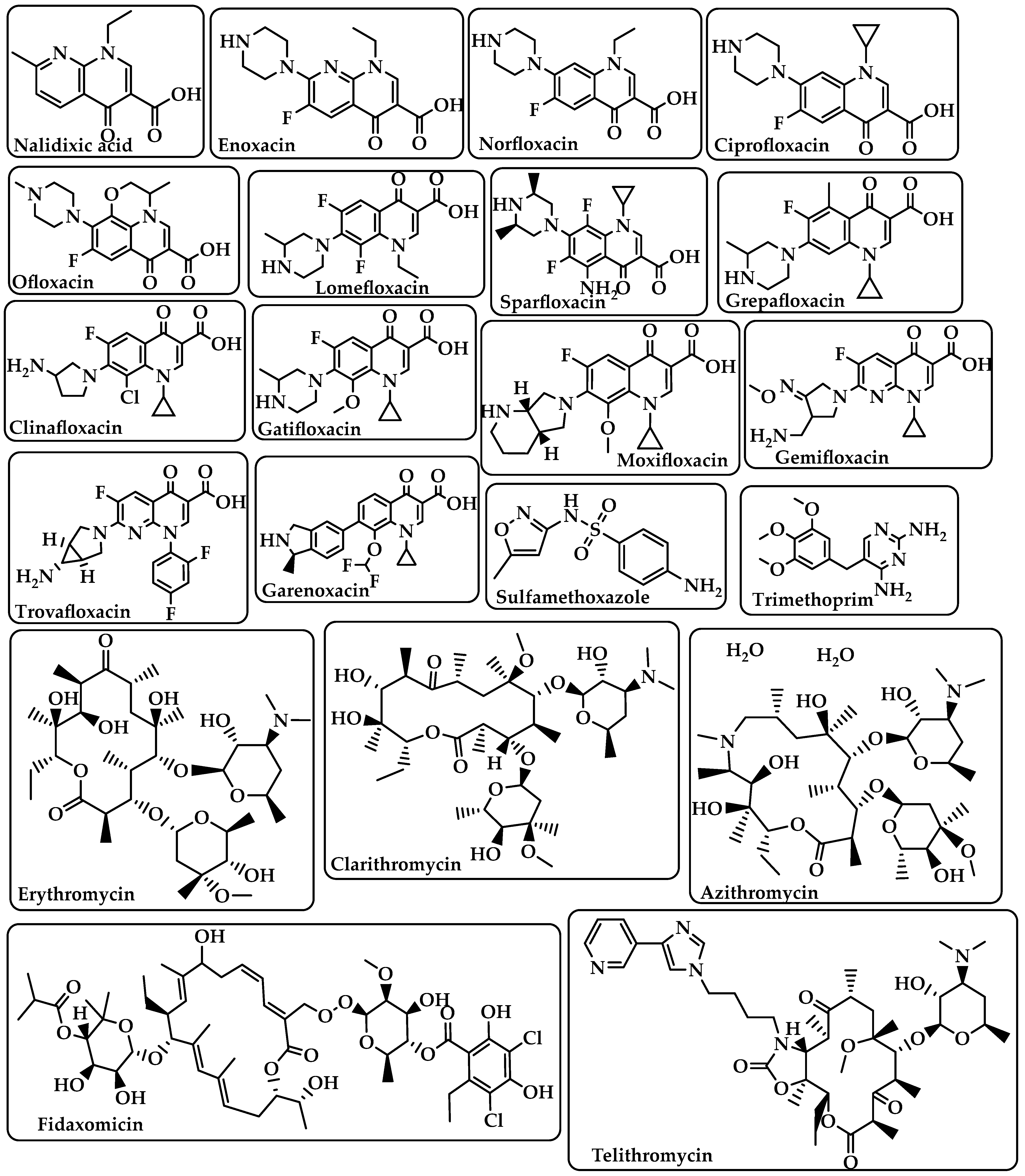{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibiotics Family | Mechanism of Action | Antibiotics |
|---|---|---|
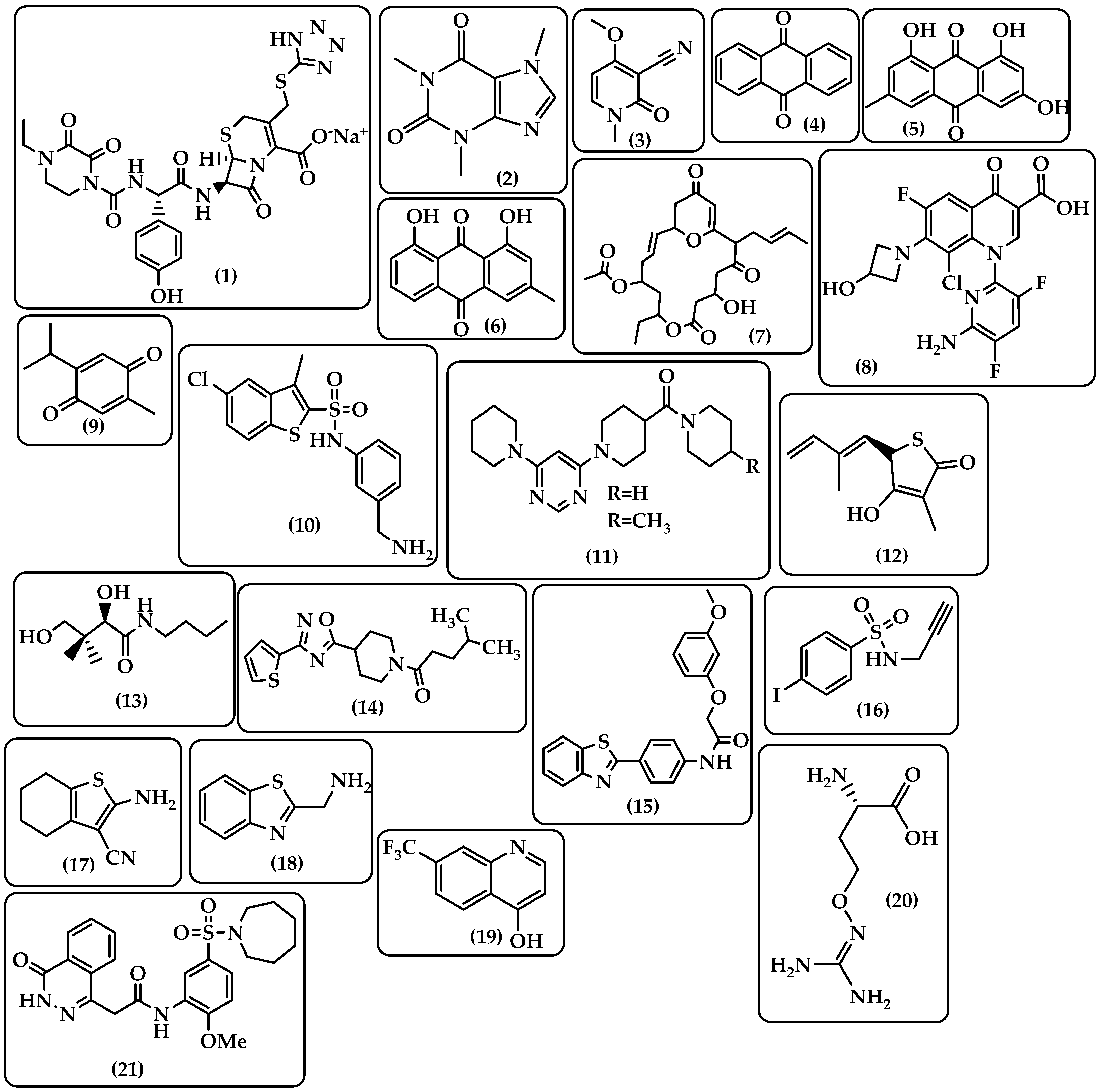
| β-lactam | Binds to the serine active site of penicillin-binding proteins (PBPs) or the allosteric site in PBP2a to inhibit bacterial cell wall peptidoglycan transpeptidation [14,15]. | Penicillins Cephalosporins Carbapenems Monocyclic β-lactams β-lactamase inhibitors (e.g., clavulanic acid) (Figure 1) |
| Glycopeptides | Interacts with the membrane-bound lipid II precursor of peptidogly and can prevent peptidoglycan from being incorporated into an essential structural cell wall component [16]. | Vancomycin Teicoplanin Telavancin Dalbavancin Oritavancin (Figure 1) |
| Lipopeptide | Carries out their action by causing Gram-positive bacteria’s cell membrane integrity to be compromised, which results in cell death [17,18]. | Polymyxins Daptomycin Amphomycin Friulimicin Ramoplanin Empedopeptin (Figure 2) |
| Rifamycins | RNA polymerase (RNAP) inhibitors are used to treat tuberculosis (TB) [19]. | Rifampin Rifabutin Rifapentine (Figure 3) |
| Aminoglycoside | By attaching to the 30S ribosome’s A-site on the 16S ribosomal RNA, they inhibit protein synthesis [20]. | Streptomycin Apramycin Tobramycin Gentamcin Amikacin Neomycin Arbekacin Plazomicin (Figure 3) |
| Fluoroquinolones | Target DNA gyrase, topoisomerase IV, and topoisomerase type II to prevent bacteria from synthesizing DNA [21]. | Nalidixic acid Enoxacin Norfloxacin Ciprofloxacin Ofloxacin Lomefloxacin Sparfloxacin Grepafloxacin Clinafloxacin Gatifloxacin Moxifloxacin Gemifloxacin Trovafloxacin Garenoxacin (Figure 4) |
| Sulfonamides–Trimethoprim | Sulfonamides interfere with the activity of the dihydropteroate synthase enzyme by competing with p-aminobenzoic acid (PABA) in the process of dihydrofolate production.The dihydrofolate reductase enzyme is inhibited by trimethoprim because it competes directly with it [22]. | Sulfamethoxazole Trimethoprim (Figure 4) |
| Macrolides | Target the nascent peptide exit tunnel (NPET) of the bacterial 50S ribosomal subunit, which is responsible for the release of newly synthesized protein from the ribosome, ultimately preventing protein synthesis [23,24]. | Erythromycin Clarithromycin Azithromycin Fidaxomicin Telithromycin (Figure 4) |
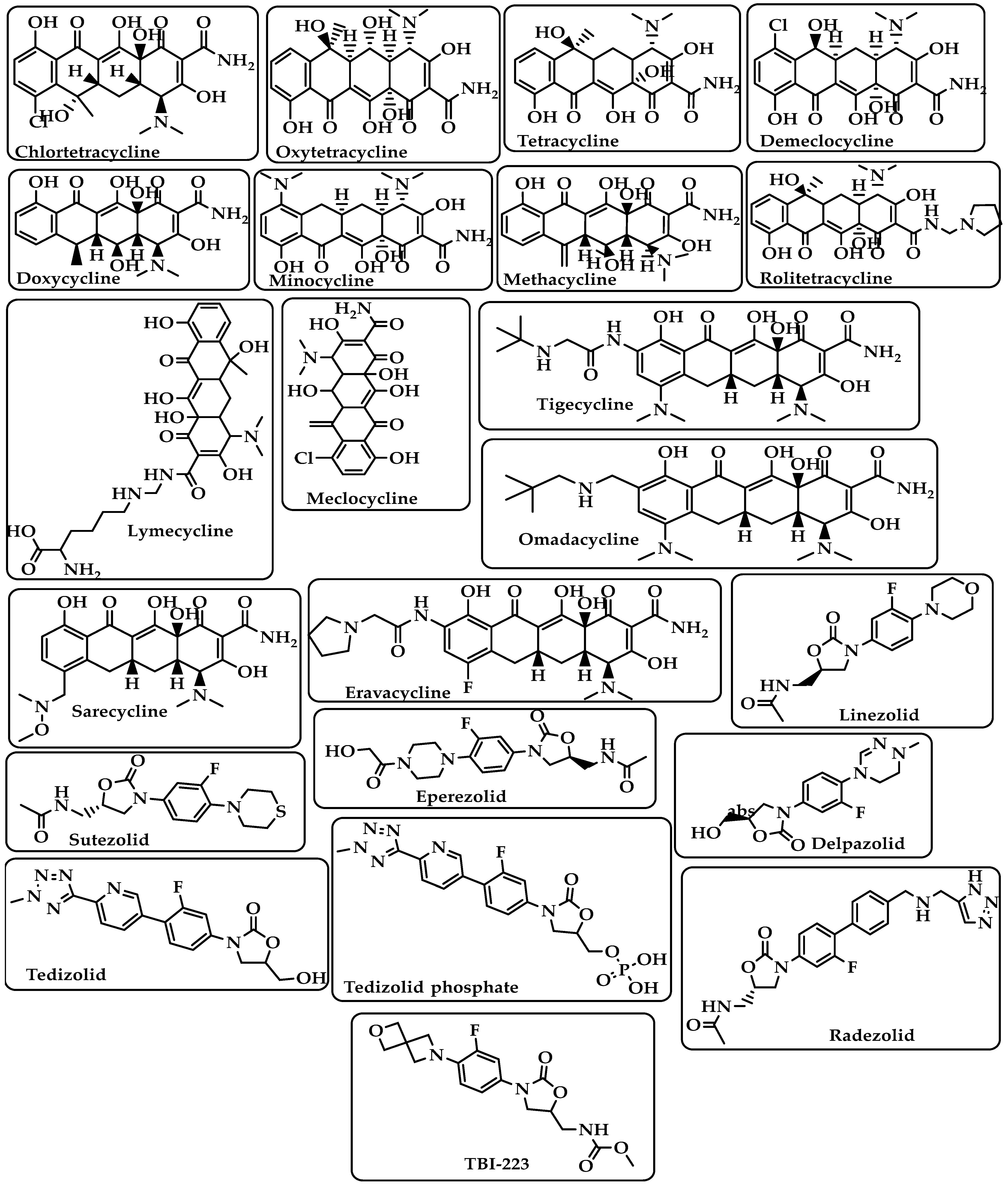
| Tetracyclines | Bind to the small subunit’s decoding site and prevent bacterial protein synthesis [25,26]. | Chlortetracycline Oxytetracycline Tetracycline Demeclocycline Doxycycline Minocycline Lymecycline Meclocycline Methacycline RolitetracyclineTigecycline Omadacycline Sarecycline Eravacycline (Figure 5) |
| Oxazolidinones | Block the translation sequence by interacting with the 50S subunit (A-site pocket) at the peptidyl transferase center (PTC) to inhibit protein synthesis [27]. | Linezolid Sutezolid Eperezolid Delpazolid Tedizolid Tedizolid phosphate Radezolid TBI-223 (Figure 5) |
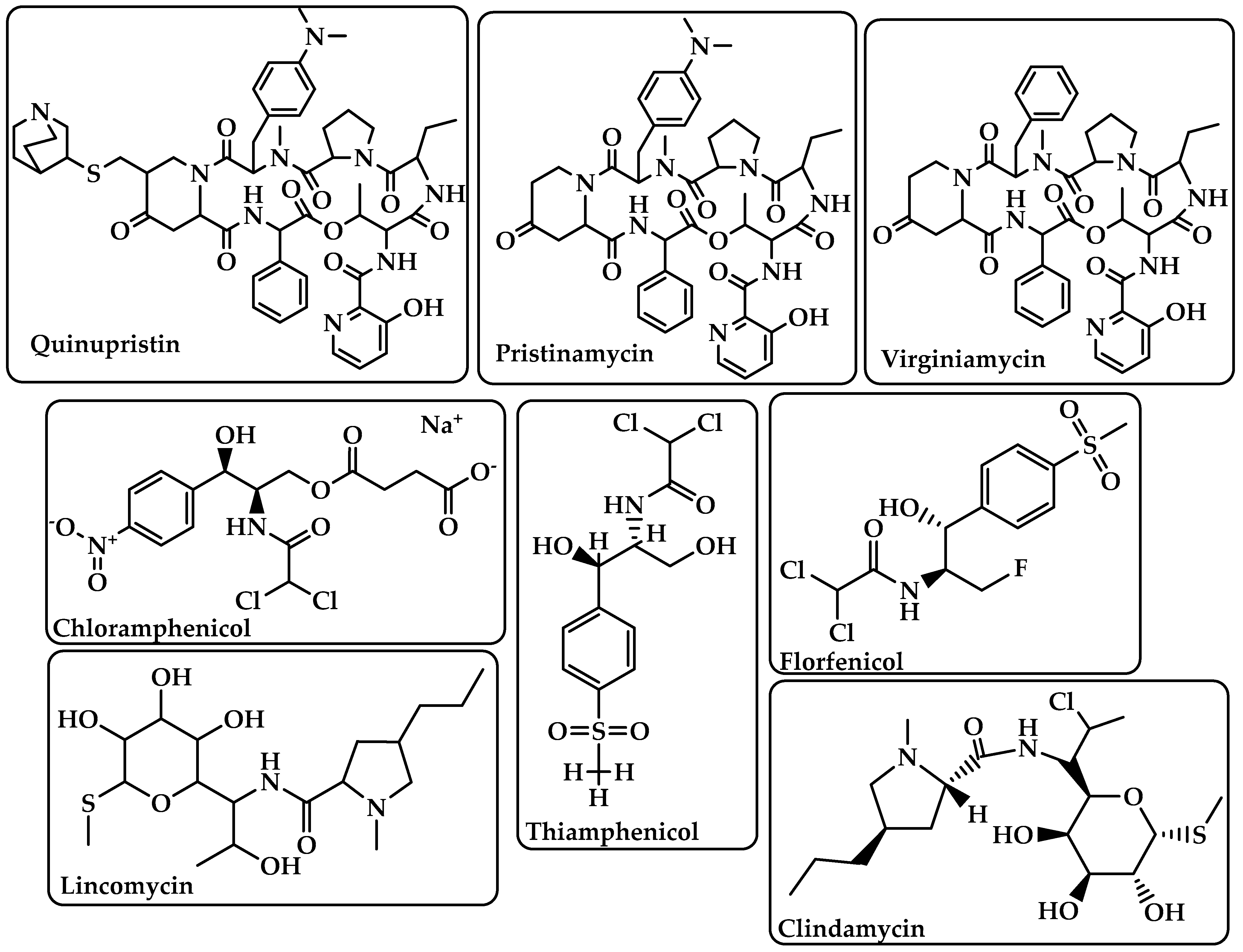
| Streptogramins | Inhibit protein synthesis during the elongation step by attaching to bacterial ribosomes [28]. The antibiotic has two unique structural groups (A and B) that cooperate to increase the affinity of group B in the nearby nascent peptide exit tunnel (NPET) when group A binds to the peptidyl transferase center (PTC) [29]. | Quinupristin Pristinamycin Virginiamycin (Figure 6) |
| Phenicoles | Inhibit protein synthesis by binding to the 50S ribosomal subunit [30]. | Chloramphenicol Thiamphenicol Florfenicol (Figure 6) |
| Lincosamides | Activate amino acid monomers by aminoacyl-tRNA, chain initiation, elongation, and termination of the formed polypeptides on the ribosome, which disrupts bacterial growth and death. These are only a few of the many processes that can be affected to prevent microbial protein synthesis [31]. | Lincomycin Clindamycin (Figure 6) |
3. Antimicrobial Resistance
4. Antibiotic Use and Resistance in Agriculture Sector
5. Novel Therapeutic Agents
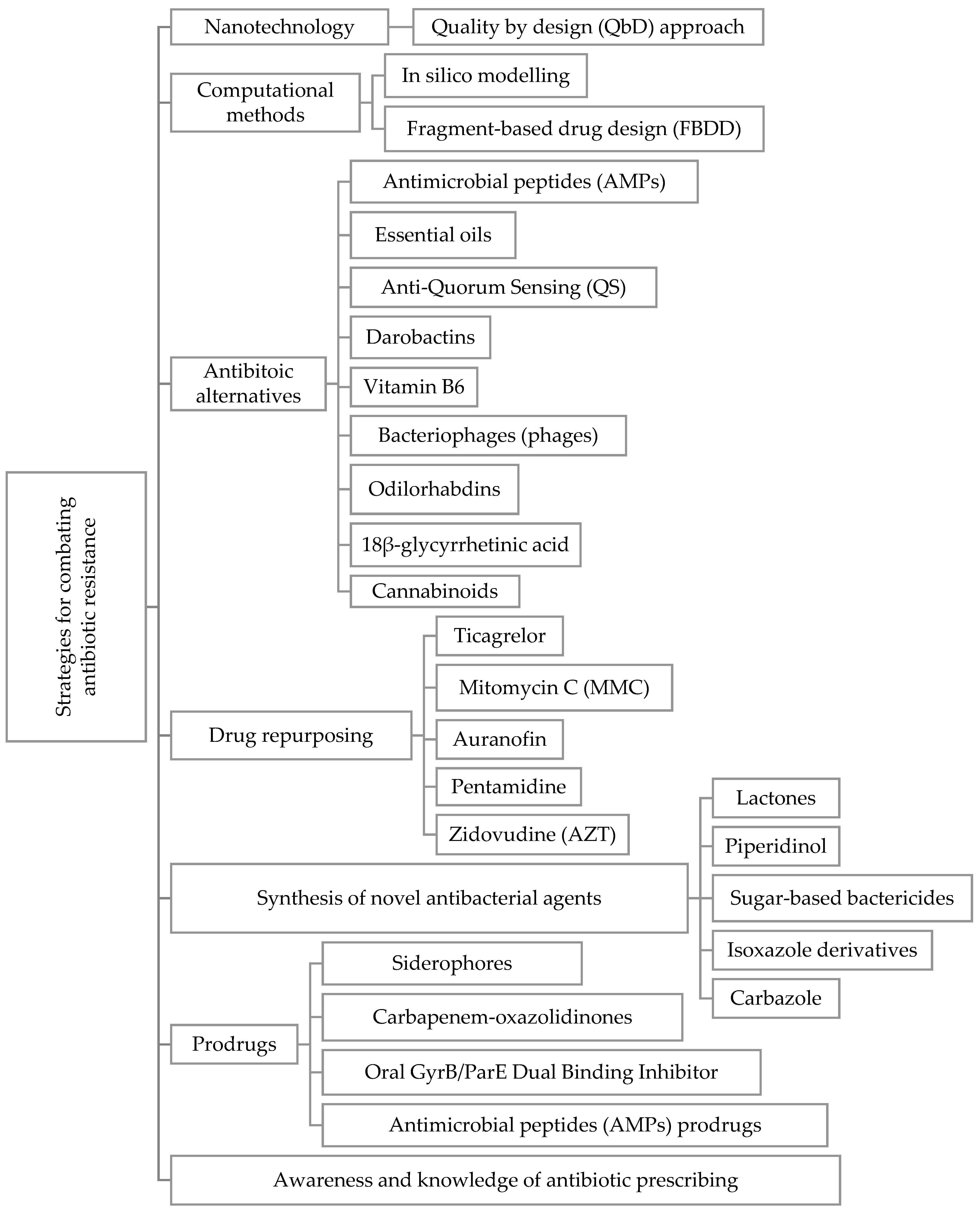
5.1. Nanotechnology in Combating Bacterial Resistance
Implementation of Quality by Design (QbD) Approach in Nano-Delivery
5.2. Computational Methods in the Development of New Antibacterial Agents
5.2.1. In Silico Modelling
5.2.2. Fragment-Based Drug Design (FBDD)
5.3. Antibiotic Alternatives
5.3.1. Antimicrobial Peptides (AMPs)
5.3.2. Essential Oils
5.3.3. Anti-Quorum Sensing (QS)
5.3.4. Vitamin B6
5.3.5. Bacteriophages (Phages)
5.3.6. Odilorhabdins (ODLs)
5.3.7. 18β-glycyrrhetinic Acid
5.3.8. Darobactins
5.3.9. Cannabinoids
5.4. Drug Repurposing
5.4.1. Ticagrelor
5.4.2. Mitomycin C (MMC)
5.4.3. Auranofin
5.4.4. Pentamidine
5.4.5. Zidovudine (AZT)
6. Synthesis of Novel Antibacterial Agents
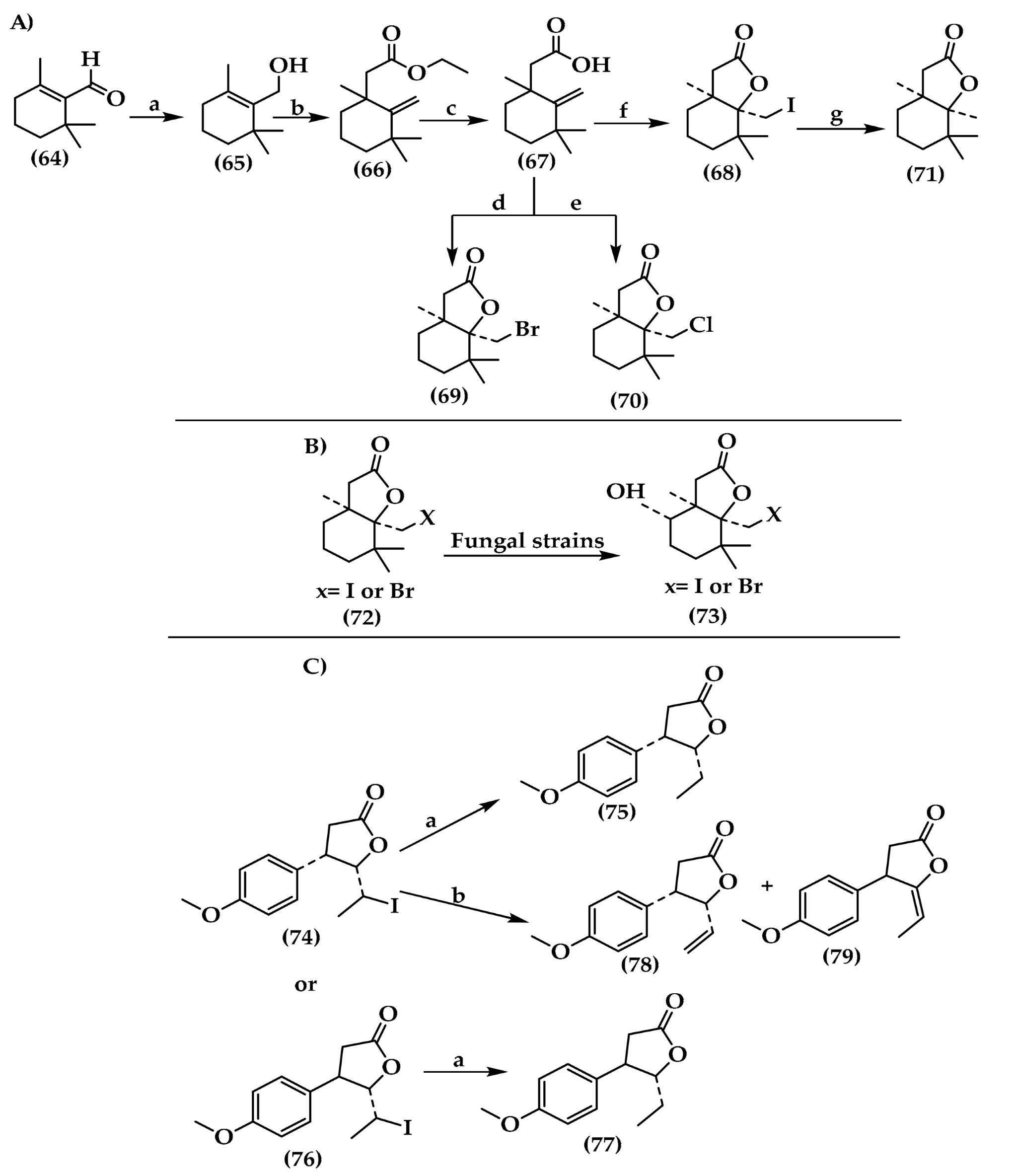
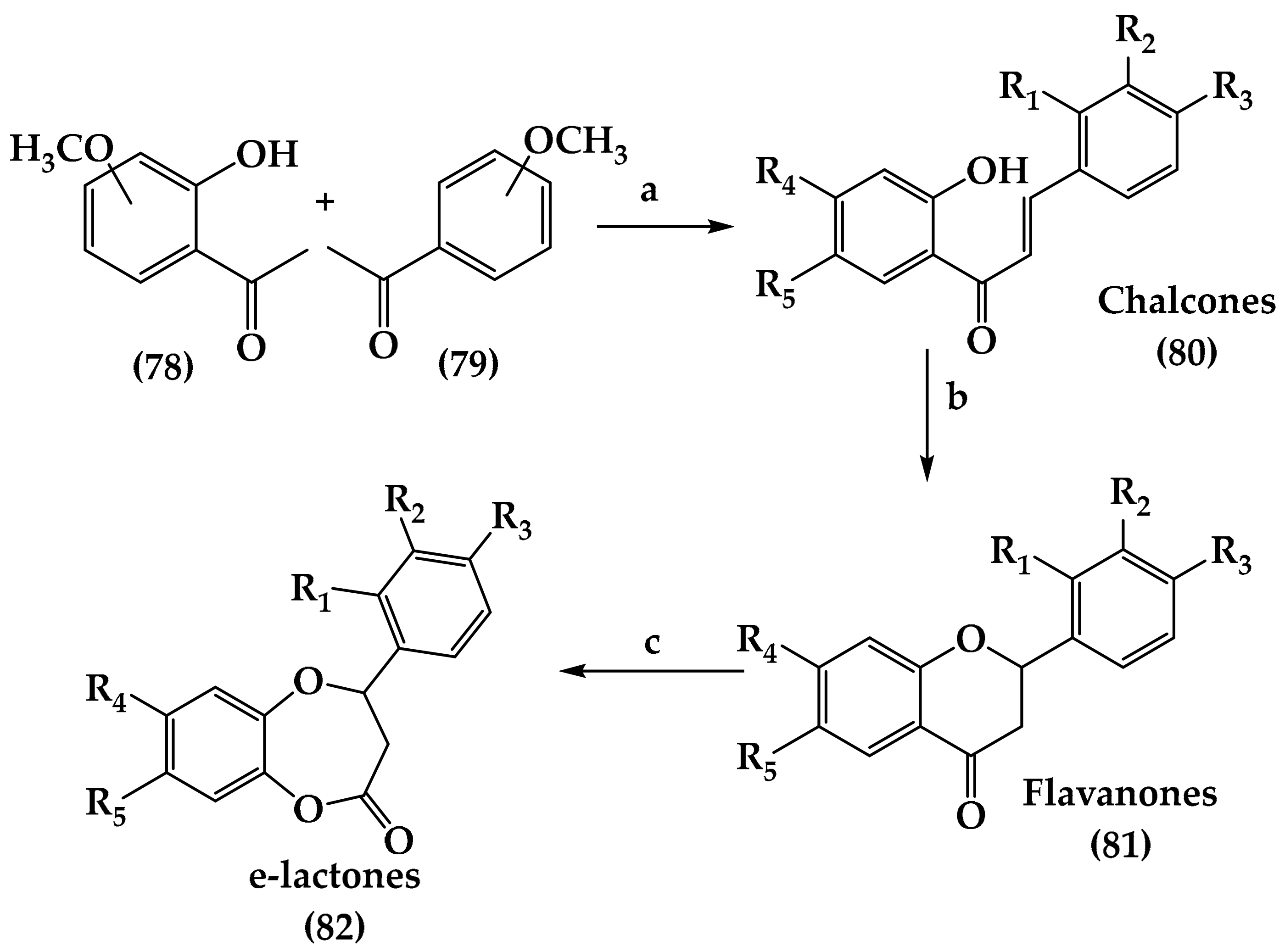
6.1. Lactones
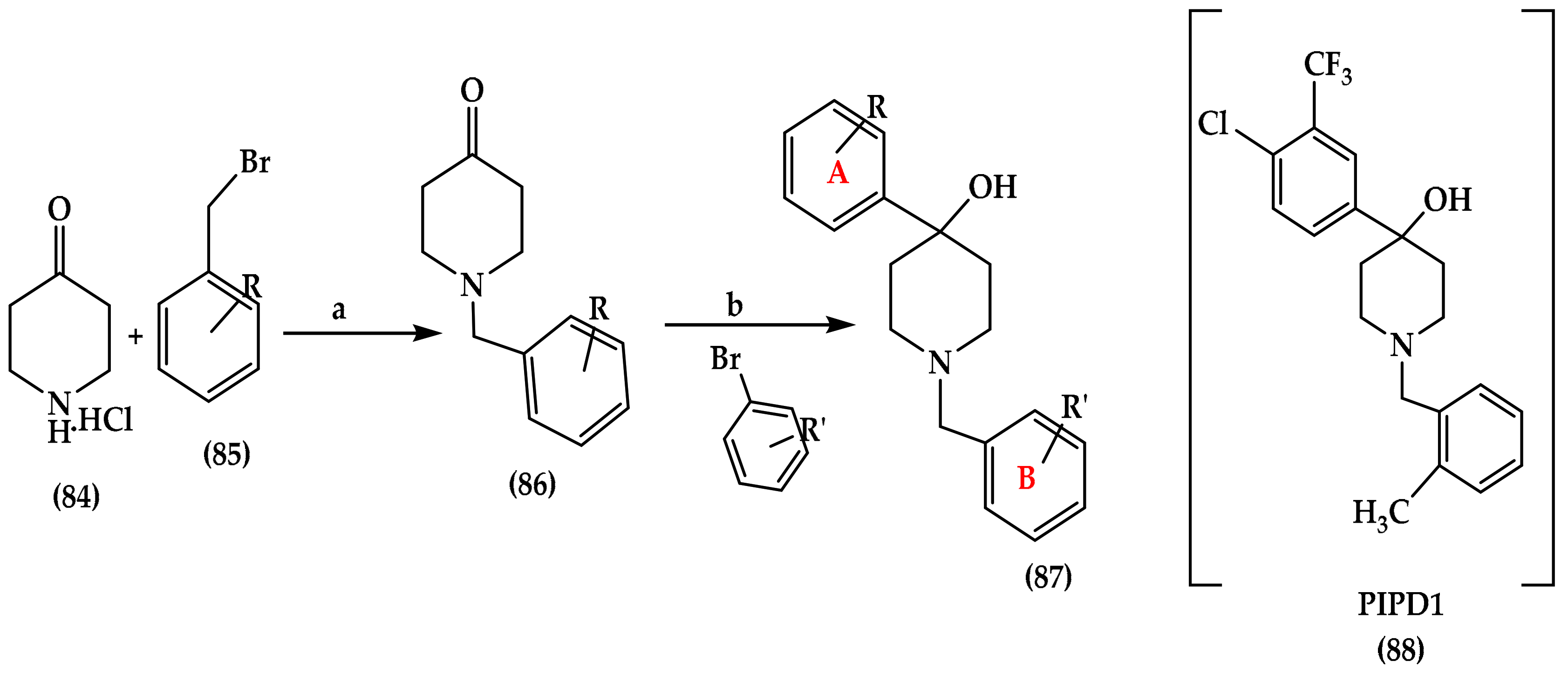
6.2. Piperidinol
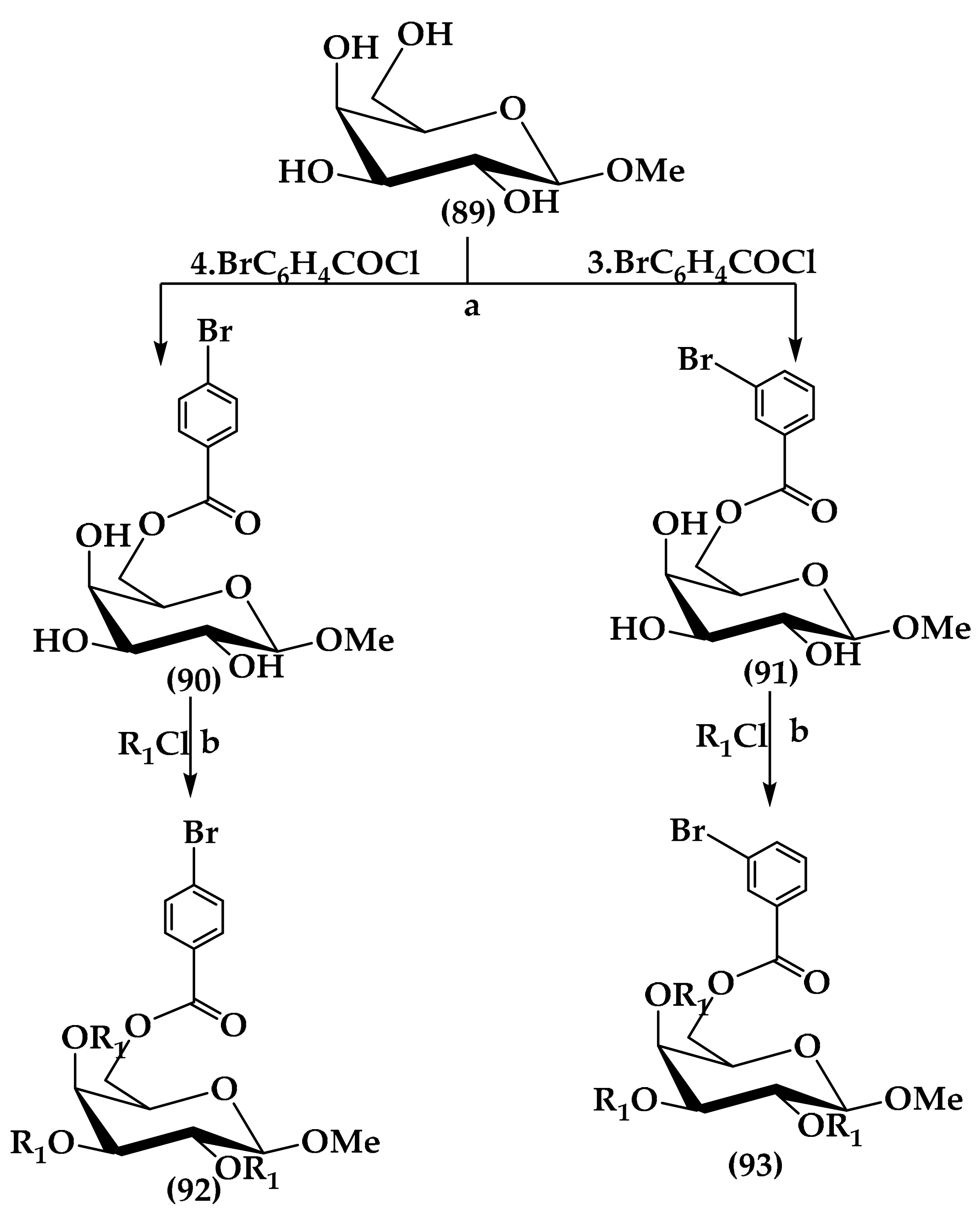
6.3. Sugar-Based Bactericides
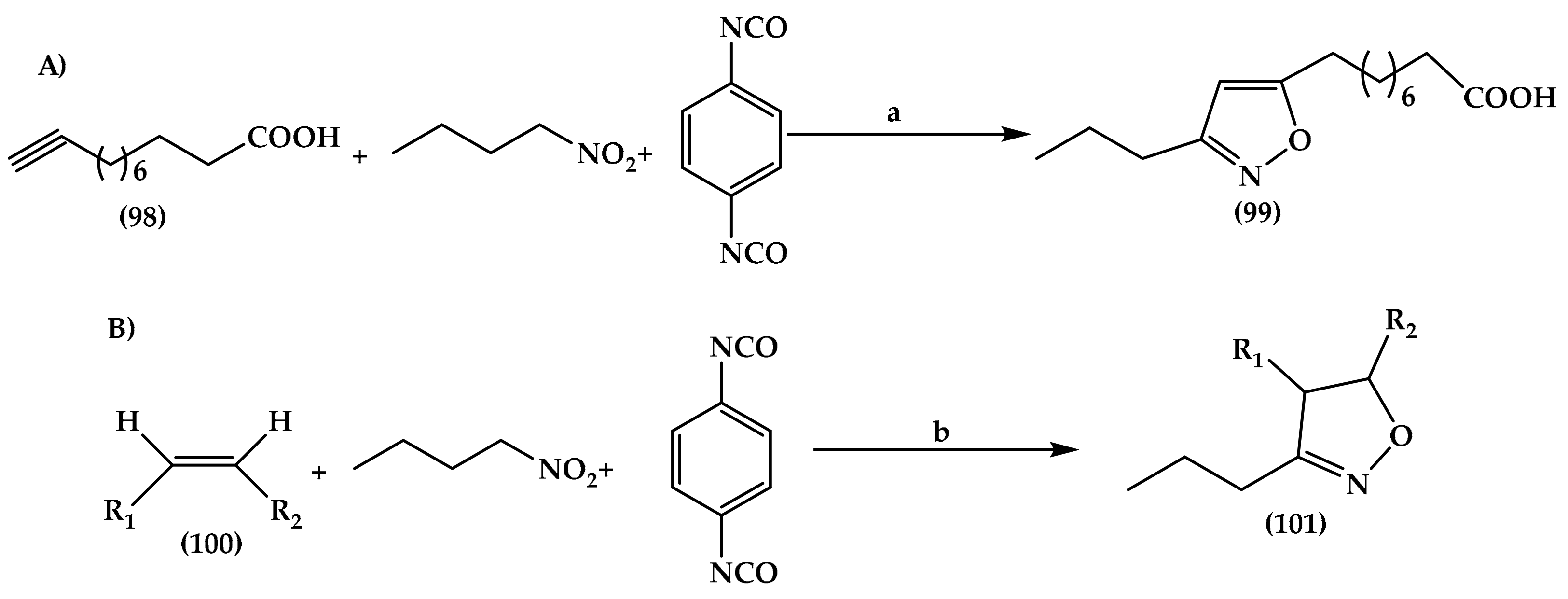
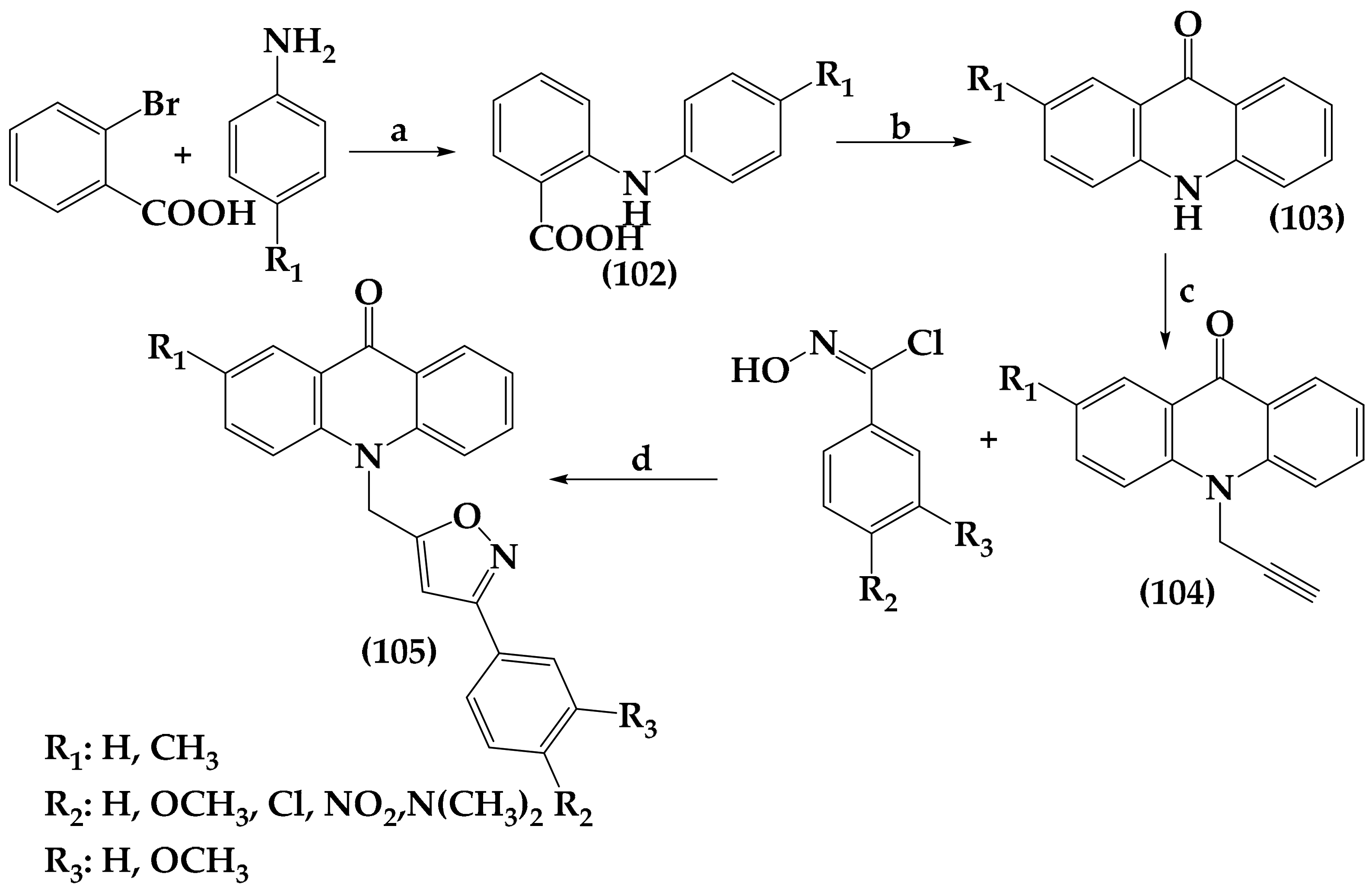
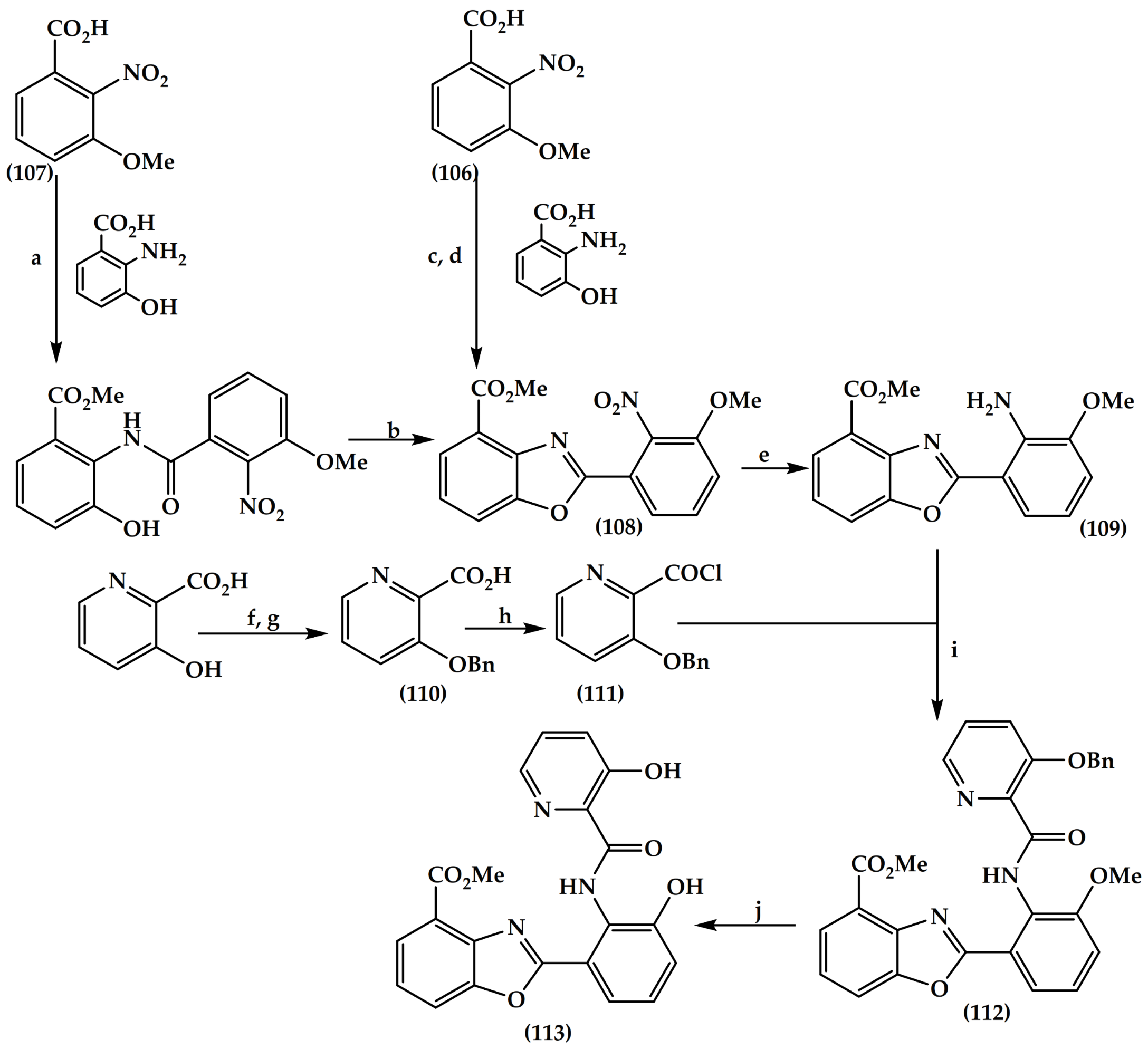
6.4. Isoxazole Derivatives
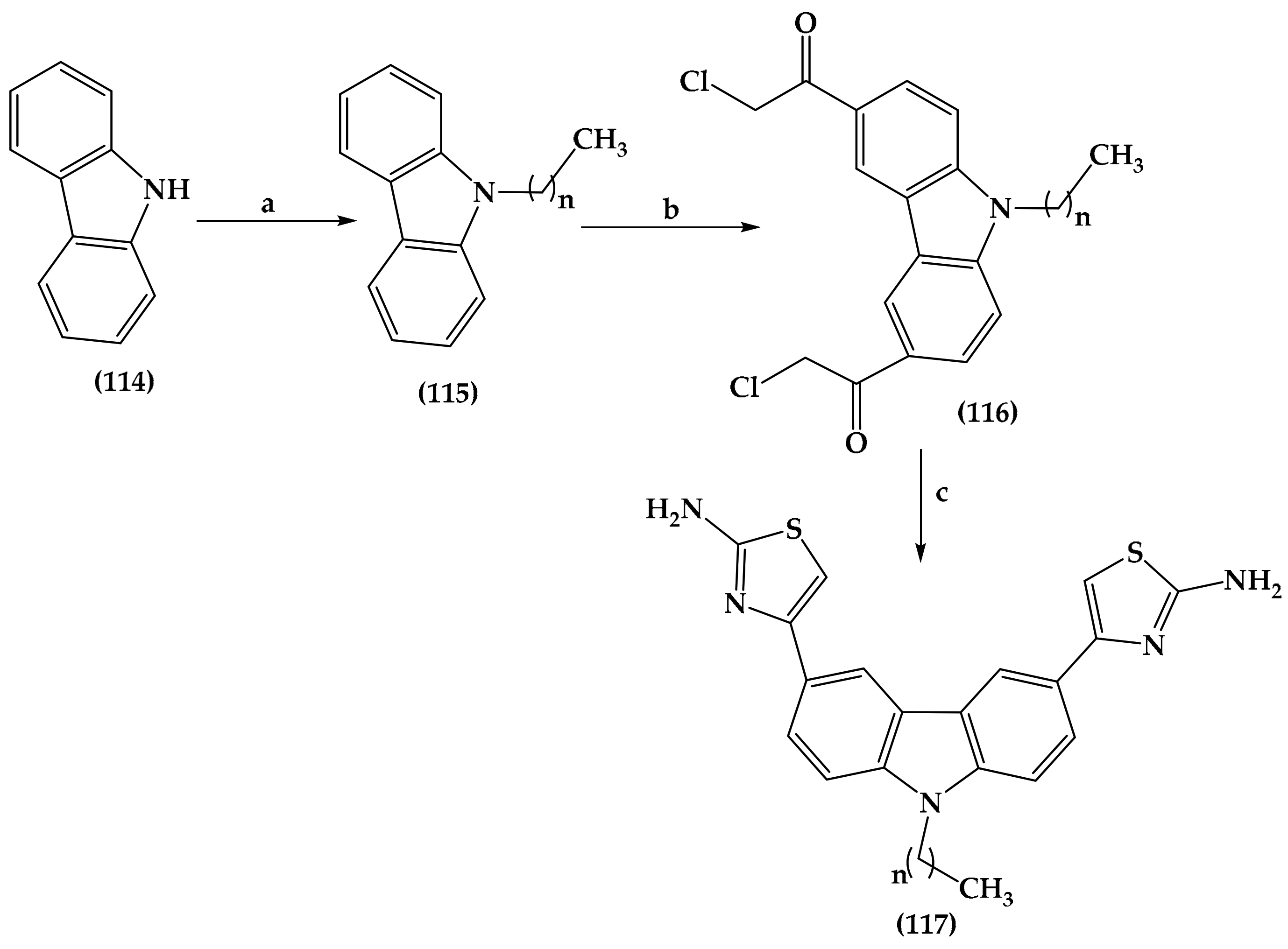
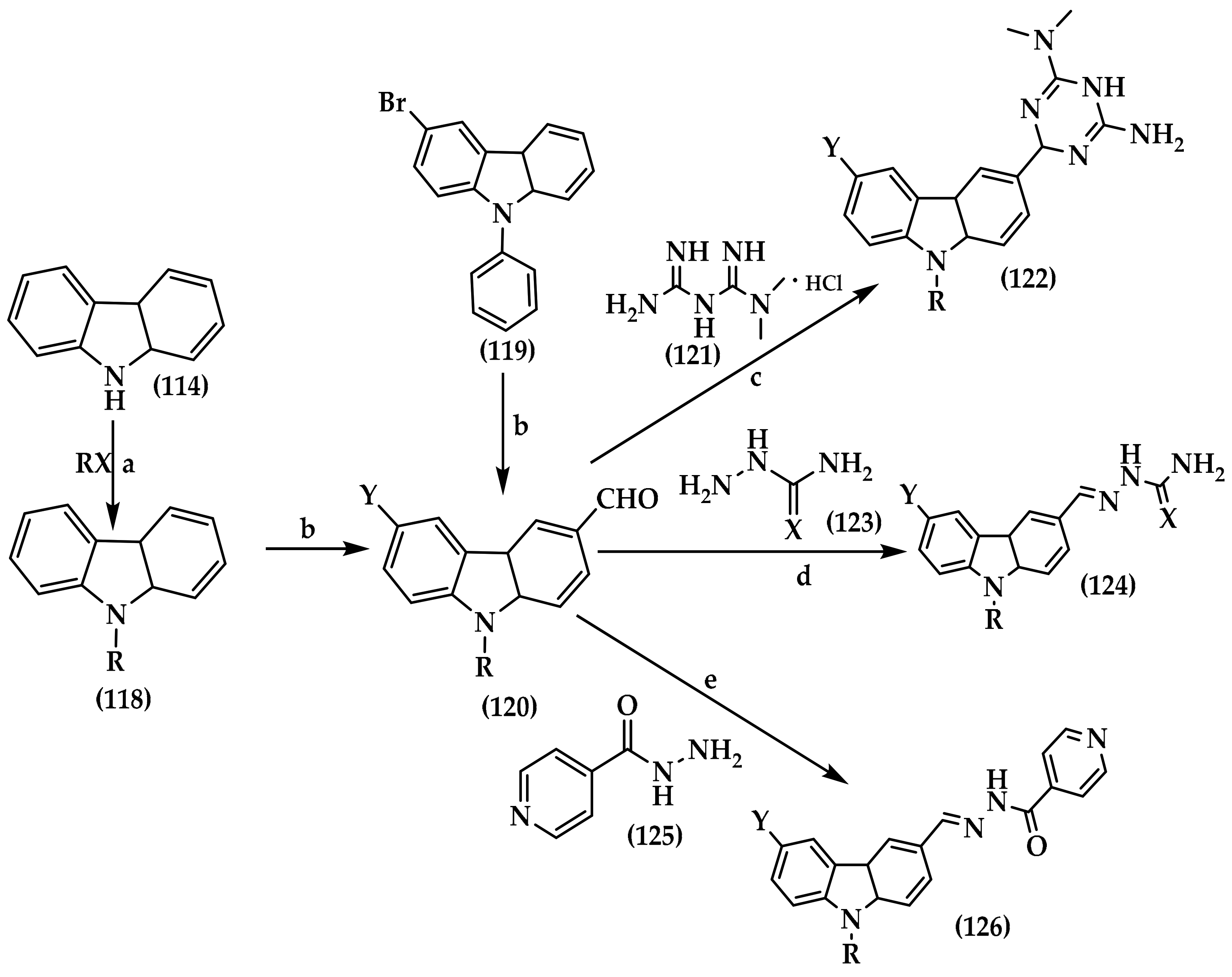
6.5. Carbazole
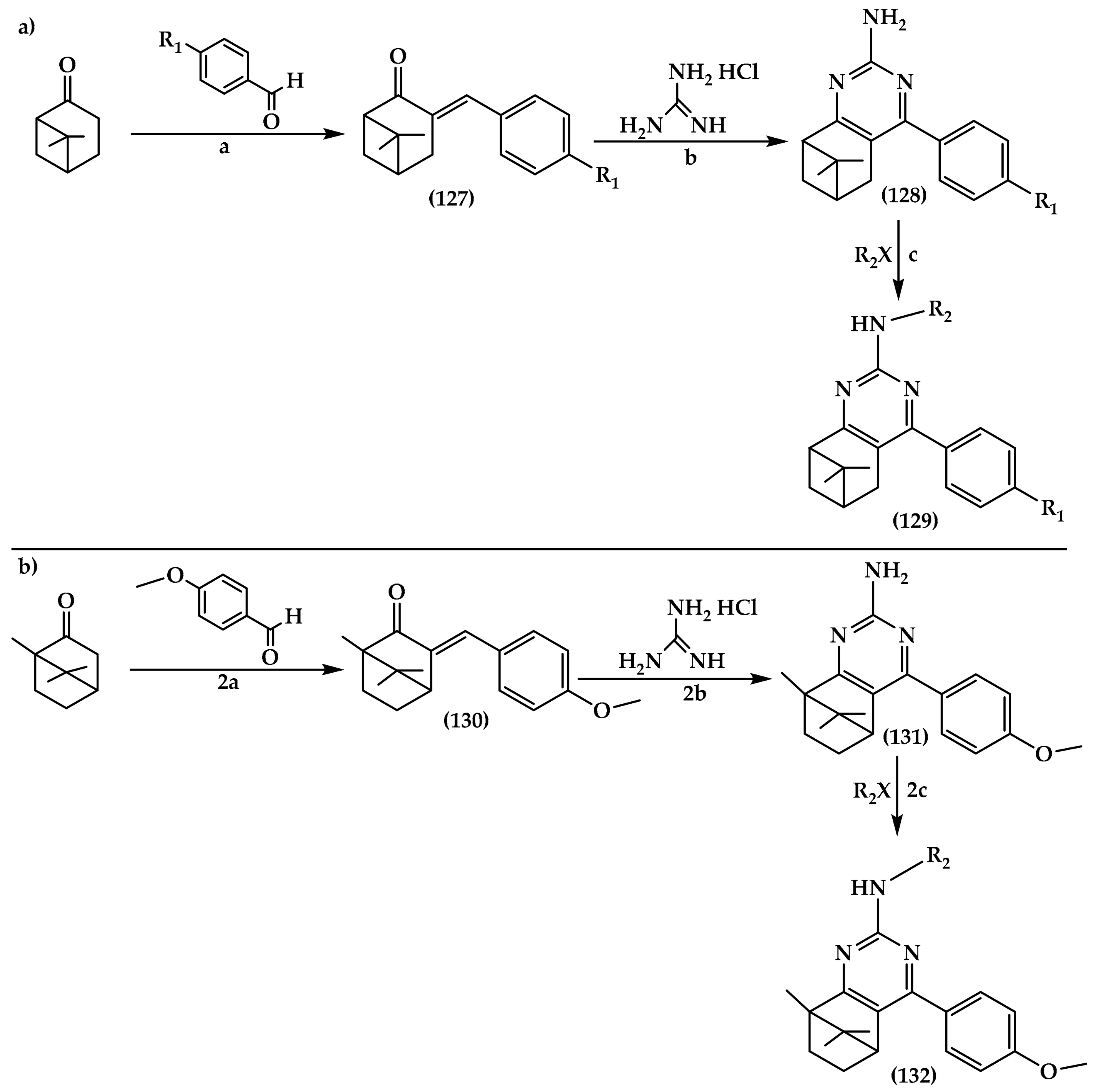
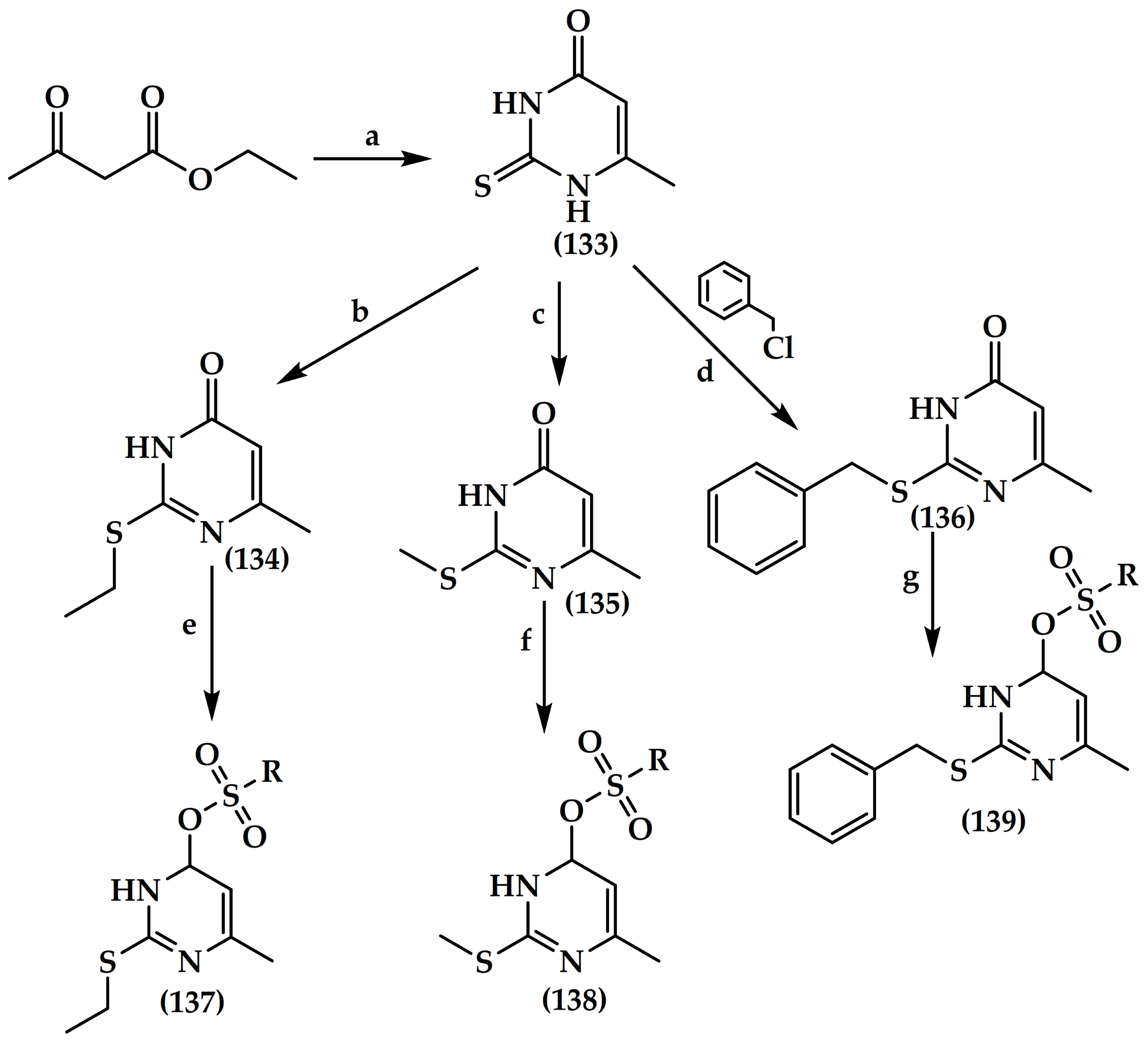
6.6. Pyrimidine Derivatives
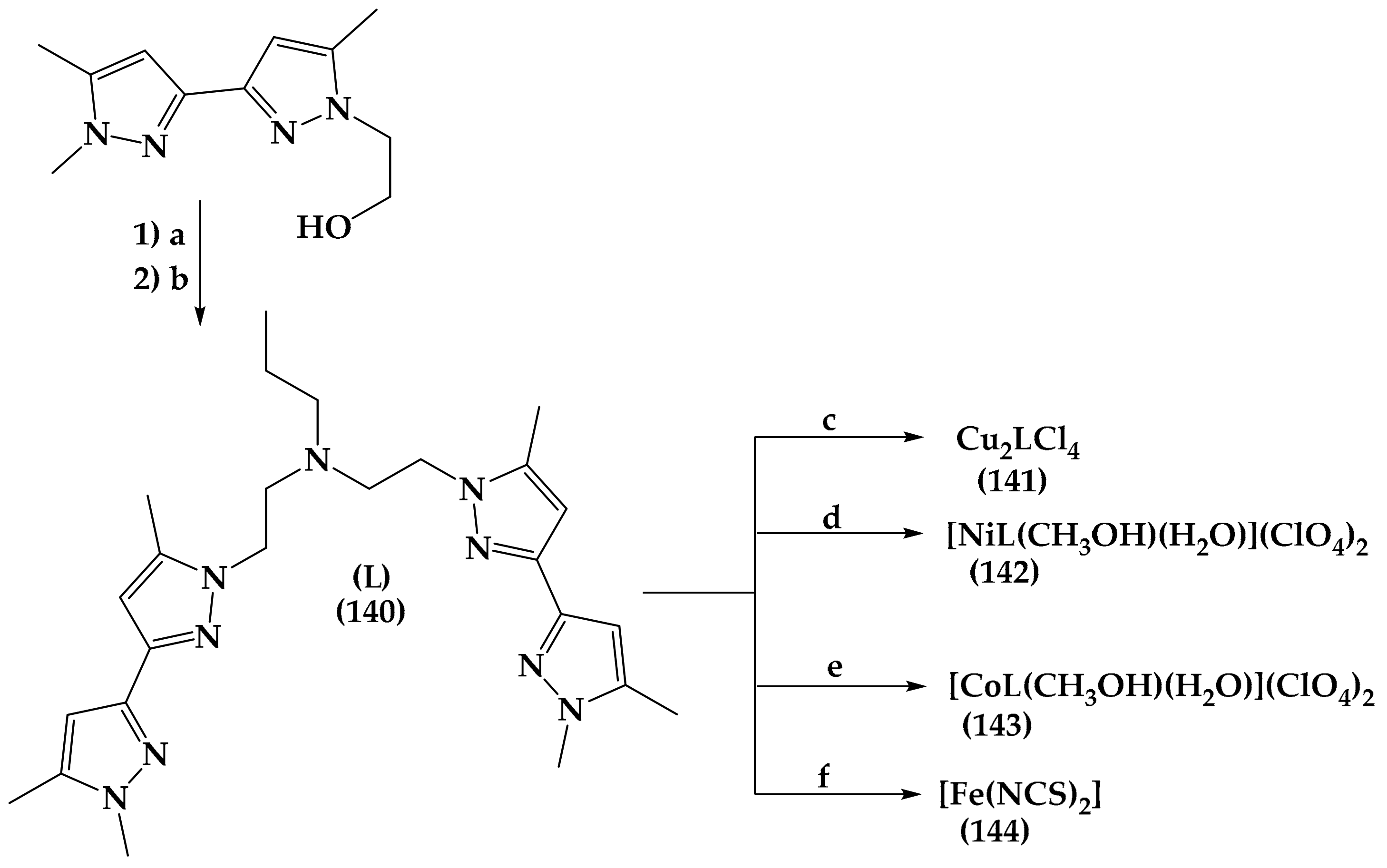
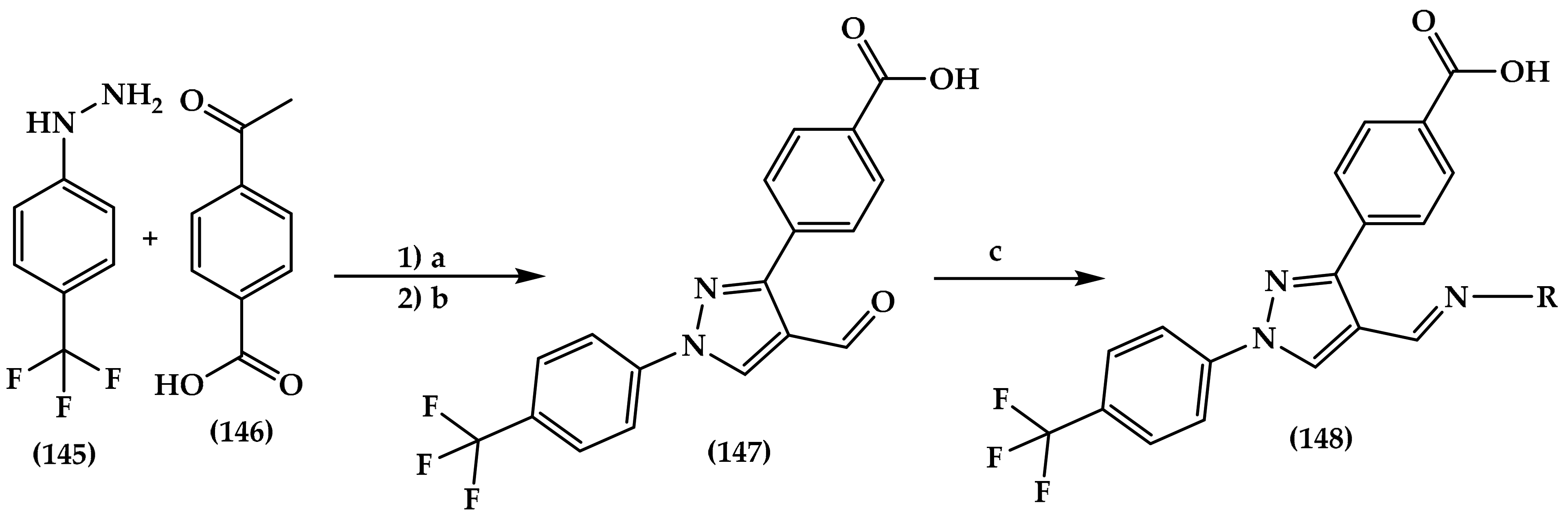
6.7. Pyrazole Derivatives
7. Prodrugs
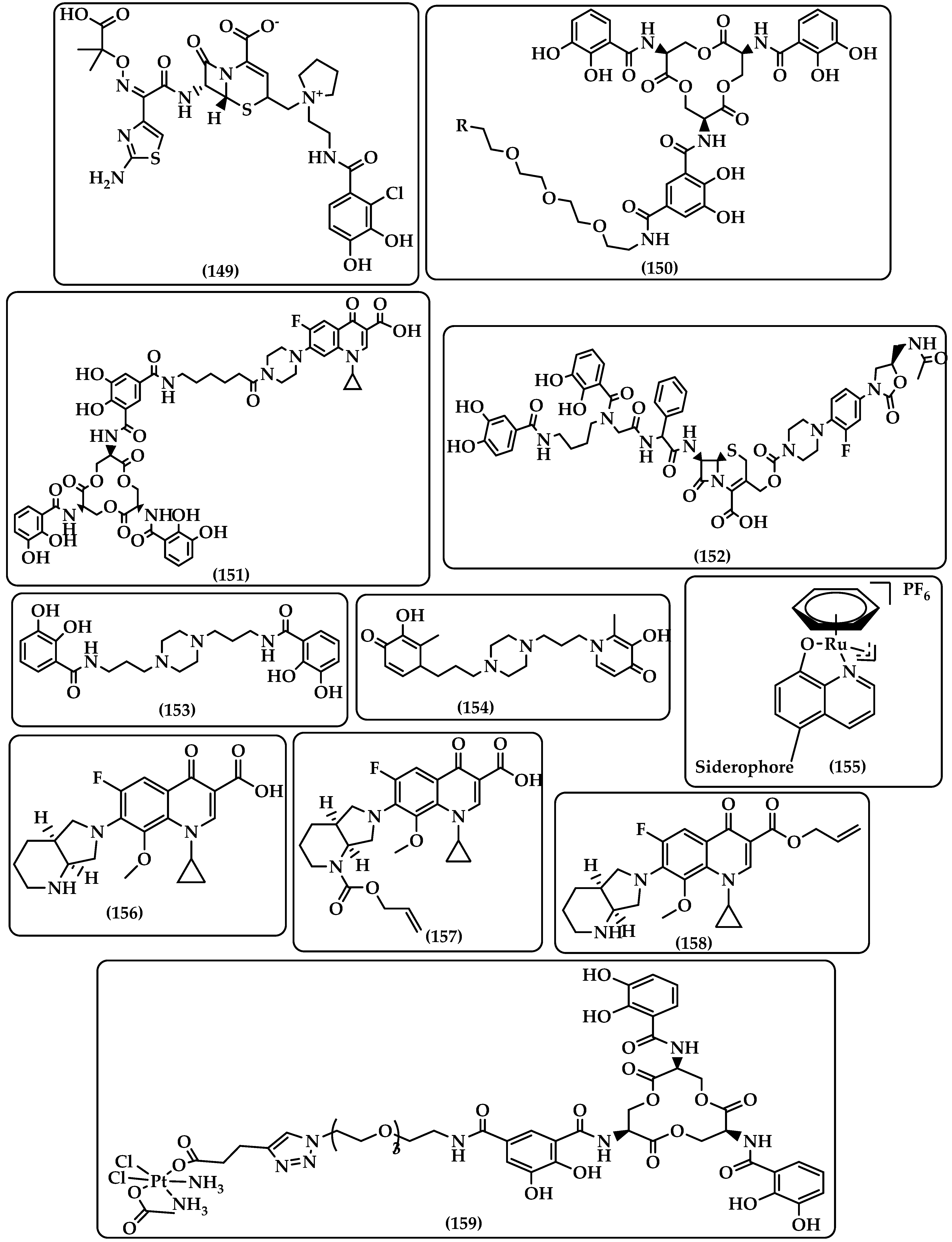
7.1. Siderophores
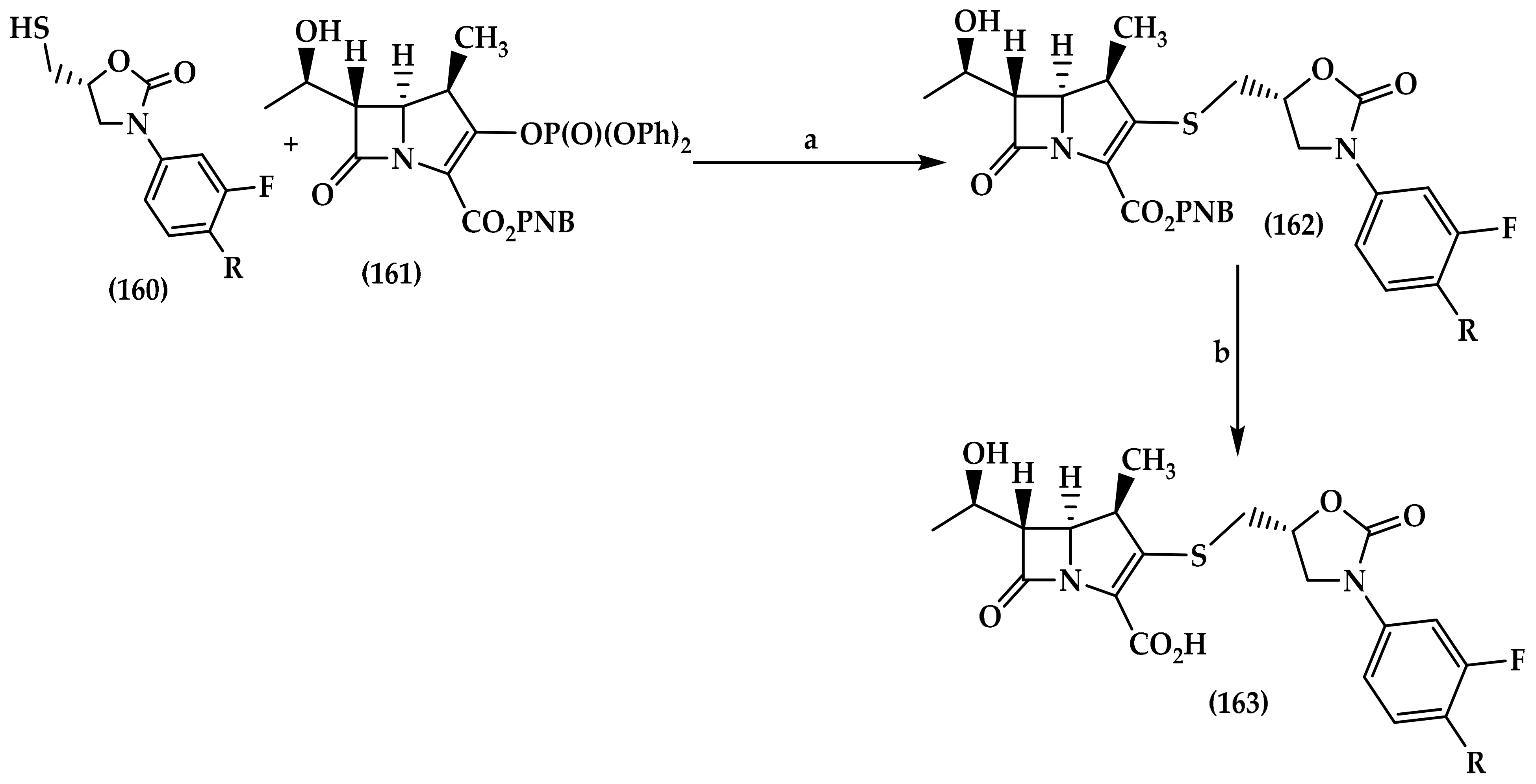
7.2. Carbapenem-Oxazolidinones
7.3. Oral GyrB/ParE Dual Binding Inhibitor
7.4. Antimicrobial Peptides (AMPs) Prodrugs
| Prodrugs | Mechanism of Action and Examples |
|---|---|
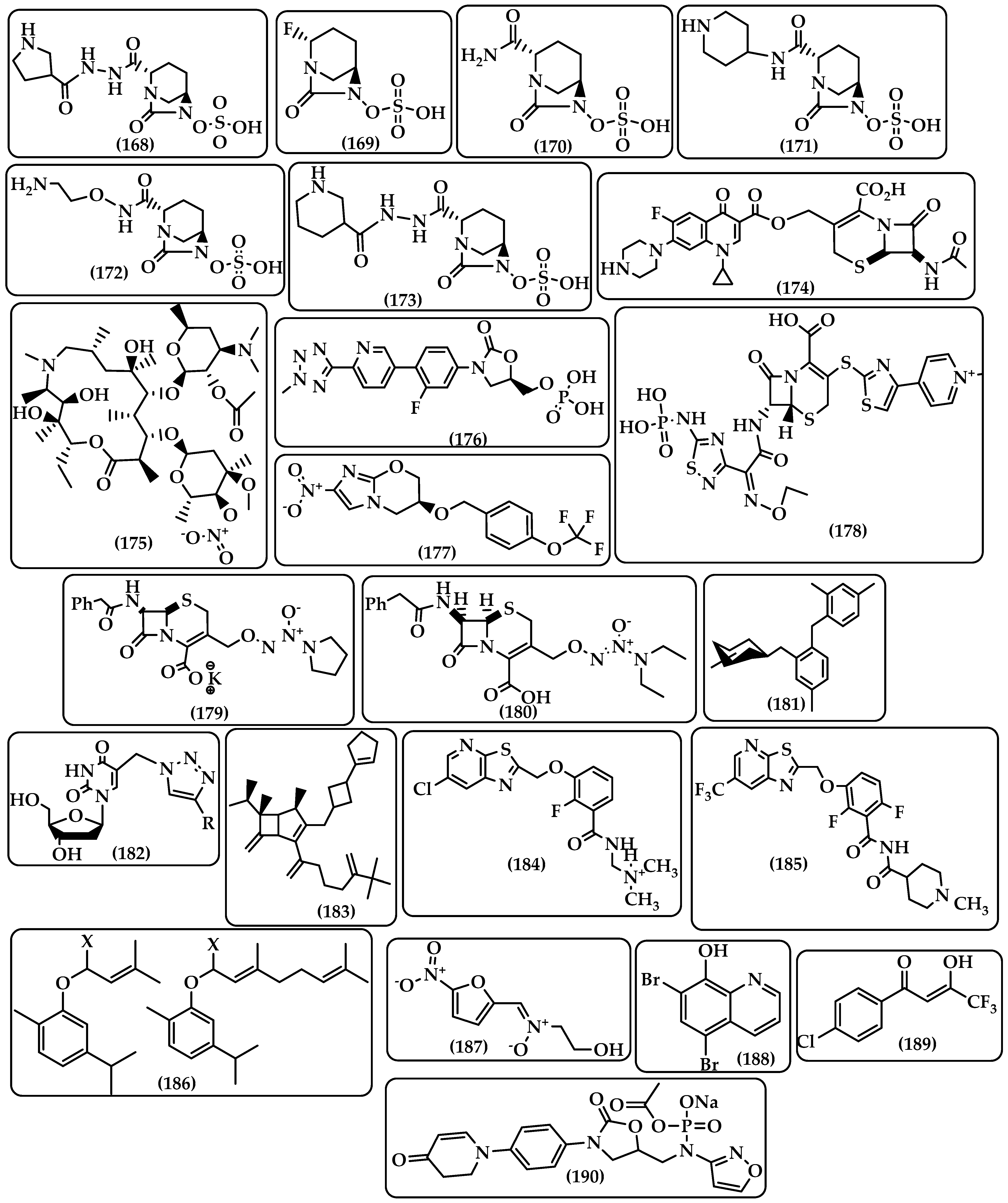
| Diazabicyclooctanones (DBOs) | The active drug is produced from DBOs, which are sulfate-containing prodrugs that are in vivo activated by esterase cleavage that intramolecularly assaults the electrophilic neopentyl methylene group [249]. DBOs function as strong inhibitors of class A and class C β-lactamases. The serine active site of the β-lactamase is targeted by an amide group on the five-membered ring of DBOs, forming a carbamoyl adduct. The effectiveness of the antibiotic can be restored by using the prodrug in conjunction with the proper oral β-lactam antibiotics [139,250,251,252]. Examples of DBOs are WCK 5153 (168), ANT3310 (169), and the following: avibactam (170), relebactam (171), nacubactam (172), zidebactam (173) (Figure 17). |
| β-Lactamase-Activated Ciprofloxacin Prodrug | A prodrug of cephalosporin and fluoroquinolone ((6R,7R) -7-Acetamido-3-(((1-cyclopropyl-6-fluoro-4-oxo-7- (piperazin-1-yl) (piperazin-1-yl) -1,4-dihydroquinoline-3-carbonyl)oxy)- methyl)-8-oxo-5-thia-1-azabicyclo [4.2.0]oct-2-ene-2-carboxylic Acid) created by Evans et al. [253] (174, Figure 17) to deliver ciprofloxacin only to bacteria that express β-lactamase. When cephalosporin is cleaved by β-lactamase, the prodrug’s 3′-cephem ester, which was created by attaching ciprofloxacin via a carboxylic acid, releases ciprofloxacin. |
| Azithromycin Prodrug CSY5669 | Both an antibiotic and an immunomodulator, azithromycin. Azithromycin prodrug (CSY5669) (175, Figure 17) was created by Saris et al. [254] to enhance the immunomodulatory properties of azithromycin by combining it with nitric oxide and acetate as immune activators. It is possible to use CSY5669 as an adjuvant drug in the treatment of pneumonia brought on by MRSA by assisting in the eradication of bacteria and limiting inflammation-associated pathology. The prodrug showed an enhancement of intracellular killing of MARSA in monocyte-derived macrophages and peripheral blood leukocytes as well as reduced inflammatory responses in mice airways in vivo. |
| Tedizolid phosphate (TR701) | Prodrug of the antibiotic oxazolidinone tedizolid (TR701) (176, Figure 17), which is used to treat bacterial skin infections. Plasma phosphatasese converts it to its active parent drug tidezolide, which is highly active in vitro against Gram-positive bacteria, including MRSA [255,256,257]. |
| Pretomanid | A prodrug of an antibiotic (177, Figure 17) that, after being converted to a desnitro derivative by Mycobacterium tuberculosis deazaflavin-dependent nitroreductase (Ddn) [258], acts by raising nitric oxide levels. To treat tuberculosis with drug resistance, it is used with bedaquiline and linezolid [259]. |
| Ceftaroline fosamil | A prodrug (178, Figure 17) that is activated by plasma phosphatase to produce ceftaroline, which is used to treat community-acquired bacterial pneumonia (CABP) and acute bacterial skin infections [260,261]. |
| Cephalosporin-3′-diazeniumdiolates (C3Ds) prodrugs | After reacting with β-lactamases and being broken down by transpeptidases, a nitric oxide (NO) donor prodrug with a β-lactam ring in its structure selectively releases NO. The diazeniumdiolate NO donor-containing PYRRO-C3D (179, Figure 17) is one of two C3Ds that are currently being developed. The second prodrug is DEA-C3D (180, Figure 17) which contains the phenacetyl side chain of cefaloram and the diazeniumdiolate NO donor. The prodrugs are a good possibility for lowering antibiotic tolerance linked to biofilms [262,263,264]. |
| Triclosan glycoside prodrugs | The identification of the bacterial enzyme glycosidase resulted in the identification of glycoside derivatives as bacterium-targeting prodrugs (181, Figure 17). Gram-positive and Gram-negative bacteria are inhibited by triclosan glycoside derivatives (α-D-glycopyranosides and β-D-glycopyranosides), which has the potential to be utilized orally for the treatment of systemic infections [265,266,267] |
| 5-Modified 2ʹ-Deoxyuridines prodrugs | The precise mechanism by which pyrimidine nucleoside derivatives work is unknown; however, some of the compounds inhibited the microbial enzyme flavin-dependent thymidylate synthase (ThyX), which is not present in humans, and others operated on mycobacterial cell wall destruction [268]. Negrya et al. [269] created carrier-linked prodrugs of 5-modified 2’-deoxyuridines (182, Figure 17) since the parent drugs, 5-dodecyloxymethyl 2’-deoxyuridine and 5- [4-decyl-(1,2,3-triazol-1-yl) methyl]-2’-deoxyuridine, were poorly soluble in water. To increase solubility, a triethylene and tetraethylene glycol moiety was linked to the 3′ and 5′ hydroxyl groups of the parent molecules using a carbonate group. |
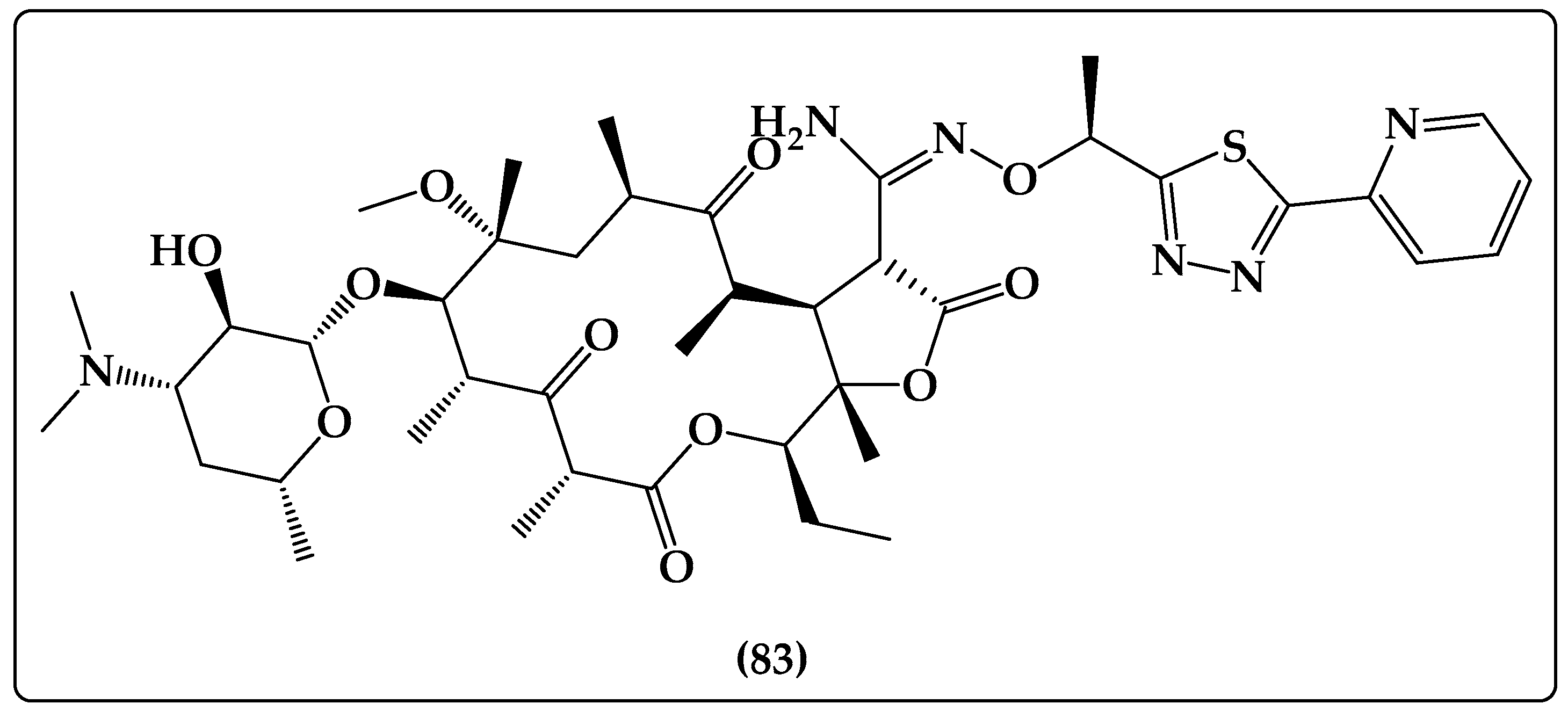
| Tebipenem pivoxil Prodrug | Tebipenem pivoxil HBr salt (183, Figure 17) is a tebipenem ester prodrug that can be taken orally and has improved bioavailability. It is now being developed to treat difficult urinary tract infections in adults. It is approved for use in Japan to treat ear, nose, throat, and respiratory infections in children [270]. |
| FtsZ-Targeting Benzamide Prodrugs | A prokaryote-specific protein called Fts-Z (Filamenting temperature-sensitive mutant Z) is involved in bacterial cell division. In order to combat methicillin-sensitive and resistant Staphylococcus aureus (MSSA and MRSA), PC190723 is a FtsZ-Targeting Benzamide the N-Mannich base prodrug TXY436 (184, Figure 17) was developed as a result of poor solubility; it has improved pharmacological characteristics but requires high effective doses. Because of this, a novel prodrug called TXA709 (185, Figure 17) was developed based on TXY436 with a CF3 group in place of the Cl on the pyridyl ring, giving it a longer half-life and higher oral bioavailability than TXY436 [271,272]. |
| Carvacrol Prodrugs | A naturally occurring monoterpene called carvacrol can damage bacterial membranes and prevent Gram-positive bacteria from forming biofilms [273]. Carvacrol prodrugs (WSCP18-19) (186, Figure 17) were created by prenylating the hydroxyl group of carvacrol due to its low water solubility and chemical stability. The prodrugs exhibit good plasma stability, minimal toxicity, and a potential antibacterial action against S. aureus and S. epidermidis [274]. |
| ADC111, ADC112 and ADC113 | Fleck et al. [275] examined thousands of chimicals in order to find non-specific molecules that prevent alamarBlue, a viability dye, from being reduced. Three prodrugs— ADC111, an analog of the nitrofuran prodrug (187), ADC112, an analog of the tilbroquinol antimicrobial (188), and ADC113, a molecule with a di-ketone functionality that is not a member of any class of recognized antimicrobials (189)—are available (Figure 17). The prodrugs have demonstrated that they are effective in killing E. coli cells [276,277]. |
| Contezolid acefosamil (CZA) prodrug | A brand-new oral oxazolidinone antibacterial medication called Contezolid (CZD) is effective against the majority of aerobic Gram-positive bacteria, including MRSA and vancomycin-resistant Enterococcus. The medication prevents the synthesis of 70S initiation complex, which is essential for bacterial reproduction [278]. As a result of its low solubility, the drug’s intravenous (IV) administration is restricted. Giving patients with diabetic foot infections more therapeutic options in hospitals and outpatient settings is therapy with IV administration followed by oral CZD [279]. Liu et al. [280] created the contezolid acefosamil (CZA) prodrug (190, Figure 17), an isoxazol-3-yl phosphoramidate derivative with excellent water solubility and good stability in pH conditions suited for IV delivery. |
8. Awareness and Knowledge of Antibiotic Prescribing
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| .OH | Hydroxyl Radicals |
| 1,3BPG | 1,3 bisphosphoglycerate |
| Acetic acid | AcOH |
| ADMET | Absorption, distribution, metabolism, excretion, toxicity |
| Ag85 | Antigen 85 |
| AHLs | N-acyl homoserine lactones |
| AIs | Auto-inducers |
| ALFs | Shrimp antilipopolysaccharide factors |
| Aluminium chloride | AlCl |
| AmB | Amphotericin B |
| AMP | Antimicrobial peptides |
| AMR | antimicrobial resistance |
| API | Active pharmaceutical ingredient |
| ASPs | Antimicrobial Stewardship Plans |
| ATP-ABC | Adenosine triphosphate-Binding Cassette |
| AZT | Zidovudine |
| BBr3 | Boron tribromide |
| BGC | Biosynthetic gene cluster |
| BioA | 7,8- diaminopelargonic acid synthase |
| BnBr | Benzyl bromide |
| C2H5I | Ethyl iodide |
| C3Ds | Cephalosporin-3′-diazeniumdiolates |
| C6H6 | Benzene |
| CABP | Community-acquired bacterial pneumonia |
| CBD | Cannabidiol |
| CBG | Cannabigerol |
| CDI | Carbonyldiimidazole |
| CEO | Cinnamon essential oil |
| CFDC | Cefiderocol |
| CH2Cl2 | Dichloromethane |
| CH3CN | Acetonitrile |
| CoAF | Cobalt nanoferrite |
| CPPs | Cell-penetrating peptides |
| CPPs | Critical process parameters |
| CQAs | Critical quality attributes |
| CR | Carbapenem resistant |
| CRAB | Carbapenem-resistant Acinetobacter baumannii |
| CRE | Carbapenem-resistant Enterobacterales |
| Cu | Copper |
| CuAF | copper nanoferrite |
| CZA | Contezolid acefosamil |
| CZD | Contezolid |
| DABCO | Triethylenediamine |
| DAR | Darobactins |
| DBOs | Diazabicyclooctanones |
| DBU | 1,8-Diazabicyclo [5.4.0]undec-7-ene |
| DBU | 1,8-Diazabicyclo(5.4.0)undec-7-ene |
| DCM | Dichloromethane |
| Ddl | D-alanyl-D-alanine synthetase |
| Ddl-B | D-alanine-D-alanine ligase |
| Ddn | Deazaflavin-dependent nitroreductase |
| DDQ | 2,3-Dichloro-5,6-dicyano-1,4-bezoquinone |
| DIPEA | N,N-Diisopropylethylamine |
| DMAP | 4-Dimethylaminopyridine |
| DMC | Dimethyl carbonate |
| DMF | Dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| DPD | (S)-4,5-dihydroxypentane-2,3-dione |
| DprE1 | Decaprenylphosphoryl-β-D-ribose 2′-epimerase |
| DS | Design space |
| DSF | Differential Scanning Fluorimetry |
| DSPG | Di-stearoyl-phosphatidylglycerol |
| E4P | D-erythrose-4-phosphate |
| E4PDH | Erythrose-4-phosphate dehydrogenase |
| ECC | Enterobacter cloacae complex |
| EOCM | Essential oil from Centipeda minima |
| EOOG | Egg oil-organogel |
| EOs | Plant essential oils |
| ESKAPE | Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species pathogens |
| Et3N | Triethylamine |
| EthR | Transcriptional repressor |
| EtOH | Ethanol |
| FBDD | Fragment-based drug design |
| FECL2 | Iron(II) chloride |
| FePPOPHydantoin | Porphyrin iron-based porous organic polymer |
| Fts-Z | Filamenting temperature-sensitive mutant Z |
| G3P | Glyceraldehyde-3-phosphate |
| GA | Glycyrrhizin |
| GC-MS | Gas chromatography-mass spectrometry |
| GRA | 18β-glycyrrhetinic acid |
| Gyr-B | DNA gyrase B subunits |
| H2CBD | 8,9-dihydrocannabidiol |
| HAM | Hamamelitannin |
| HCl | Hydrochloric acid |
| HGT | Horizontal gene transfer |
| HTS | High throughput screening |
| I2 | Iodine |
| InhA | 2-trans-enoyl-acyl carrier protein reductase |
| i-Pr2Net | N,N-Diisopropylethylamine |
| K2CO3 | Potassium carbonate |
| KI | Potassium iodide |
| KNCS | Potassium thiocyanate |
| KOH | Potassium hydroxide |
| Lf | Lactoferrin |
| LiOH | Lithium hydroxide |
| MATE | Multidrug and Toxic Compound Extrusion |
| MBC | Minimum bactericidal concentration |
| m-CPBA | meta-Chloroperoxybenzoic acid |
| MD | Molecular dynamics |
| MDR | Multidrug resistance |
| MEI | Iodomethane |
| MeOH, | Methanol |
| MIC | Minimum inhibitory concentration |
| MMC | Mitomycin C |
| MRSA | Methicillin-resistant S. aureus |
| Ms | Saturation magnetization |
| MSSA | Methicillin-sensitive Staphylococcus aureus |
| mtk-QSBER | multitasking model for quantitative-structure biological effect relationships |
| NaBH4 | Sodium borohydride |
| NaBH4 | Sodium borohydride |
| NaH | Sodium hydride |
| NaHCO3 | Sodium bicarbonate |
| NAOCH3 | Sodium methoxide |
| NAOH | Sodium hydroxide |
| NBS | N-Bromosuccinimide |
| n-Bu3SnH. | Tributyltin hydride |
| n-BuLi | n-Butllithium |
| NCS | N-Chlorosuccinimide |
| NIR | Near-infrared |
| NMR | Nuclear magnetic resonance |
| NO | Nitric oxide |
| NPET | Nascent peptide exit tunnel |
| NPs | Nanoparticles |
| NRPSs | non-ribosomal peptide synthetases |
| ODLs | Odilorhabdins |
| PABA | p-aminobenzoic acid |
| PACE | Proteobacterial Antimicrobial Compound Efflux |
| PBPs | Penicillin-binding proteins |
| PDA@FeS NPs | ferrous sulfide-polydopamine nanoparticles |
| Pd-C | Palladium on carbon |
| PE | Phosphatidyl-ethanolamine |
| PEITC | Phenethyl isothiocyanate |
| PGLEO | Psidium guajava (guava) leaf essential oil |
| PIPD1 | Piperidinol-containing molecule |
| PJI | Prosthetic joint infection |
| PKSs | Polyketide synthases |
| POCl3 | Phosphoryl chloride |
| PPA | Polyphosphoric acid |
| PTC | Peptidyl transferase center |
| QbD | Quality by design |
| QS | Quorum Sensing |
| RA | Risk Assessment |
| RIF-BSA-NPs | Rifampicin-loaded bovine serum albumin nanoparticles |
| RNAP | RNA polymerase |
| RND | Resistance Nodulation Division |
| ROS | Reactive oxygen species |
| ROS | Reactive oxygen species () |
| rt. | Room temperature |
| SAM (RaS) | Radical S-adenosylmethionine |
| SAR | Structure-activity relationships |
| Silver oxide | Ag2O |
| SMR | Small Multidrug Resistance |
| SSD | Silver Sulphadiazine |
| TB | Tuberculosis |
| TBAB | Tetra-n- butylammonium bromide |
| T-BUOH | Tert-Butyl alcohol |
| T-BUOK | Potassium tert-butoxide |
| TCA | Tricarboxylic acid cycle |
| Tf | Transferrin |
| THBTP | Tetrahydro-1-benzothiophene |
| THC | Trans-Δ-9-tetrahydrocannabinol |
| THF | Tetrahydrofuran |
| ThyX | Thymidylate synthase |
| TLM | Thiolactomycin |
| TPP | Target product profile |
| TQ | Thymoquinone |
| TriBE inhibitor | Pyrimidoindole inhibitor |
| TRPPs | Tetracycline ribosomal protection proteins |
| WHO | World Health Organization |
| XDR | extensively drug resistant |
| β-MGP | β-D-galactopyranoside |
References
- Adedeji, W.A. The Treasure Called Antibiotics. Ann. Ib. Postgrad. Med. 2016, 14, 56–57. [Google Scholar] [PubMed]
- Breijyeh, Z.; Jubeh, B.; Karaman, R. Resistance of Gram-Negative Bacteria to Current Antibacterial Agents and Approaches to Resolve It. Molecules 2020, 25, 1340. [Google Scholar] [CrossRef] [PubMed]
- Karaman, R.; Jubeh, B.; Breijyeh, Z. Resistance of Gram-Positive Bacteria to Current Antibacterial Agents and Overcoming Approaches. Molecules 2020, 25, 2888. [Google Scholar] [CrossRef] [PubMed]
- Antimicrobial Resistance, C. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Asokan, G.V.; Ramadhan, T.; Ahmed, E.; Sanad, H. WHO Global Priority Pathogens List: A Bibliometric Analysis of Medline-PubMed for Knowledge Mobilization to Infection Prevention and Control Practices in Bahrain. Oman Med. J. 2019, 34, 184–193. [Google Scholar] [CrossRef]
- WHO. Priority Pathogens List of Antibiotic-Resistant Bacteria for Which New Antibiotics are Urgently Needed; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Shrivastava, S.; Shrivastava, P.; Ramasamy, J. World health organization releases global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics. J. Med. Soc. 2018, 32, 76. [Google Scholar] [CrossRef]
- Mancuso, G.; Midiri, A.; Gerace, E.; Biondo, C. Bacterial Antibiotic Resistance: The Most Critical Pathogens. Pathogens 2021, 10, 1310. [Google Scholar] [CrossRef]
- Mulani, M.S.; Kamble, E.E.; Kumkar, S.N.; Tawre, M.S.; Pardesi, K.R. Emerging Strategies to Combat ESKAPE Pathogens in the Era of Antimicrobial Resistance: A Review. Front. Microbiol. 2019, 10, 539. [Google Scholar] [CrossRef]
- De Oliveira, D.M.P.; Forde, B.M.; Kidd, T.J.; Harris, P.N.A.; Schembri, M.A.; Beatson, S.A.; Paterson, D.L.; Walker, M.J. Antimicrobial Resistance in ESKAPE Pathogens. Clin. Microbiol. Rev. 2020, 33, e00181-19. [Google Scholar] [CrossRef]
- Qadri, H.; Shah, A.H.; Mir, M. Novel Strategies to Combat the Emerging Drug Resistance in Human Pathogenic Microbes. Curr. Drug Targets 2021, 22, 1424–1436. [Google Scholar] [CrossRef]
- Kohanski, M.A.; Dwyer, D.J.; Collins, J.J. How antibiotics kill bacteria: From targets to networks. Nat. Rev. Microbiol. 2010, 8, 423–435. [Google Scholar] [CrossRef]
- Calhoun, C.; Wermuth, H.R.; Hall, G.A. Antibiotics; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Bush, K.; Bradford, P.A. beta-Lactams and beta-Lactamase Inhibitors: An Overview. Cold Spring Harb. Perspect. Med. 2016, 6, a025247. [Google Scholar] [CrossRef]
- Eyler, R.F.; Shvets, K. Clinical Pharmacology of Antibiotics. Clin. J. Am. Soc. Nephrol. Cjasn 2019, 14, 1080–1090. [Google Scholar] [CrossRef]
- Blaskovich, M.A.T.; Hansford, K.A.; Butler, M.S.; Jia, Z.; Mark, A.E.; Cooper, M.A. Developments in Glycopeptide Antibiotics. Acs Infect. Dis. 2018, 4, 715–735. [Google Scholar] [CrossRef]
- Bionda, N.; Pitteloud, J.P.; Cudic, P. Cyclic lipodepsipeptides: A new class of antibacterial agents in the battle against resistant bacteria. Future Med. Chem. 2013, 5, 1311–1330. [Google Scholar] [CrossRef]
- Schneider, T.; Muller, A.; Miess, H.; Gross, H. Cyclic lipopeptides as antibacterial agents—Potent antibiotic activity mediated by intriguing mode of actions. Int. J. Med. Microbiol. Ijmm 2014, 304, 37–43. [Google Scholar] [CrossRef]
- Adams, R.A.; Leon, G.; Miller, N.M.; Reyes, S.P.; Thantrong, C.H.; Thokkadam, A.M.; Lemma, A.S.; Sivaloganathan, D.M.; Wan, X.; Brynildsen, M.P. Rifamycin antibiotics and the mechanisms of their failure. J. Antibiot. 2021, 74, 786–798. [Google Scholar] [CrossRef]
- Krause, K.M.; Serio, A.W.; Kane, T.R.; Connolly, L.E. Aminoglycosides: An Overview. Cold Spring Harb. Perspect. Med. 2016, 6, a027029. [Google Scholar] [CrossRef]
- Pham, T.D.M.; Ziora, Z.M.; Blaskovich, M.A.T. Quinolone antibiotics. Medchemcomm 2019, 10, 1719–1739. [Google Scholar] [CrossRef]
- Kemnic, T.R.; Coleman, M. Trimethoprim Sulfamethoxazole; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Patel, P.H.; Hashmi, M.F. Macrolides; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Vazquez-Laslop, N.; Mankin, A.S. How Macrolide Antibiotics Work. Trends Biochem. Sci. 2018, 43, 668–684. [Google Scholar] [CrossRef] [PubMed]
- Rusu, A.; Buta, E.L. The Development of Third-Generation Tetracycline Antibiotics and New Perspectives. Pharmaceutics 2021, 13, 2085. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, F.; Starosta, A.L.; Arenz, S.; Sohmen, D.; Donhofer, A.; Wilson, D.N. Tetracycline antibiotics and resistance mechanisms. Biol. Chem. 2014, 395, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Foti, C.; Piperno, A.; Scala, A.; Giuffre, O. Oxazolidinone Antibiotics: Chemical, Biological and Analytical Aspects. Molecules 2021, 26, 4280. [Google Scholar] [CrossRef] [PubMed]
- Bonfiglio, G.; Furneri, P.M. Novel streptogramin antibiotics. Expert Opin. Investig. Drugs 2001, 10, 185–198. [Google Scholar] [CrossRef]
- Li, Q.; Pellegrino, J.; Lee, D.J.; Tran, A.A.; Chaires, H.A.; Wang, R.; Park, J.E.; Ji, K.; Chow, D.; Zhang, N.; et al. Synthetic group A streptogramin antibiotics that overcome Vat resistance. Nature 2020, 586, 145–150. [Google Scholar] [CrossRef]
- Oong, G.C.; Tadi, P. Chloramphenicol; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Spizek, J.; Rezanka, T. Lincosamides: Chemical structure, biosynthesis, mechanism of action, resistance, and applications. Biochem Pharm. 2017, 133, 20–28. [Google Scholar] [CrossRef]
- WHO. World Health Organization Ten Threats to Global Health in 2019; WHO: Geneva, Switzerland, 2019. [Google Scholar]
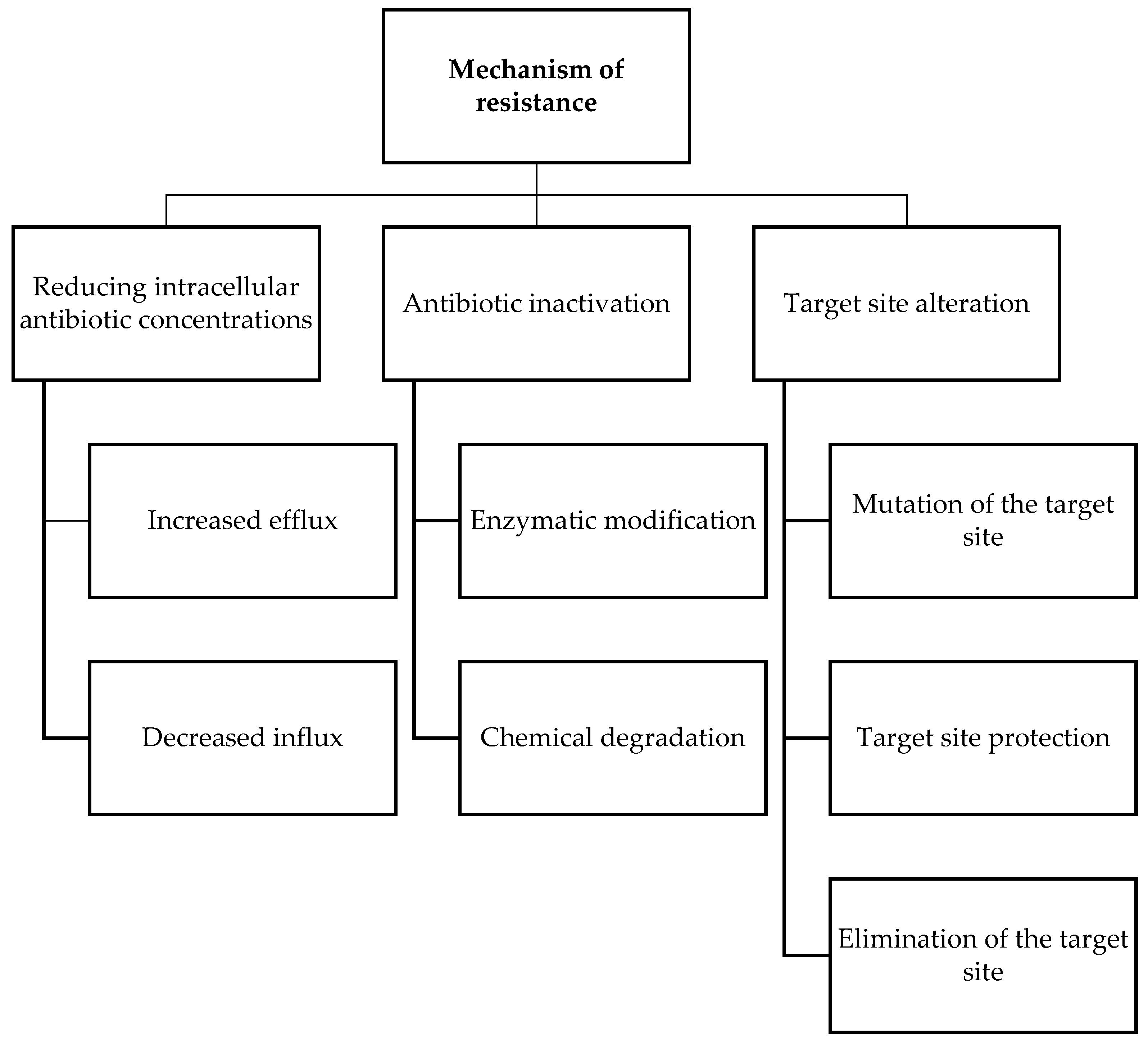
- Reygaert, W.C. An overview of the antimicrobial resistance mechanisms of bacteria. Aims Microbiol. 2018, 4, 482–501. [Google Scholar] [CrossRef]
- Francine, P. Systems Biology: New Insight into Antibiotic Resistance. Microorganisms 2022, 10, 2362. [Google Scholar] [CrossRef]
- Chancey, S.T.; Zahner, D.; Stephens, D.S. Acquired inducible antimicrobial resistance in Gram-positive bacteria. Future Microbiol. 2012, 7, 959–978. [Google Scholar] [CrossRef]
- Mahon, C.R.; Lehman, D.C. Textbook of Diagnostic Microbiology—E-Book; Elsevier Health Sciences: USA, 2022. [Google Scholar]
- Sultan, I.; Rahman, S.; Jan, A.T.; Siddiqui, M.T.; Mondal, A.H.; Haq, Q.M.R. Antibiotics, Resistome and Resistance Mechanisms: A Bacterial Perspective. Front. Microbiol. 2018, 9, 2066. [Google Scholar] [CrossRef]
- Ndagi, U.; Falaki, A.A.; Abdullahi, M.; Lawal, M.M.; Soliman, M.E. Antibiotic resistance: Bioinformatics-based understanding as a functional strategy for drug design. Rsc Adv. 2020, 10, 18451–18468. [Google Scholar] [CrossRef]
- Kabra, R.; Chauhan, N.; Kumar, A.; Ingale, P.; Singh, S. Efflux pumps and antimicrobial resistance: Paradoxical components in systems genomics. Prog. Biophys. Mol. Biol. 2019, 141, 15–24. [Google Scholar] [CrossRef]
- Ogawara, H. Comparison of Antibiotic Resistance Mechanisms in Antibiotic-Producing and Pathogenic Bacteria. Molecules 2019, 24, 3430. [Google Scholar] [CrossRef]
- Spengler, G.; Kincses, A.; Gajdacs, M.; Amaral, L. New Roads Leading to Old Destinations: Efflux Pumps as Targets to Reverse Multidrug Resistance in Bacteria. Molecules 2017, 22, 468. [Google Scholar] [CrossRef]
- Munita, J.M.; Arias, C.A. Mechanisms of Antibiotic Resistance. Microbiol. Spectr. 2016, 4, 481–511. [Google Scholar] [CrossRef]
- Schaenzer, A.J.; Wright, G.D. Antibiotic Resistance by Enzymatic Modification of Antibiotic Targets. Trends Mol. Med. 2020, 26, 768–782. [Google Scholar] [CrossRef]
- Wilson, D.N.; Hauryliuk, V.; Atkinson, G.C.; O’Neill, A.J. Target protection as a key antibiotic resistance mechanism. Nat. Rev. Microbiol. 2020, 18, 637–648. [Google Scholar] [CrossRef]
- Manyi-Loh, C.; Mamphweli, S.; Meyer, E.; Okoh, A. Antibiotic Use in Agriculture and Its Consequential Resistance in Environmental Sources: Potential Public Health Implications. Molecules 2018, 23, 795. [Google Scholar] [CrossRef]
- Ayukekbong, J.A.; Ntemgwa, M.; Atabe, A.N. The threat of antimicrobial resistance in developing countries: Causes and control strategies. Antimicrob. Resist. Infect. Control 2017, 6, 47. [Google Scholar] [CrossRef]
- Finley, R.L.; Collignon, P.; Larsson, D.G.; McEwen, S.A.; Li, X.Z.; Gaze, W.H.; Reid-Smith, R.; Timinouni, M.; Graham, D.W.; Topp, E. The scourge of antibiotic resistance: The important role of the environment. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2013, 57, 704–710. [Google Scholar] [CrossRef]
- Guetiya Wadoum, R.E.; Zambou, N.F.; Anyangwe, F.F.; Njimou, J.R.; Coman, M.M.; Verdenelli, M.C.; Cecchini, C.; Silvi, S.; Orpianesi, C.; Cresci, A.; et al. Abusive use of antibiotics in poultry farming in Cameroon and the public health implications. Br. Poult. Sci. 2016, 57, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Baynes, R.E.; Dedonder, K.; Kissell, L.; Mzyk, D.; Marmulak, T.; Smith, G.; Tell, L.; Gehring, R.; Davis, J.; Riviere, J.E. Health concerns and management of select veterinary drug residues. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2016, 88, 112–122. [Google Scholar] [CrossRef]
- Maron, D.F.; Smith, T.J.; Nachman, K.E. Restrictions on antimicrobial use in food animal production: An international regulatory and economic survey. Glob. Health 2013, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Gillings, M.R. Evolutionary consequences of antibiotic use for the resistome, mobilome and microbial pangenome. Front. Microbiol. 2013, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Dolliver, H.; Gupta, S. Antibiotic losses in leaching and surface runoff from manure-amended agricultural land. J. Environ. Qual. 2008, 37, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. Mmbr 2010, 74, 417–433. [Google Scholar] [CrossRef]
- Phillips, I.; Casewell, M.; Cox, T.; De Groot, B.; Friis, C.; Jones, R.; Nightingale, C.; Preston, R.; Waddell, J. Does the use of antibiotics in food animals pose a risk to human health? A critical review of published data. J. Antimicrob. Chemother. 2004, 53, 28–52. [Google Scholar] [CrossRef]
- Marshall, B.M.; Levy, S.B. Food animals and antimicrobials: Impacts on human health. Clin. Microbiol. Rev. 2011, 24, 718–733. [Google Scholar] [CrossRef]
- Moudgil, P.; Bedi, J.S.; Moudgil, A.D.; Gill, J.P.S.; Aulakh, R.S. Emerging issue of antibiotic resistance from food producing animals in India: Perspective and legal framework. Food Rev. Int. 2017, 34, 447–462. [Google Scholar] [CrossRef]
- Sharma, C.; Rokana, N.; Chandra, M.; Singh, B.P.; Gulhane, R.D.; Gill, J.P.S.; Ray, P.; Puniya, A.K.; Panwar, H. Antimicrobial Resistance: Its Surveillance, Impact, and Alternative Management Strategies in Dairy Animals. Front. Vet. Sci. 2017, 4, 237. [Google Scholar] [CrossRef]
- Ying, G.G.; He, L.Y.; Ying, A.J.; Zhang, Q.Q.; Liu, Y.S.; Zhao, J.L. China Must Reduce Its Antibiotic Use. Environ. Sci. Technol. 2017, 51, 1072–1073. [Google Scholar] [CrossRef]
- Marquardt, R.R.; Li, S. Antimicrobial resistance in livestock: Advances and alternatives to antibiotics. Anim. Front. Rev. Mag. Anim. Agric. 2018, 8, 30–37. [Google Scholar] [CrossRef]
- Rahman, M.R.T.; Fliss, I.; Biron, E. Insights in the Development and Uses of Alternatives to Antibiotic Growth Promoters in Poultry and Swine Production. Antibiotics 2022, 11, 766. [Google Scholar] [CrossRef]
- Mubeen, B.; Ansar, A.N.; Rasool, R.; Ullah, I.; Imam, S.S.; Alshehri, S.; Ghoneim, M.M.; Alzarea, S.I.; Nadeem, M.S.; Kazmi, I. Nanotechnology as a Novel Approach in Combating Microbes Providing an Alternative to Antibiotics. Antibiotics 2021, 10, 1473. [Google Scholar] [CrossRef]
- Vimbela, G.V.; Ngo, S.M.; Fraze, C.; Yang, L.; Stout, D.A. Antibacterial properties and toxicity from metallic nanomaterials. Int. J. Nanomed. 2017, 12, 3941–3965. [Google Scholar] [CrossRef]
- Zhou, Y.; Kong, Y.; Kundu, S.; Cirillo, J.D.; Liang, H. Antibacterial activities of gold and silver nanoparticles against Escherichia coli and bacillus Calmette-Guérin. J. Nanobiotechnology 2012, 10, 9. [Google Scholar] [CrossRef]
- Khan, I.; Saeed, K.; Khan, I. Nanoparticles: Properties, applications and toxicities. Arab. J. Chem. 2019, 12, 908–931. [Google Scholar] [CrossRef]
- Zhang, N.; Xiong, G.; Liu, Z. Toxicity of metal-based nanoparticles: Challenges in the nano era. Front. Bioeng. Biotechnol. 2022, 10, 1001572. [Google Scholar] [CrossRef]
- Yadav, N.; Kumar, U.; Chauhan, V.S. Conformationally restricted, dipeptide-based, self-assembled nanoparticles for efficient vancomycin delivery. Nanomedicine 2023. [Google Scholar] [CrossRef]
- Mohammadinejat, M.; Sepahi, A.A.; Alipour, E. Antibacterial and Anti-Biofilm Activities of Silver Nano Particles Conjugated to Chitosan Against Multi-Drug Resistant Bacteria. Clin. Lab. 2023, 69. [Google Scholar] [CrossRef]
- Xu, N.; Huang, Q.; Shi, L.; Wang, J.; Li, X.; Guo, W.; Yan, D.; Ni, T.; Yang, Z.; Yan, Y. A bioinspired polydopamine-FeS nanocomposite with high antimicrobial efficiency via NIR-mediated Fenton reaction. Dalton Trans. 2023, 52, 1687–1701. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Q.; Qu, X.; Zhang, Q.; Zhang, X. A metalloporphyrin and hydantoin functionalized nanozyme with synergistically enhanced bacterial inhibition. Biomater. Sci. 2023, 11, 1785–1796. [Google Scholar] [CrossRef] [PubMed]
- El-Bassuony, A.A.H.; Gamal, W.M.; Abdelsalam, H.K. Impact of different magnetic materials added to silver-magnetite nanoparticles on the structural, magnetic and antimicrobial properties. Eur. Phys. J. Spec. Top. 2023, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lei, R.; Hou, J.; Chen, Q.; Yuan, W.; Cheng, B.; Sun, Y.; Jin, Y.; Ge, L.; Ben-Sasson, S.A.; Chen, J.; et al. Self-Assembling Myristoylated Human alpha-Defensin 5 as a Next-Generation Nanobiotics Potentiates Therapeutic Efficacy in Bacterial Infection. Acs Nano 2018, 12, 5284–5296. [Google Scholar] [CrossRef] [PubMed]
- Mangoni, A.A.; Tuccinardi, T.; Collina, S.; Vanden Eynde, J.J.; Munoz-Torrero, D.; Karaman, R.; Siciliano, C.; de Sousa, M.E.; Prokai-Tatrai, K.; Rautio, J.; et al. Breakthroughs in Medicinal Chemistry: New Targets and Mechanisms, New Drugs, New Hopes-3. Molecules 2018, 23, 130. [Google Scholar] [CrossRef]
- Yu, L.X.; Amidon, G.; Khan, M.A.; Hoag, S.W.; Polli, J.; Raju, G.K.; Woodcock, J. Understanding pharmaceutical quality by design. Aaps J. 2014, 16, 771–783. [Google Scholar] [CrossRef]
- Woodcock, J. The concept of pharmaceutical quality. Am. Pharm. Rev. 2004, 7, 1–3. [Google Scholar]
- Joshi, M.; Yadav, K.S.; Prabhakar, B. Quality by Design Approach for Development and Optimization of Rifampicin Loaded Bovine Serum Albumin Nanoparticles and Characterization. Curr. Drug Deliv. 2021, 18, 1338–1351. [Google Scholar] [CrossRef]
- Thakur, K.; Mahajan, A.; Sharma, G.; Singh, B.; Raza, K.; Chhibber, S.; Katare, O.P. Implementation of Quality by Design (QbD) approach in development of silver sulphadiazine loaded egg oil organogel: An improved dermatokinetic profile and therapeutic efficacy in burn wounds. Int. J. Pharm. 2020, 576, 118977. [Google Scholar] [CrossRef]
- Ghodake, V.; Vishwakarma, J.; Vavilala, S.L.; Patravale, V. Cefoperazone sodium liposomal formulation to mitigate P. aeruginosa biofilm in Cystic fibrosis infection: A QbD approach. Int. J. Pharm. 2020, 587, 119696. [Google Scholar] [CrossRef]
- Liu, H.; Rivnay, B.; Avery, K.; Myung, J.H.; Kozak, D.; Landrau, N.; Nivorozhkin, A.; Ashraf, M.; Yoon, S. Optimization of the manufacturing process of a complex amphotericin B liposomal formulation using quality by design approach. Int. J. Pharm. 2020, 585, 119473. [Google Scholar] [CrossRef]
- Manteghi, R.; Pallagi, E.; Olajos, G.; Csoka, I. Pegylation and formulation strategy of Anti-Microbial Peptide (AMP) according to the quality by design approach. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2020, 144, 105197. [Google Scholar] [CrossRef]
- Ekins, S.; Mestres, J.; Testa, B. In silico pharmacology for drug discovery: Methods for virtual ligand screening and profiling. Br. J. Pharmacol. 2007, 152, 9–20. [Google Scholar] [CrossRef]
- Colquitt, R.B.; Colquhoun, D.A.; Thiele, R.H. In silico modelling of physiologic systems. Best Pract. Res. Clin. Anaesthesiol. 2011, 25, 499–510. [Google Scholar] [CrossRef]
- Oyama, L.B.; Olleik, H.; Teixeira, A.C.N.; Guidini, M.M.; Pickup, J.A.; Hui, B.Y.P.; Vidal, N.; Cookson, A.R.; Vallin, H.; Wilkinson, T.; et al. In silico identification of two peptides with antibacterial activity against multidrug-resistant Staphylococcus aureus. Npj Biofilms Microbiomes 2022, 8, 58. [Google Scholar] [CrossRef]
- Masalha, M.; Rayan, M.; Adawi, A.; Abdallah, Z.; Rayan, A. Capturing antibacterial natural products with in silico techniques. Mol. Med. Rep. 2018, 18, 763–770. [Google Scholar] [CrossRef]
- Alhadrami, H.A.; Abdulaal, W.H.; Hassan, H.M.; Alhakamy, N.A.; Sayed, A.M. In Silico-Based Discovery of Natural Anthraquinones with Potential against Multidrug-Resistant E. coli. Pharmaceuticals 2022, 15, 86. [Google Scholar] [CrossRef]
- Ali, H.M. In-silico investigation of a novel inhibitors against the antibiotic-resistant Neisseria gonorrhoeae bacteria. Saudi J. Biol. Sci. 2022, 29, 103424. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Cordeiro, M.N. Multitasking models for quantitative structure-biological effect relationships: Current status and future perspectives to speed up drug discovery. Expert Opin. Drug Discov. 2015, 10, 245–256. [Google Scholar] [CrossRef]
- Qureshi, K.A.; Imtiaz, M.; Parvez, A.; Rai, P.K.; Jaremko, M.; Emwas, A.H.; Bholay, A.D.; Fatmi, M.Q. In Vitro and In Silico Approaches for the Evaluation of Antimicrobial Activity, Time-Kill Kinetics, and Anti-Biofilm Potential of Thymoquinone (2-Methyl-5-propan-2-ylcyclohexa-2,5-diene-1,4-dione) against Selected Human Pathogens. Antibiotics 2022, 11, 79. [Google Scholar] [CrossRef]
- Muller, E.; Hotzel, H.; Linde, J.; Hanel, I.; Tomaso, H. Antimicrobial Resistance and in silico Virulence Profiling of Aliarcobacter butzleri Strains From German Water Poultry. Front. Microbiol. 2020, 11, 617685. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, M.; Mendes, V.; Vistal, R.G.; Dias, D.M.G.; Zahorszka, M.; Mikusova, K.; Kordulakova, J.; Coyne, A.G.; Blundell, T.L.; Abell, C. Fragment-Based Design of Mycobacterium tuberculosis InhA Inhibitors. J. Med. Chem. 2020, 63, 4749–4761. [Google Scholar] [CrossRef] [PubMed]
- Gutschow, M.; Eynde, J.J.V.; Jampilek, J.; Kang, C.; Mangoni, A.A.; Fossa, P.; Karaman, R.; Trabocchi, A.; Scott, P.J.H.; Reynisson, J.; et al. Breakthroughs in Medicinal Chemistry: New Targets and Mechanisms, New Drugs, New Hopes-7. Molecules 2020, 25, 2968. [Google Scholar] [CrossRef] [PubMed]
- Mallakuntla, M.K.; Togre, N.S.; Santos, D.B.; Tiwari, S. Implications of Fragment-Based Drug Discovery in Tuberculosis and HIV. Pharmaceuticals 2022, 15, 1415. [Google Scholar] [CrossRef]
- Manina, G.; Pasca, M.R.; Buroni, S.; De Rossi, E.; Riccardi, G. Decaprenylphosphoryl-beta-D-ribose 2’-epimerase from Mycobacterium tuberculosis is a magic drug target. Curr. Med. Chem. 2010, 17, 3099–3108. [Google Scholar] [CrossRef]
- Borthwick, J.A.; Alemparte, C.; Wall, I.; Whitehurst, B.C.; Argyrou, A.; Burley, G.; de Dios-Anton, P.; Guijarro, L.; Monteiro, M.C.; Ortega, F.; et al. Mycobacterium tuberculosis Decaprenylphosphoryl-beta-d-ribose Oxidase Inhibitors: Expeditious Reconstruction of Suboptimal Hits into a Series with Potent in Vivo Activity. J. Med. Chem. 2020, 63, 2557–2576. [Google Scholar] [CrossRef]
- Kapilashrami, K.; Bommineni, G.R.; Machutta, C.A.; Kim, P.; Lai, C.T.; Simmerling, C.; Picart, F.; Tonge, P.J. Thiolactomycin-based beta-ketoacyl-AcpM synthase A (KasA) inhibitors: Fragment-based inhibitor discovery using transient one-dimensional nuclear overhauser effect NMR spectroscopy. J. Biol. Chem. 2013, 288, 6045–6052. [Google Scholar] [CrossRef]
- Villemagne, B.; Flipo, M.; Blondiaux, N.; Crauste, C.; Malaquin, S.; Leroux, F.; Piveteau, C.; Villeret, V.; Brodin, P.; Villoutreix, B.O.; et al. Ligand efficiency driven design of new inhibitors of Mycobacterium tuberculosis transcriptional repressor EthR using fragment growing, merging, and linking approaches. J. Med. Chem. 2014, 57, 4876–4888. [Google Scholar] [CrossRef]
- Scheich, C.; Puetter, V.; Schade, M. Novel small molecule inhibitors of MDR Mycobacterium tuberculosis by NMR fragment screening of antigen 85C. J. Med. Chem. 2010, 53, 8362–8367. [Google Scholar] [CrossRef]
- Mendes, V.; Blundell, T.L. Targeting tuberculosis using structure-guided fragment-based drug design. Drug Discov. Today 2017, 22, 546–554. [Google Scholar] [CrossRef]
- Dai, R.; Wilson, D.J.; Geders, T.W.; Aldrich, C.C.; Finzel, B.C. Inhibition of Mycobacterium tuberculosis transaminase BioA by aryl hydrazines and hydrazides. Chembiochem A Eur. J. Chem. Biol. 2014, 15, 575–586. [Google Scholar] [CrossRef]
- Gupta, P.; Thomas, S.E.; Zaidan, S.A.; Pasillas, M.A.; Cory-Wright, J.; Sebastian-Perez, V.; Burgess, A.; Cattermole, E.; Meghir, C.; Abell, C.; et al. A fragment-based approach to assess the ligandability of ArgB, ArgC, ArgD and ArgF in the L-arginine biosynthetic pathway of Mycobacterium tuberculosis. Comput. Struct. Biotechnol. J. 2021, 19, 3491–3506. [Google Scholar] [CrossRef]
- Whitehouse, A.J.; Libardo, M.D.J.; Kasbekar, M.; Brear, P.D.; Fischer, G.; Thomas, C.J.; Barry, C.E., 3rd; Boshoff, H.I.M.; Coyne, A.G.; Abell, C. Targeting of Fumarate Hydratase from Mycobacterium tuberculosis Using Allosteric Inhibitors with a Dimeric-Binding Mode. J. Med. Chem. 2019, 62, 10586–10604. [Google Scholar] [CrossRef]
- Kasbekar, M.; Fischer, G.; Mott, B.T.; Yasgar, A.; Hyvonen, M.; Boshoff, H.I.; Abell, C.; Barry, C.E., 3rd; Thomas, C.J. Selective small molecule inhibitor of the Mycobacterium tuberculosis fumarate hydratase reveals an allosteric regulatory site. Proc. Natl. Acad. Sci. USA 2016, 113, 7503–7508. [Google Scholar] [CrossRef]
- Lazzaro, B.P.; Zasloff, M.; Rolff, J. Antimicrobial peptides: Application informed by evolution. Science 2020, 368, eaau5480. [Google Scholar] [CrossRef]
- Wiesner, J.; Vilcinskas, A. Antimicrobial peptides: The ancient arm of the human immune system. Virulence 2010, 1, 440–464. [Google Scholar] [CrossRef]
- Graf, M.; Wilson, D.N. Intracellular Antimicrobial Peptides Targeting the Protein Synthesis Machinery. Adv. Exp. Med. Biol. 2019, 1117, 73–89. [Google Scholar] [CrossRef]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef]
- Wang, K.; Dang, W.; Yan, J.; Chen, R.; Liu, X.; Yan, W.; Zhang, B.; Xie, J.; Zhang, J.; Wang, R. Membrane perturbation action mode and structure-activity relationships of Protonectin, a novel antimicrobial peptide from the venom of the neotropical social wasp Agelaia pallipes pallipes. Antimicrob. Agents Chemother. 2013, 57, 4632–4639. [Google Scholar] [CrossRef]
- Wang, K.; Yan, J.; Chen, R.; Dang, W.; Zhang, B.; Zhang, W.; Song, J.; Wang, R. Membrane-active action mode of polybia-CP, a novel antimicrobial peptide isolated from the venom of Polybia paulista. Antimicrob. Agents Chemother. 2012, 56, 3318–3323. [Google Scholar] [CrossRef]
- Campoccia, D.; Montanaro, L.; Ravaioli, S.; Mariani, V.; Bottau, G.; De Donno, A.; Arciola, C.R. Antibacterial Activity on Orthopedic Clinical Isolates and Cytotoxicity of the Antimicrobial Peptide Dadapin-1. Int. J. Mol. Sci. 2023, 24, 779. [Google Scholar] [CrossRef] [PubMed]
- Boullet, H.; Bentot, F.; Hequet, A.; Ganem-Elbaz, C.; Bechara, C.; Pacreau, E.; Launay, P.; Sagan, S.; Jolivalt, C.; Lacombe, C.; et al. Small AntiMicrobial Peptide With in Vivo Activity Against Sepsis. Molecules 2019, 24, 1702. [Google Scholar] [CrossRef] [PubMed]
- Mangoni, A.A.; Eynde, J.J.V.; Jampilek, J.; Hadjipavlou-Litina, D.; Liu, H.; Reynisson, J.; Sousa, M.E.; Gomes, P.A.C.; Prokai-Tatrai, K.; Tuccinardi, T.; et al. Breakthroughs in Medic inal Chemistry: New Targets and Mechanisms, New Drugs, New Hopes-5. Molecules 2019, 24, 130. [Google Scholar] [CrossRef]
- Matos, G.M.; Garcia-Teodoro, B.; Martins, C.P.; Schmitt, P.; Guzman, F.; de Freitas, A.C.O.; Stoco, P.H.; Ferreira, F.A.; Stadnik, M.J.; Robl, D.; et al. Antimicrobial Spectrum of Activity and Mechanism of Action of Linear Alpha-Helical Peptides Inspired by Shrimp Anti-Lipopolysaccharide Factors. Biomolecules 2023, 13, 150. [Google Scholar] [CrossRef] [PubMed]
- Zharkova, M.S.; Komlev, A.S.; Filatenkova, T.A.; Sukhareva, M.S.; Vladimirova, E.V.; Trulioff, A.S.; Orlov, D.S.; Dmitriev, A.V.; Afinogenova, A.G.; Spiridonova, A.A.; et al. Combined Use of Antimicrobial Peptides with Antiseptics against Multidrug-Resistant Bacteria: Pros and Cons. Pharmaceutics 2023, 15, 291. [Google Scholar] [CrossRef]
- Arakal, B.S.; Whitworth, D.E.; James, P.E.; Rowlands, R.; Madhusoodanan, N.P.T.; Baijoo, M.R.; Livingstone, P.G. In Silico and In Vitro Analyses Reveal Promising Antimicrobial Peptides from Myxobacteria. Probiotics Antimicrob. Proteins 2023, 15, 202–214. [Google Scholar] [CrossRef]
- Chen, Y.C.; Qiu, W.; Zhang, W.; Zhang, J.; Chen, R.; Chen, F.; Wang, K.J. A Novel Antimicrobial Peptide Sp-LECin with Broad-Spectrum Antimicrobial Activity and Anti-Pseudomonas aeruginosa Infection in Zebrafish. Int. J. Mol. Sci. 2022, 24, 267. [Google Scholar] [CrossRef]
- Angane, M.; Swift, S.; Huang, K.; Butts, C.A.; Quek, S.Y. Essential Oils and Their Major Components: An Updated Review on Antimicrobial Activities, Mechanism of Action and Their Potential Application in the Food Industry. Foods 2022, 11, 464. [Google Scholar] [CrossRef]
- Tofah, M.L.; Mseddi, K.; Al-Abbasi, O.K.; Ben Yazid, A.; Khechine, A.; Gdoura, R.; Khannous, L. A New Lavender (Lavandula multifida L.) Ecotype from Arid Tunisia, with Differential Essential Oil Composition and Higher Antimicrobial Potential. Life 2022, 13, 103. [Google Scholar] [CrossRef]
- Su, F.; Yang, G.; Hu, D.; Ruan, C.; Wang, J.; Zhang, Y.; Zhu, Q. Chemical Composition, Antibacterial and Antioxidant Activities of Essential Oil from Centipeda minima. Molecules 2023, 28, 824. [Google Scholar] [CrossRef]
- Alam, A.; Jawaid, T.; Alsanad, S.M.; Kamal, M.; Balaha, M.F. Composition, Antibacterial Efficacy, and Anticancer Activity of Essential Oil Extracted from Psidium guajava (L.) Leaves. Plants 2023, 12, 246. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhao, Y.; Chen, X.; Li, W.; Wang, L.; Li, W.; Du, J.; Zhang, S. Effects of cinnamon essential oil on the physiological metabolism of Salmonella enteritidis. Front. Microbiol. 2022, 13, 1035894. [Google Scholar] [CrossRef]
- Tsai, C.S.; Winans, S.C. LuxR-type quorum-sensing regulators that are detached from common scents. Mol. Microbiol. 2010, 77, 1072–1082. [Google Scholar] [CrossRef]
- Stotani, S.; Gatta, V.; Medda, F.; Padmanaban, M.; Karawajczyk, A.; Tammela, P.; Giordanetto, F.; Tzalis, D.; Collina, S. A Versatile Strategy for the Synthesis of 4,5-Dihydroxy-2,3-Pentanedione (DPD) and Related Compounds as Potential Modulators of Bacterial Quorum Sensing. Molecules 2018, 23, 2545. [Google Scholar] [CrossRef]
- Kiran, M.D.; Adikesavan, N.V.; Cirioni, O.; Giacometti, A.; Silvestri, C.; Scalise, G.; Ghiselli, R.; Saba, V.; Orlando, F.; Shoham, M.; et al. Discovery of a quorum-sensing inhibitor of drug-resistant staphylococcal infections by structure-based virtual screening. Mol. Pharmacol. 2008, 73, 1578–1586. [Google Scholar] [CrossRef]
- Brackman, G.; Breyne, K.; De Rycke, R.; Vermote, A.; Van Nieuwerburgh, F.; Meyer, E.; Van Calenbergh, S.; Coenye, T. The Quorum Sensing Inhibitor Hamamelitannin Increases Antibiotic Susceptibility of Staphylococcus aureus Biofilms by Affecting Peptidoglycan Biosynthesis and eDNA Release. Sci. Rep. 2016, 6, 20321. [Google Scholar] [CrossRef]
- Vermote, A.; Brackman, G.; Risseeuw, M.D.; Vanhoutte, B.; Cos, P.; Van Hecke, K.; Breyne, K.; Meyer, E.; Coenye, T.; Van Calenbergh, S. Hamamelitannin Analogues that Modulate Quorum Sensing as Potentiators of Antibiotics against Staphylococcus aureus. Angew. Chem. 2016, 55, 6551–6555. [Google Scholar] [CrossRef]
- Ragab, A.; Fouad, S.A.; Ammar, Y.A.; Aboul-Magd, D.S.; Abusaif, M.S. Antibiofilm and Anti-Quorum-Sensing Activities of Novel Pyrazole and Pyrazolo[1,5-a]pyrimidine Derivatives as Carbonic Anhydrase I and II Inhibitors: Design, Synthesis, Radiosterilization, and Molecular Docking Studies. Antibiotics 2023, 12, 128. [Google Scholar] [CrossRef]
- Zhang, Y.; Dong, L.; Sun, L.; Hu, X.; Wang, X.; Nie, T.; Li, X.; Wang, P.; Pang, P.; Pang, J.; et al. ML364 exerts the broad-spectrum antivirulence effect by interfering with the bacterial quorum sensing system. Front. Microbiol. 2022, 13, 980217. [Google Scholar] [CrossRef]
- Hurtova, M.; Kanova, K.; Dobiasova, S.; Holasova, K.; Cakova, D.; Hoang, L.; Biedermann, D.; Kuzma, M.; Cvacka, J.; Kren, V.; et al. Selectively Halogenated Flavonolignans-Preparation and Antibacterial Activity. Int. J. Mol. Sci. 2022, 23, 15121. [Google Scholar] [CrossRef]
- Nimma, R.; Kumar, A.; Gani, Z.; Gahlawat, A.; Dilawari, R.; Rohilla, R.K.; Kumbhar, H.; Garg, P.; Chopra, S.; Raje, M.; et al. Characterization of the enzymatic and multifunctional properties of Acinetobacter baumannii erythrose-4-phosphate dehydrogenase (E4PDH). Microb. Pathog. 2023, 175, 105992. [Google Scholar] [CrossRef] [PubMed]
- Barra, A.L.C.; Dantas, L.O.C.; Morao, L.G.; Gutierrez, R.F.; Polikarpov, I.; Wrenger, C.; Nascimento, A.S. Essential Metabolic Routes as a Way to ESKAPE From Antibiotic Resistance. Front. Public Health 2020, 8, 26. [Google Scholar] [CrossRef] [PubMed]
- Kuranova, N.N.; Yarullin, D.N.; Zavalishin, M.N.; Gamov, G.A. Complexation of Gold(III) with Pyridoxal 5’-Phosphate-Derived Hydrazones in Aqueous Solution. Molecules 2022, 27, 7346. [Google Scholar] [CrossRef] [PubMed]
- Jevtovic, V.; Alshammari, N.; Latif, S.; Alsukaibi, A.K.D.; Humaidi, J.; Alanazi, T.Y.A.; Abdulaziz, F.; Matalka, S.I.; Pantelic, N.D.; Markovic, M.; et al. Synthesis, Crystal Structure, Theoretical Calculations, Antibacterial Activity, Electrochemical Behavior, and Molecular Docking of Ni(II) and Cu(II) Complexes with Pyridoxal-Semicarbazone. Molecules 2022, 27, 6322. [Google Scholar] [CrossRef] [PubMed]
- Scaccaglia, M.; Rega, M.; Vescovi, M.; Pinelli, S.; Tegoni, M.; Bacci, C.; Pelosi, G.; Bisceglie, F. Gallium(III)-pyridoxal thiosemicarbazone derivatives as nontoxic agents against Gram-negative bacteria. Met. Integr. Biometal Sci. 2022, 14, mfac070. [Google Scholar] [CrossRef]
- Hobbs, Z.; Abedon, S.T. Diversity of phage infection types and associated terminology: The problem with ‘Lytic or lysogenic’. Fems Microbiol. Lett. 2016, 363, fnw047. [Google Scholar] [CrossRef]
- Viertel, T.M.; Ritter, K.; Horz, H.P. Viruses versus bacteria-novel approaches to phage therapy as a tool against multidrug-resistant pathogens. J. Antimicrob. Chemother. 2014, 69, 2326–2336. [Google Scholar] [CrossRef]
- Alexyuk, P.; Bogoyavlenskiy, A.; Alexyuk, M.; Akanova, K.; Moldakhanov, Y.; Berezin, V. Isolation and Characterization of Lytic Bacteriophages Active against Clinical Strains of E. coli and Development of a Phage Antimicrobial Cocktail. Viruses 2022, 14, 2381. [Google Scholar] [CrossRef]
- Egido, J.E.; Toner-Bartelds, C.; Costa, A.R.; Brouns, S.J.J.; Rooijakkers, S.H.M.; Bardoel, B.W.; Haas, P.J. Monitoring phage-induced lysis of gram-negatives in real time using a fluorescent DNA dye. Sci. Rep. 2023, 13, 856. [Google Scholar] [CrossRef]
- Berdy, J. Bioactive microbial metabolites. J. Antibiot. 2005, 58, 1–26. [Google Scholar] [CrossRef]
- Pantel, L.; Florin, T.; Dobosz-Bartoszek, M.; Racine, E.; Sarciaux, M.; Serri, M.; Houard, J.; Campagne, J.M.; de Figueiredo, R.M.; Midrier, C.; et al. Odilorhabdins, Antibacterial Agents that Cause Miscoding by Binding at a New Ribosomal Site. Mol. Cell 2018, 70, 83–94.e87. [Google Scholar] [CrossRef]
- Mangoni, A.A.; Guillou, C.; Vanden Eynde, J.J.; Hulme, C.; Jampilek, J.; Li, W.; Prokai-Tatrai, K.; Rautio, J.; Collina, S.; Tuccinardi, T.; et al. Breakthroughs in Medicinal Chemistry: New Targets and Mechanisms, New Drugs, New Hopes(-)4. Molecules 2018, 24, 130. [Google Scholar] [CrossRef]
- Racine, E.; Nordmann, P.; Pantel, L.; Sarciaux, M.; Serri, M.; Houard, J.; Villain-Guillot, P.; Demords, A.; Vingsbo Lundberg, C.; Gualtieri, M. In Vitro and In Vivo Characterization of NOSO-502, a Novel Inhibitor of Bacterial Translation. Antimicrob. Agents Chemother. 2018, 62, e01016-18. [Google Scholar] [CrossRef]
- Pantel, L.; Guerin, F.; Serri, M.; Gravey, F.; Houard, J.; Maurent, K.; Attwood, M.; Noel, A.; MacGowan, A.; Racine, E.; et al. Exploring Cluster-Dependent Antibacterial Activities and Resistance Pathways of NOSO-502 and Colistin against Enterobacter cloacae Complex Species. Antimicrob. Agents Chemother. 2022, 66, e0077622. [Google Scholar] [CrossRef]
- Pastorino, G.; Cornara, L.; Soares, S.; Rodrigues, F.; Oliveira, M. Liquorice (Glycyrrhiza glabra): A phytochemical and pharmacological review. Phytother. Res. Ptr 2018, 32, 2323–2339. [Google Scholar] [CrossRef]
- Zhao, Y.; Su, X. Antibacterial activity of 18beta-glycyrrhetinic acid against Neisseria gonorrhoeae in vitro. Biochem. Biophys. Rep. 2023, 33, 101427. [Google Scholar] [CrossRef]
- Vanden Eynde, J.J.; Mangoni, A.A.; Rautio, J.; Leprince, J.; Azuma, Y.T.; Garcia-Sosa, A.T.; Hulme, C.; Jampilek, J.; Karaman, R.; Li, W.; et al. Breakthroughs in Medicinal Chemistry: New Targets and Mechanisms, New Drugs, New Hopes-6. Molecules 2019, 25, 119. [Google Scholar] [CrossRef]
- Imai, Y.; Meyer, K.J.; Iinishi, A.; Favre-Godal, Q.; Green, R.; Manuse, S.; Caboni, M.; Mori, M.; Niles, S.; Ghiglieri, M.; et al. A new antibiotic selectively kills Gram-negative pathogens. Nature 2019, 576, 459–464. [Google Scholar] [CrossRef]
- Wuisan, Z.G.; Kresna, I.D.M.; Bohringer, N.; Lewis, K.; Schaberle, T.F. Optimization of heterologous Darobactin A expression and identification of the minimal biosynthetic gene cluster. Metab. Eng. 2021, 66, 123–136. [Google Scholar] [CrossRef]
- Bohringer, N.; Green, R.; Liu, Y.; Mettal, U.; Marner, M.; Modaresi, S.M.; Jakob, R.P.; Wuisan, Z.G.; Maier, T.; Iinishi, A.; et al. Mutasynthetic Production and Antimicrobial Characterization of Darobactin Analogs. Microbiol. Spectr. 2021, 9, e0153521. [Google Scholar] [CrossRef]
- Seyfert, C.E.; Porten, C.; Yuan, B.; Deckarm, S.; Panter, F.; Bader, C.D.; Coetzee, J.; Deschner, F.; Tehrani, K.; Higgins, P.G.; et al. Darobactins Exhibiting Superior Antibiotic Activity by Cryo-EM Structure Guided Biosynthetic Engineering. Angew. Chem. 2023, 62, e202214094. [Google Scholar] [CrossRef] [PubMed]
- Luther, A.; Urfer, M.; Zahn, M.; Muller, M.; Wang, S.Y.; Mondal, M.; Vitale, A.; Hartmann, J.B.; Sharpe, T.; Monte, F.L.; et al. Chimeric peptidomimetic antibiotics against Gram-negative bacteria. Nature 2019, 576, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Hart, E.M.; Mitchell, A.M.; Konovalova, A.; Grabowicz, M.; Sheng, J.; Han, X.; Rodriguez-Rivera, F.P.; Schwaid, A.G.; Malinverni, J.C.; Balibar, C.J.; et al. A small-molecule inhibitor of BamA impervious to efflux and the outer membrane permeability barrier. Proc. Natl. Acad. Sci. USA 2019, 116, 21748–21757. [Google Scholar] [CrossRef]
- Peterson, J.H.; Doyle, M.T.; Bernstein, H.D. Small Molecule Antibiotics Inhibit Distinct Stages of Bacterial Outer Membrane Protein Assembly. mBio 2022, 13, e0228622. [Google Scholar] [CrossRef] [PubMed]
- Breijyeh, Z.; Jubeh, B.; Bufo, S.A.; Karaman, R.; Scrano, L. Cannabis: A Toxin-Producing Plant with Potential Therapeutic Uses. Toxins 2021, 13, 117. [Google Scholar] [CrossRef]
- Blaskovich, M.A.T.; Kavanagh, A.M.; Elliott, A.G.; Zhang, B.; Ramu, S.; Amado, M.; Lowe, G.J.; Hinton, A.O.; Pham, D.M.T.; Zuegg, J.; et al. The antimicrobial potential of cannabidiol. Commun. Biol. 2021, 4, 7. [Google Scholar] [CrossRef]
- Kosgodage, U.S.; Matewele, P.; Awamaria, B.; Kraev, I.; Warde, P.; Mastroianni, G.; Nunn, A.V.; Guy, G.W.; Bell, J.D.; Inal, J.M.; et al. Cannabidiol Is a Novel Modulator of Bacterial Membrane Vesicles. Front. Cell. Infect. Microbiol. 2019, 9, 324. [Google Scholar] [CrossRef]
- Luz-Veiga, M.; Amorim, M.; Pinto-Ribeiro, I.; Oliveira, A.L.S.; Silva, S.; Pimentel, L.L.; Rodriguez-Alcala, L.M.; Madureira, R.; Pintado, M.; Azevedo-Silva, J.; et al. Cannabidiol and Cannabigerol Exert Antimicrobial Activity without Compromising Skin Microbiota. Int. J. Mol. Sci. 2023, 24, 2389. [Google Scholar] [CrossRef]
- Wu, Q.; Guo, M.; Zou, L.; Wang, Q.; Xia, Y. 8,9-Dihydrocannabidiol, an Alternative of Cannabidiol, Its Preparation, Antibacterial and Antioxidant Ability. Molecules 2023, 28, 445. [Google Scholar] [CrossRef]
- Gildea, L.; Ayariga, J.A.; Xu, J.; Villafane, R.; Robertson, B.K.; Samuel-Foo, M.; Ajayi, O.S. Cannabis sativa CBD Extract Exhibits Synergy with Broad-Spectrum Antibiotics against Salmonella enterica subsp. Enterica serovar typhimurium. Microorganisms 2022, 10, 2360. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, Y.R.; He, Y.H.; Ding, Y.Y.; An, J.X.; Zhang, Z.J.; Zhao, W.B.; Hu, Y.M.; Liu, Y.Q. Drug repurposing strategy part 1: From approved drugs to agri-bactericides leads. J. Antibiot. 2023, 76, 27–51. [Google Scholar] [CrossRef]
- Tobinick, E.L. The value of drug repositioning in the current pharmaceutical market. Drug News Perspect. 2009, 22, 119–125. [Google Scholar] [CrossRef]
- Boyd, N.K.; Teng, C.; Frei, C.R. Brief Overview of Approaches and Challenges in New Antibiotic Development: A Focus On Drug Repurposing. Front. Cell. Infect. Microbiol. 2021, 11, 684515. [Google Scholar] [CrossRef]
- Roundtable on Translating Genomic-Based Research for Health; Board on Health Sciences Policy; Institute of Medicine. Drug Repurposing and Repositioning: Workshop Summary; National Academies Press: Washington, DC, USA, 2014. [Google Scholar]
- Konreddy, A.K.; Rani, G.U.; Lee, K.; Choi, Y. Recent Drug-Repurposing-Driven Advances in the Discovery of Novel Antibiotics. Curr. Med. Chem. 2019, 26, 5363–5388. [Google Scholar] [CrossRef]
- Springthorpe, B.; Bailey, A.; Barton, P.; Birkinshaw, T.N.; Bonnert, R.V.; Brown, R.C.; Chapman, D.; Dixon, J.; Guile, S.D.; Humphries, R.G.; et al. From ATP to AZD6140: The discovery of an orally active reversible P2Y12 receptor antagonist for the prevention of thrombosis. Bioorg Med. Chem. Lett. 2007, 17, 6013–6018. [Google Scholar] [CrossRef]
- Wallentin, L.; Becker, R.C.; Budaj, A.; Cannon, C.P.; Emanuelsson, H.; Held, C.; Horrow, J.; Husted, S.; James, S.; Katus, H.; et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N. Engl. J. Med. 2009, 361, 1045–1057. [Google Scholar] [CrossRef]
- Sexton, T.R.; Zhang, G.; Macaulay, T.E.; Callahan, L.A.; Charnigo, R.; Vsevolozhskaya, O.A.; Li, Z.; Smyth, S. Ticagrelor Reduces Thromboinflammatory Markers in Patients With Pneumonia. Jacc. Basic Transl. Sci. 2018, 3, 435–449. [Google Scholar] [CrossRef]
- Lancellotti, P.; Musumeci, L.; Jacques, N.; Servais, L.; Goffin, E.; Pirotte, B.; Oury, C. Antibacterial Activity of Ticagrelor in Conventional Antiplatelet Dosages Against Antibiotic-Resistant Gram-Positive Bacteria. Jama Cardiol. 2019, 4, 596–599. [Google Scholar] [CrossRef]
- Teng, R.; Oliver, S.; Hayes, M.A.; Butler, K. Absorption, distribution, metabolism, and excretion of ticagrelor in healthy subjects. Drug Metab. Dispos. Biol. Fate Chem. 2010, 38, 1514–1521. [Google Scholar] [CrossRef]
- Pant, N.; Miranda-Hernandez, S.; Rush, C.; Warner, J.; Eisen, D.P. Non-Antimicrobial Adjuvant Therapy Using Ticagrelor Reduced Biofilm-Related Staphylococcus aureus Prosthetic Joint Infection. Front. Pharmacol. 2022, 13, 927783. [Google Scholar] [CrossRef]
- Phanchana, M.; Phetruen, T.; Harnvoravongchai, P.; Raksat, P.; Ounjai, P.; Chankhamhaengdecha, S.; Janvilisri, T. Repurposing a platelet aggregation inhibitor ticagrelor as an antimicrobial against Clostridioides difficile. Sci. Rep. 2020, 10, 6497. [Google Scholar] [CrossRef] [PubMed]
- Bradner, W.T. Mitomycin C: A clinical update. Cancer Treat. Rev. 2001, 27, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Kwan, B.W.; Chowdhury, N.; Wood, T.K. Combatting bacterial infections by killing persister cells with mitomycin C. Environ. Microbiol. 2015, 17, 4406–4414. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Muniz, M.Y.; Lopez-Jacome, L.E.; Hernandez-Duran, M.; Franco-Cendejas, R.; Licona-Limon, P.; Ramos-Balderas, J.L.; Martinez-Vazquez, M.; Belmont-Diaz, J.A.; Wood, T.K.; Garcia-Contreras, R. Repurposing the anticancer drug mitomycin C for the treatment of persistent Acinetobacter baumannii infections. Int. J. Antimicrob. Agents 2017, 49, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Pacios, O.; Fernandez-Garcia, L.; Bleriot, I.; Blasco, L.; Gonzalez-Bardanca, M.; Lopez, M.; Fernandez-Cuenca, F.; Oteo, J.; Pascual, A.; Martinez-Martinez, L.; et al. Enhanced Antibacterial Activity of Repurposed Mitomycin C and Imipenem in Combination with the Lytic Phage vB_KpnM-VAC13 against Clinical Isolates of Klebsiella pneumoniae. Antimicrob. Agents Chemother. 2021, 65, e0090021. [Google Scholar] [CrossRef]
- Roder, C.; Thomson, M.J. Auranofin: Repurposing an old drug for a golden new age. Drugs RD 2015, 15, 13–20. [Google Scholar] [CrossRef]
- She, P.; Zhou, L.; Li, S.; Liu, Y.; Xu, L.; Chen, L.; Luo, Z.; Wu, Y. Synergistic Microbicidal Effect of Auranofin and Antibiotics Against Planktonic and Biofilm-Encased S. aureus and E. faecalis. Front. Microbiol. 2019, 10, 2453. [Google Scholar] [CrossRef]
- Kim, H.R.; Eom, Y.B. Auranofin promotes antibacterial effect of doripenem against carbapenem-resistant Acinetobacter baumannii. J. Appl. Microbiol. 2022, 133, 1422–1433. [Google Scholar] [CrossRef]
- Chen, H.; Yang, N.; Yu, L.; Li, J.; Zhang, H.; Zheng, Y.; Xu, M.; Liu, Y.; Yang, Y.; Li, J. Synergistic Microbicidal Effect of AUR and PEITC Against Staphylococcus aureus Skin Infection. Front. Cell. Infect. Microbiol. 2022, 12, 927289. [Google Scholar] [CrossRef]
- Hutton, M.L.; Pehlivanoglu, H.; Vidor, C.J.; James, M.L.; Thomson, M.J.; Lyras, D. Repurposing auranofin as a Clostridioides difficile therapeutic. J. Antimicrob. Chemother. 2020, 75, 409–417. [Google Scholar] [CrossRef]
- Huang, T.L.; Mayence, A.; Vanden Eynde, J.J. Some non-conventional biomolecular targets for diamidines. A short survey. Bioorganic Med. Chem. 2014, 22, 1983–1992. [Google Scholar] [CrossRef]
- Stokes, J.M.; MacNair, C.R.; Ilyas, B.; French, S.; Cote, J.P.; Bouwman, C.; Farha, M.A.; Sieron, A.O.; Whitfield, C.; Coombes, B.K.; et al. Pentamidine sensitizes Gram-negative pathogens to antibiotics and overcomes acquired colistin resistance. Nat. Microbiol. 2017, 2, 17028. [Google Scholar] [CrossRef]
- Munoz-Torrero, D.; Mangoni, A.A.; Guillou, C.; Collina, S.; Vanden Eynde, J.J.; Rautio, J.; Keseru, G.M.; Hulme, C.; Chibale, K.; Luque, F.J.; et al. Breakthroughs in Medicinal Chemistry: New Targets and Mechanisms, New Drugs, New Hopes. Molecules 2017, 22, 743. [Google Scholar] [CrossRef]
- Yu, Y.; Zhao, H.; Lin, J.; Li, Z.; Tian, G.; Yang, Y.Y.; Yuan, P.; Ding, X. Repurposing Non-Antibiotic Drugs Auranofin and Pentamidine in Combination to Combat Multidrug-Resistant Gram-Negative Bacteria. Int. J. Antimicrob. Agents 2022, 59, 106582. [Google Scholar] [CrossRef]
- Mitsuya, H.; Weinhold, K.J.; Furman, P.A.; St Clair, M.H.; Lehrman, S.N.; Gallo, R.C.; Bolognesi, D.; Barry, D.W.; Broder, S. 3’-Azido-3’-deoxythymidine (BW A509U): An antiviral agent that inhibits the infectivity and cytopathic effect of human T-lymphotropic virus type III/lymphadenopathy-associated virus in vitro. Proc. Natl. Acad. Sci. USA 1985, 82, 7096–7100. [Google Scholar] [CrossRef]
- Furman, P.A.; Fyfe, J.A.; St Clair, M.H.; Weinhold, K.; Rideout, J.L.; Freeman, G.A.; Lehrman, S.N.; Bolognesi, D.P.; Broder, S.; Mitsuya, H. Phosphorylation of 3’-azido-3’-deoxythymidine and selective interaction of the 5'-triphosphate with human immunodeficiency virus reverse transcriptase. Proc. Natl. Acad. Sci. USA 1986, 83, 8333–8337. [Google Scholar] [CrossRef]
- Doleans-Jordheim, A.; Bergeron, E.; Bereyziat, F.; Ben-Larbi, S.; Dumitrescu, O.; Mazoyer, M.A.; Morfin, F.; Dumontet, C.; Freney, J.; Jordheim, L.P. Zidovudine (AZT) has a bactericidal effect on enterobacteria and induces genetic modifications in resistant strains. Eur. J. Clin. Microbiol. Infect. Dis. Off. Publ. Eur. Soc. Clin. Microbiol. 2011, 30, 1249–1256. [Google Scholar] [CrossRef]
- Beach, J.W. Chemotherapeutic agents for human immunodeficiency virus infection: Mechanism of action, pharmacokinetics, metabolism, and adverse reactions. Clin. Ther. 1998, 20, 2–25. [Google Scholar] [CrossRef]
- Elwell, L.P.; Ferone, R.; Freeman, G.A.; Fyfe, J.A.; Hill, J.A.; Ray, P.H.; Richards, C.A.; Singer, S.C.; Knick, V.B.; Rideout, J.L.; et al. Antibacterial activity and mechanism of action of 3’-azido-3’-deoxythymidine (BW A509U). Antimicrob. Agents Chemother. 1987, 31, 274–280. [Google Scholar] [CrossRef]
- Lewin, C.S.; Amyes, S.G. Conditions required for the antibacterial activity of zidovudine. Eur. J. Clin. Microbiol. Infect. Dis. Off. Publ. Eur. Soc. Clin. Microbiol. 1989, 8, 737–741. [Google Scholar] [CrossRef]
- Ng, S.M.S.; Sioson, J.S.P.; Yap, J.M.; Ng, F.M.; Ching, H.S.V.; Teo, J.W.P.; Jureen, R.; Hill, J.; Chia, C.S.B. Repurposing Zidovudine in combination with Tigecycline for treating carbapenem-resistant Enterobacteriaceae infections. Eur. J. Clin. Microbiol. Infect. Dis. Off. Publ. Eur. Soc. Clin. Microbiol. 2018, 37, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Mazur, M.; Maslowiec, D. Antimicrobial Activity of Lactones. Antibiotics 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Mazur, M.; Gladkowski, W.; Podkowik, M.; Bania, J.; Nawrot, J.; Bialonska, A.; Wawrzenczyk, C. Lactones 43. New biologically active lactones: Beta-cyclocitral derivatives. Pest Manag. Sci. 2014, 70, 286–294. [Google Scholar] [CrossRef]
- Mazur, M.; Gladkowski, W.; Srcek, V.G.; Radosevic, K.; Maciejewska, G.; Wawrzenczyk, C. Regio- and enantioselective microbial hydroxylation and evaluation of cytotoxic activity of beta-cyclocitral-derived halolactones. PLoS ONE 2017, 12, e0183429. [Google Scholar] [CrossRef] [PubMed]
- Mazur, M.; Skrobiszewski, A.; Gladkowski, W.; Podkowik, M.; Bania, J.; Nawrot, J.; Klejdysz, T.; Wawrzenczyk, C. Lactones 46. Synthesis, antifeedant and antibacterial activity of gamma-lactones with a p-methoxyphenyl substituent. Pest Manag. Sci. 2016, 72, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Gladkowski, W.; Siepka, M.; Janeczko, T.; Kostrzewa-Suslow, E.; Poplonski, J.; Mazur, M.; Zarowska, B.; Laba, W.; Maciejewska, G.; Wawrzenczyk, C. Synthesis and Antimicrobial Activity of Methoxy- Substituted gamma-Oxa-epsilon-lactones Derived from Flavanones. Molecules 2019, 24, 4151. [Google Scholar] [CrossRef]
- Flamm, R.K.; Rhomberg, P.R.; Sader, H.S. In Vitro Activity of the Novel Lactone Ketolide Nafithromycin (WCK 4873) against Contemporary Clinical Bacteria from a Global Surveillance Program. Antimicrob. Agents Chemother. 2017, 61, e01230-17. [Google Scholar] [CrossRef]
- Iwanowski, P.; Bhatia, A.; Gupta, M.; Patel, A.; Chavan, R.; Yeole, R.; Friedland, D. Safety, tolerability and pharmacokinetics of oral nafithromycin (WCK4873) after single or multiple doses and effects of food on single-dose bioavailability in healthy adult subjects. Antimicrob. Agents Chemother. 2019, 63, e01253-19. [Google Scholar] [CrossRef]
- Dupont, C.; Chen, Y.; Xu, Z.; Roquet-Baneres, F.; Blaise, M.; Witt, A.K.; Dubar, F.; Biot, C.; Guerardel, Y.; Maurer, F.P.; et al. A piperidinol-containing molecule is active against Mycobacterium tuberculosis by inhibiting the mycolic acid flippase activity of MmpL3. J. Biol. Chem. 2019, 294, 17512–17523. [Google Scholar] [CrossRef]
- de Ruyck, J.; Dupont, C.; Lamy, E.; Le Moigne, V.; Biot, C.; Guerardel, Y.; Herrmann, J.L.; Blaise, M.; Grassin-Delyle, S.; Kremer, L.; et al. Structure-Based Design and Synthesis of Piperidinol-Containing Molecules as New Mycobacterium abscessus Inhibitors. ChemistryOpen 2020, 9, 351–365. [Google Scholar] [CrossRef]
- Islam, M.; Arifuzzaman, A.; Rahman, M.; Rahman, M.A.; Kawsar, S.M.A.K. Novel methyl 4,6-O-benzylidene-α-D-glucopyranoside derivatives: Synthesis, structural characterization and evaluation of antibacterial activities. Hacettepe J. Biol. Chem. 2019, 47, 153–164. [Google Scholar] [CrossRef]
- Kawsar, S.M.; Faruk, M.O.; Rahman, M.S.; Fujii, Y.; Ozeki, Y. Regioselective synthesis, characterization, and antimicrobial activities of some new monosaccharide derivatives. Sci. Pharm. 2014, 82, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Ahmmed, F.; Islam, A.U.; Mukhrish, Y.E.; Bakri, Y.E.; Ahmad, S.; Ozeki, Y.; Kawsar, S.M.A. Efficient Antibacterial/Antifungal Activities: Synthesis, Molecular Docking, Molecular Dynamics, Pharmacokinetic, and Binding Free Energy of Galactopyranoside Derivatives. Molecules 2022, 28, 219. [Google Scholar] [CrossRef]
- Dias, F.R.F.; Novais, J.S.; Devillart, T.; da Silva, W.A.; Ferreira, M.O.; Loureiro, R.S.; Campos, V.R.; Ferreira, V.F.; de Souza, M.; Castro, H.C.; et al. Synthesis and antimicrobial evaluation of amino sugar-based naphthoquinones and isoquinoline-5,8-diones and their halogenated compounds. Eur. J. Med. Chem. 2018, 156, 1–12. [Google Scholar] [CrossRef]
- Dias, C.; Pais, J.P.; Nunes, R.; Blazquez-Sanchez, M.T.; Marques, J.T.; Almeida, A.F.; Serra, P.; Xavier, N.M.; Vila-Vicosa, D.; Machuqueiro, M.; et al. Sugar-based bactericides targeting phosphatidylethanolamine-enriched membranes. Nat. Commun. 2018, 9, 4857. [Google Scholar] [CrossRef]
- Puerto, A.S.; Fernandez, J.G.; del Castillo Jde, D.; Pino, M.J.; Angulo, G.P. In vitro activity of beta-lactam and non-beta-lactam antibiotics in extended-spectrum beta-lactamase-producing clinical isolates of Escherichia coli. Diagn. Microbiol. Infect. Dis. 2006, 54, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Ahmad, A.; Varshney, H.; Rauf, A.; Rehan, M.; Subbarao, N.; Khan, A.U. Designing and synthesis of novel antimicrobial heterocyclic analogs of fatty acids. Eur. J. Med. Chem. 2013, 70, 887–900. [Google Scholar] [CrossRef]
- Aarjane, M.; Slassi, S.; Tazi, B.; Amine, A. Synthesis and biological evaluation of novel isoxazole derivatives from acridone. Arch. Der Pharm. 2020, 354, e2000261. [Google Scholar] [CrossRef]
- Tipparaju, S.K.; Joyasawal, S.; Pieroni, M.; Kaiser, M.; Brun, R.; Kozikowski, A.P. In pursuit of natural product leads: Synthesis and biological evaluation of 2-[3-hydroxy-2-[(3-hydroxypyridine-2-carbonyl)amino]phenyl]benzoxazole-4-carboxylic acid (A-33853) and its analogues: Discovery of N-(2-benzoxazol-2-ylphenyl)benzamides as novel antileishmanial chemotypes. J. Med. Chem. 2008, 51, 7344–7347. [Google Scholar] [CrossRef]
- Bashir, M.; Bano, A.; Ijaz, A.S.; Chaudhary, B.A. Recent Developments and Biological Activities of N-Substituted Carbazole Derivatives: A Review. Molecules 2015, 20, 13496–13517. [Google Scholar] [CrossRef]
- Markad, S.B.; Argade, N.P. Diversity oriented convergent access for collective total synthesis of bioactive multifunctional carbazole alkaloids: Synthesis of carbazomycin A, carbazomycin B, hyellazole, chlorohyellazole, and clausenaline D. Org. Lett. 2014, 16, 5470–5473. [Google Scholar] [CrossRef]
- Addla, D.; Wen, S.-Q.; Gao, W.-W.; Maddili, S.K.; Zhang, L.; Zhou, C.-H. Design, synthesis, and biological evaluation of novel carbazole aminothiazoles as potential DNA-targeting antimicrobial agents. MedChemComm 2016, 7, 1988–1994. [Google Scholar] [CrossRef]
- Zhang, Y.; Tangadanchu, V.K.R.; Cheng, Y.; Yang, R.G.; Lin, J.M.; Zhou, C.H. Potential Antimicrobial Isopropanol-Conjugated Carbazole Azoles as Dual Targeting Inhibitors of Enterococcus faecalis. Acs Med. Chem. Lett. 2018, 9, 244–249. [Google Scholar] [CrossRef]
- Zhang, F.F.; Gan, L.L.; Zhou, C.H. Synthesis, antibacterial and antifungal activities of some carbazole derivatives. Bioorg. Med. Chem. Lett. 2010, 20, 1881–1884. [Google Scholar] [CrossRef]
- Xue, Y.J.; Li, M.Y.; Jin, X.J.; Zheng, C.J.; Piao, H.R. Design, synthesis and evaluation of carbazole derivatives as potential antimicrobial agents. J. Enzym. Inhib. Med. Chem. 2021, 36, 295–306. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, Y.; Wang, S.; Wu, H. Synthesis and Biological Evaluation of Novel Pyrimidine Amine Derivatives Bearing Bicyclic Monoterpene Moieties. Molecules 2022, 27, 8104. [Google Scholar] [CrossRef]
- Li, C.; Liu, Y.; Ren, X.; Tan, Y.; Jin, L.; Zhou, X. Design, Synthesis and Bioactivity of Novel Pyrimidine Sulfonate Esters Containing Thioether Moiety. Int. J. Mol. Sci. 2023, 24, 4691. [Google Scholar] [CrossRef]
- Nakahara, Y.; Toda, T.; Matsunami, A.; Kayaki, Y.; Kuwata, S. Protic NNN and NCN Pincer-Type Ruthenium Complexes Featuring (Trifluoromethyl)pyrazole Arms: Synthesis and Application to Catalytic Hydrogen Evolution from Formic Acid. Chem. Asian J. 2018, 13, 73–80. [Google Scholar] [CrossRef]
- Karrouchi, K.; Radi, S.; Ramli, Y.; Taoufik, J.; Mabkhot, Y.N.; Al-Aizari, F.A.; Ansar, M. Synthesis and Pharmacological Activities of Pyrazole Derivatives: A Review. Molecules 2018, 23, 134. [Google Scholar] [CrossRef]
- Sayed, G.H.; Azab, M.E.; Anwer, K.E.; Raouf, M.A.; Negm, N.A. Pyrazole, pyrazolone and enaminonitrile pyrazole derivatives: Synthesis, characterization and potential in corrosion inhibition and antimicrobial applications. J. Mol. Liq. 2018, 252, 329–338. [Google Scholar] [CrossRef]
- Chkirate, K.; Fettach, S.; Karrouchi, K.; Sebbar, N.K.; Essassi, E.M.; Mague, J.T.; Radi, S.; El Abbes Faouzi, M.; Adarsh, N.N.; Garcia, Y. Novel Co(II) and Cu(II) coordination complexes constructed from pyrazole-acetamide: Effect of hydrogen bonding on the self assembly process and antioxidant activity. J. Inorg. Biochem. 2019, 191, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Kulmaczewski, R.; Bamiduro, F.; Shahid, N.; Cespedes, O.; Halcrow, M.A. Structural Transformations and Spin-Crossover in [FeL(2) ](2+) Salts (L=4-tert-Butylsulfanyl-2,6-dipyrazol-1-ylpyridine): The Influence of Bulky Ligand Substituents. Chemistry 2021, 27, 2082–2092. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.J.; Lutz, S.; O’Kane, C.; Zeller, M.; Chen, C.H.; Al Assil, T.; Lee, W.T. Synthesis and characterization of an iron complex bearing a hemilabile NNN-pincer for catalytic hydrosilylation of organic carbonyl compounds. Dalton Trans. 2018, 47, 3243–3247. [Google Scholar] [CrossRef] [PubMed]
- Draoui, Y.; Radi, S.; Tanan, A.; Oulmidi, A.; Miras, H.N.; Benabbes, R.; Ouahhoudo, S.; Mamri, S.; Rotaru, A.; Garcia, Y. Novel family of bis-pyrazole coordination complexes as potent antibacterial and antifungal agents. Rsc Adv. 2022, 12, 17755–17764. [Google Scholar] [CrossRef]
- Abula, A.; Xu, Z.; Zhu, Z.; Peng, C.; Chen, Z.; Zhu, W.; Aisa, H.A. Substitution Effect of the Trifluoromethyl Group on the Bioactivity in Medicinal Chemistry: Statistical Analysis and Energy Calculations. J. Chem. Inf. Modeling 2020, 60, 6242–6250. [Google Scholar] [CrossRef]
- Alkhaibari, I.; Kc, H.R.; Angappulige, D.H.; Gilmore, D.; Alam, M.A. Novel pyrazoles as potent growth inhibitors of staphylococci, enterococci and Acinetobacter baumannii bacteria. Future Med. Chem. 2022, 14, 233–244. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Karaman, R. Enzyme Models-From Catalysis to Prodrugs. Molecules 2021, 26, 3248. [Google Scholar] [CrossRef]
- Jubeh, B.; Breijyeh, Z.; Karaman, R. Antibacterial Prodrugs to Overcome Bacterial Resistance. Molecules 2020, 25, 1543. [Google Scholar] [CrossRef]
- Mislin, G.L.; Schalk, I.J. Siderophore-dependent iron uptake systems as gates for antibiotic Trojan horse strategies against Pseudomonas aeruginosa. Met. Integr. Biometal Sci. 2014, 6, 408–420. [Google Scholar] [CrossRef]
- Wu, J.Y.; Srinivas, P.; Pogue, J.M. Cefiderocol: A Novel Agent for the Management of Multidrug-Resistant Gram-Negative Organisms. Infect. Dis. Ther. 2020, 9, 17–40. [Google Scholar] [CrossRef]
- Daoud, L.; Al-Marzooq, F.; Moubareck, C.A.; Ghazawi, A.; Collyns, T. Elucidating the effect of iron acquisition systems in Klebsiella pneumoniae on susceptibility to the novel siderophore-cephalosporin cefiderocol. PLoS ONE 2022, 17, e0277946. [Google Scholar] [CrossRef]
- Sato, T.; Yamawaki, K. Cefiderocol: Discovery, Chemistry, and In Vivo Profiles of a Novel Siderophore Cephalosporin. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2019, 69, S538–S543. [Google Scholar] [CrossRef]
- Hennigar, S.R.; McClung, J.P. Nutritional Immunity: Starving Pathogens of Trace Minerals. Am. J. Lifestyle Med. 2016, 10, 170–173. [Google Scholar] [CrossRef]
- Zheng, T.; Nolan, E.M. Enterobactin-mediated delivery of beta-lactam antibiotics enhances antibacterial activity against pathogenic Escherichia coli. J. Am. Chem. Soc. 2014, 136, 9677–9691. [Google Scholar] [CrossRef]
- Neumann, W.; Sassone-Corsi, M.; Raffatellu, M.; Nolan, E.M. Esterase-Catalyzed Siderophore Hydrolysis Activates an Enterobactin-Ciprofloxacin Conjugate and Confers Targeted Antibacterial Activity. J. Am. Chem. Soc. 2018, 140, 5193–5201. [Google Scholar] [CrossRef]
- Liu, R.; Miller, P.A.; Vakulenko, S.B.; Stewart, N.K.; Boggess, W.C.; Miller, M.J. A Synthetic Dual Drug Sideromycin Induces Gram-Negative Bacteria To Commit Suicide with a Gram-Positive Antibiotic. J. Med. Chem. 2018, 61, 3845–3854. [Google Scholar] [CrossRef]
- Loupias, P.; Dechamps-Olivier, I.; Dupont, L.; Vanlemmens, P.; Mullie, C.; Taudon, N.; Bouchut, A.; Dassonville-Klimpt, A.; Sonnet, P. Study of Iron Piperazine-Based Chelators as Potential Siderophore Mimetics. Pharmaceuticals 2019, 12, 160. [Google Scholar] [CrossRef]
- Southwell, J.W.; Herman, R.; Raines, D.J.; Clarke, J.E.; Boswald, I.; Dreher, T.; Gutenthaler, S.M.; Schubert, N.; Seefeldt, J.; Metzler-Nolte, N.; et al. Siderophore-Linked Ruthenium Catalysts for Targeted Allyl Ester Prodrug Activation within Bacterial Cells. Chemistry 2022, 29, e202202536. [Google Scholar] [CrossRef]
- Guo, C.; Nolan, E.M. Heavy-Metal Trojan Horse: Enterobactin-Directed Delivery of Platinum(IV) Prodrugs to Escherichia coli. J. Am. Chem. Soc. 2022, 144, 12756–12768. [Google Scholar] [CrossRef]
- Wang, J.; Huang, J.; Wu, Y.; Hai, L.; Wang, G. Design and Synthesis of Novel Thioether-linked Carbapenem-Oxazolidinone Hybrids as Potential Antimicrobial Agents. Lett. Org. Chem. 2008, 5, 336–341. [Google Scholar] [CrossRef]
- Sissi, C.; Palumbo, M. In front of and behind the replication fork: Bacterial type IIA topoisomerases. Cell. Mol. Life Sci. 2010, 67, 2001–2024. [Google Scholar] [CrossRef]
- Oblak, M.; Kotnik, M.; Solmajer, T. Discovery and development of ATPase inhibitors of DNA gyrase as antibacterial agents. Curr. Med. Chem. 2007, 14, 2033–2047. [Google Scholar] [CrossRef] [PubMed]
- Tari, L.W.; Li, X.; Trzoss, M.; Bensen, D.C.; Chen, Z.; Lam, T.; Zhang, J.; Lee, S.J.; Hough, G.; Phillipson, D.; et al. Tricyclic GyrB/ParE (TriBE) inhibitors: A new class of broad-spectrum dual-targeting antibacterial agents. PLoS ONE 2013, 8, e84409. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Russo, R.; Westfall, L.; Shrestha, R.; Zimmerman, M.; Dartois, V.; Kurepina, N.; Kreiswirth, B.; Singleton, E.; Li, S.G.; et al. A Novel Oral GyrB/ParE Dual Binding Inhibitor Effective against Multidrug-Resistant Neisseria gonorrhoeae and Other High-Threat Pathogens. Antimicrob. Agents Chemother. 2022, 66, e0041422. [Google Scholar] [CrossRef] [PubMed]
- Forde, E.; Shafiy, G.; Fitzgerald-Hughes, D.; Stromstedt, A.A.; Devocelle, M. Action of antimicrobial peptides and their prodrugs on model and biological membranes. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2018, 24, e3086. [Google Scholar] [CrossRef] [PubMed]
- Desgranges, S.; Ruddle, C.C.; Burke, L.P.; McFadden, T.M.; O’Brien, J.E.; Fitzgerald-Hughes, D.; Humphreys, H.; Smyth, T.P.; Devocelle, M. β-Lactam-host defence peptide conjugates as antibiotic prodrug candidates targeting resistant bacteria. Rsc Adv. 2012, 2, 2480. [Google Scholar] [CrossRef]
- Du, X.; Xia, C.; Shen, J.; Wu, B.; Shen, Z. Characterization of florfenicol resistance among calf pathogenic Escherichia coli. Fems Microbiol. Lett. 2004, 236, 183–189. [Google Scholar] [CrossRef]
- Schwarz, S.; Kehrenberg, C.; Doublet, B.; Cloeckaert, A. Molecular basis of bacterial resistance to chloramphenicol and florfenicol. Fems Microbiol. Rev. 2004, 28, 519–542. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, C.P.; Chen, S.Q.; Liu, M.J.; Zhang, L.; Lin, S.Y.; Shu, G.; Yuan, Z.X.; Lin, J.C.; Peng, G.N.; et al. Poloxamer modified florfenicol instant microparticles for improved oral bioavailability. Colloids Surf. BBiointerfaces 2020, 193, 111078. [Google Scholar] [CrossRef]
- Li, Z.; Yang, Y.J.; Qin, Z.; Li, S.H.; Bai, L.X.; Li, J.Y.; Liu, X.W. Florfenicol-Polyarginine Conjugates Exhibit Promising Antibacterial Activity Against Resistant Strains. Front. Chem. 2022, 10, 921091. [Google Scholar] [CrossRef]
- Gordon, E.M.; Duncton, M.A.J.; Gallop, M.A. Orally Absorbed Derivatives of the beta-Lactamase Inhibitor Avibactam. Design of Novel Prodrugs of Sulfate Containing Drugs. J. Med. Chem. 2018, 61, 10340–10344. [Google Scholar] [CrossRef]
- Coleman, K. Diazabicyclooctanes (DBOs): A potent new class of non-beta-lactam beta-lactamase inhibitors. Curr. Opin. Microbiol. 2011, 14, 550–555. [Google Scholar] [CrossRef]
- Lagacé-Wiens, P.; Walkty, A.; Karlowsky, J.A. Ceftazidime–avibactam: An evidence-based review of its pharmacology and potential use in the treatment of Gram-negative bacterial infections. Core Evid. 2014, 9, 13. [Google Scholar] [CrossRef]
- Rajavel, M.; Kumar, V.; Nguyen, H.; Wyatt, J.; Marshall, S.H.; Papp-Wallace, K.M.; Deshpande, P.; Bhavsar, S.; Yeole, R.; Bhagwat, S.; et al. Structural Characterization of Diazabicyclooctane beta-Lactam "Enhancers" in Complex with Penicillin-Binding Proteins PBP2 and PBP3 of Pseudomonas aeruginosa. mBio 2021, 12, e03058-20. [Google Scholar] [CrossRef]
- Evans, L.E.; Krishna, A.; Ma, Y.; Webb, T.E.; Marshall, D.C.; Tooke, C.L.; Spencer, J.; Clarke, T.B.; Armstrong, A.; Edwards, A.M. Exploitation of Antibiotic Resistance as a Novel Drug Target: Development of a beta-Lactamase-Activated Antibacterial Prodrug. J. Med. Chem. 2019, 62, 4411–4425. [Google Scholar] [CrossRef]
- Saris, A.; Qin, W.; van Linge, C.C.A.; Reijnders, T.D.Y.; Florquin, S.; Burnet, M.; Strass, S.; de Vos, A.F.; van der Poll, T. The Azithromycin Pro-Drug CSY5669 Boosts Bacterial Killing While Attenuating Lung Inflammation Associated with Pneumonia Caused by Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2022, 66, e0229821. [Google Scholar] [CrossRef]
- Kanafani, Z.A.; Corey, G.R. Tedizolid (TR-701): A new oxazolidinone with enhanced potency. Expert Opin. Investig. Drugs 2012, 21, 515–522. [Google Scholar] [CrossRef]
- Shaw, K.; Poppe, S.; Schaadt, R.; Brown-Driver, V.; Finn, J.; Pillar, C.; Shinabarger, D.; Zurenko, G. In vitro activity of TR-700, the antibacterial moiety of the prodrug TR-701, against linezolid-resistant strains. Antimicrob. Agents Chemother. 2008, 52, 4442–4447. [Google Scholar] [CrossRef]
- Barber, K.E.; Smith, J.R.; Raut, A.; Rybak, M.J. Evaluation of tedizolid against Staphylococcus aureus and enterococci with reduced susceptibility to vancomycin, daptomycin or linezolid. J. Antimicrob. Chemother. 2015, 71, 152–155. [Google Scholar] [CrossRef]
- Lyons, M.A. Modeling and Simulation of Pretomanid Pharmacokinetics in Pulmonary Tuberculosis Patients. Antimicrob. Agents Chemother. 2018, 62, e02359-17. [Google Scholar] [CrossRef]
- Conradie, F.; Diacon, A.H.; Ngubane, N.; Howell, P.; Everitt, D.; Crook, A.M.; Mendel, C.M.; Egizi, E.; Moreira, J.; Timm, J.; et al. Treatment of Highly Drug-Resistant Pulmonary Tuberculosis. N. Engl. J. Med. 2020, 382, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Najjar, A.; Najjar, A.; Karaman, R. Newly Developed Prodrugs and Prodrugs in Development; an Insight of the Recent Years. Molecules 2020, 25. [Google Scholar] [CrossRef] [PubMed]
- Girish, C.; Balakrishnan, S. Ceftaroline fosamil: A novel anti-Methicillin-resistant Staphylococcus aureus cephalosporin. J. Pharmacol. Pharmacother. 2011, 2, 209–211. [Google Scholar] [CrossRef] [PubMed]
- Allan, R.N.; Kelso, M.J.; Rineh, A.; Yepuri, N.R.; Feelisch, M.; Soren, O.; Brito-Mutunayagam, S.; Salib, R.J.; Stoodley, P.; Clarke, S.C. Cephalosporin-NO-donor prodrug PYRRO-C3D shows β-lactam-mediated activity against Streptococcus pneumoniae biofilms. Nitric Oxide Biol. Chem. 2017, 65, 43–49. [Google Scholar] [CrossRef]
- Soren, O.; Rineh, A.; Silva, D.G.; Cai, Y.; Howlin, R.P.; Allan, R.N.; Feelisch, M.; Davies, J.C.; Connett, G.J.; Faust, S.N. Cephalosporin nitric oxide-donor prodrug DEA-C3D disperses biofilms formed by clinical cystic fibrosis isolates of Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2019, 75, 117–125. [Google Scholar] [CrossRef]
- Barraud, N.; Kardak, B.G.; Yepuri, N.R.; Howlin, R.P.; Webb, J.S.; Faust, S.N.; Kjelleberg, S.; Rice, S.A.; Kelso, M.J. Cephalosporin-3’-diazeniumdiolates: Targeted NO-donor prodrugs for dispersing bacterial biofilms. Angew. Chem. 2012, 51, 9057–9060. [Google Scholar] [CrossRef]
- Kampfer, P.; Rauhoff, O.; Dott, W. Glycosidase profiles of members of the family Enterobacteriaceae. J. Clin. Microbiol. 1991, 29, 2877–2879. [Google Scholar] [CrossRef]
- Levy, C.W.; Roujeinikova, A.; Sedelnikova, S.; Baker, P.J.; Stuitje, A.R.; Slabas, A.R.; Rice, D.W.; Rafferty, J.B. Molecular basis of triclosan activity. Nature 1999, 398, 383–384. [Google Scholar] [CrossRef]
- Howse, G.L.; Bovill, R.A.; Stephens, P.J.; Osborn, H.M.I. Synthesis and antibacterial profiles of targeted triclosan derivatives. Eur. J. Med. Chem. 2019, 162, 51–58. [Google Scholar] [CrossRef]
- Khandazhinskaya, A.L.; Alexandrova, L.A.; Matyugina, E.S.; Solyev, P.N.; Efremenkova, O.V.; Buckheit, K.W.; Wilkinson, M.; Buckheit, R.W., Jr.; Chernousova, L.N.; Smirnova, T.G.; et al. Novel 5’-Norcarbocyclic Pyrimidine Derivatives as Antibacterial Agents. Molecules 2018, 23, 3069. [Google Scholar] [CrossRef]
- Negrya, S.D.; Jasko, M.V.; Solyev, P.N.; Karpenko, I.L.; Efremenkova, O.V.; Vasilyeva, B.F.; Sumarukova, I.G.; Kochetkov, S.N.; Alexandrova, L.A. Synthesis of water-soluble prodrugs of 5-modified 2’-deoxyuridines and their antibacterial activity. J. Antibiot. 2020, 73, 236–246. [Google Scholar] [CrossRef]
- McEntee, L.; Johnson, A.; Farrington, N.; Unsworth, J.; Dane, A.; Jain, A.; Cotroneo, N.; Critchley, I.; Melnick, D.; Parr, T.; et al. Pharmacodynamics of Tebipenem: New Options for Oral Treatment of Multidrug-Resistant Gram-Negative Infections. Antimicrob. Agents Chemother. 2019, 63, e00603-19. [Google Scholar] [CrossRef]
- Kaul, M.; Mark, L.; Zhang, Y.; Parhi, A.K.; LaVoie, E.J.; Pilch, D.S. An FtsZ-targeting prodrug with oral antistaphylococcal efficacy in vivo. Antimicrob. Agents Chemother. 2013, 57, 5860–5869. [Google Scholar] [CrossRef]
- Kaul, M.; Mark, L.; Zhang, Y.; Parhi, A.K.; Lyu, Y.L.; Pawlak, J.; Saravolatz, S.; Saravolatz, L.D.; Weinstein, M.P.; LaVoie, E.J. TXA709, an FtsZ-targeting benzamide prodrug with improved pharmacokinetics and enhanced in vivo efficacy against methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2015, 59, 4845–4855. [Google Scholar] [CrossRef]
- Marinelli, L.; Di Stefano, A.; Cacciatore, I. Carvacrol and its derivatives as antibacterial agents. Phytochem. Rev. 2018, 17, 903–921. [Google Scholar] [CrossRef]
- Marinelli, L.; Fornasari, E.; Eusepi, P.; Ciulla, M.; Genovese, S.; Epifano, F.; Fiorito, S.; Turkez, H.; Ortucu, S.; Mingoia, M.; et al. Carvacrol prodrugs as novel antimicrobial agents. Eur. J. Med. Chem. 2019, 178, 515–529. [Google Scholar] [CrossRef]
- Fleck, L.E.; North, E.J.; Lee, R.E.; Mulcahy, L.R.; Casadei, G.; Lewis, K. A screen for and validation of prodrug antimicrobials. Antimicrob. Agents Chemother. 2014, 58, 1410–1419. [Google Scholar] [CrossRef]
- Guay, D.R. An update on the role of nitrofurans in the management of urinary tract infections. Drugs 2001, 61, 353–364. [Google Scholar] [CrossRef]
- Gershon, H.; Parmegiani, R. Antimicrobial activity of 8-quinolinol, its salts with salicylic acid and 3-hydroxy-2-naphthoic acid, and the respective copper (II) chelates in liquid culture. Appl. Microbiol. 1963, 11, 62–65. [Google Scholar] [CrossRef]
- Hoy, S.M. Contezolid: First Approval. Drugs 2021, 81, 1587–1591. [Google Scholar] [CrossRef]
- Bulitta, J.B.; Hafkin, B.; Fang, E. 1118. Population Pharmacokinetics of Contezolid Acefosamil and Contezolid - Rationale for a Safe and Effective Loading Dose Regimen. Open Forum Infect. Dis. 2021, 8 (Suppl. S1), S651. [Google Scholar] [CrossRef]
- Liu, J.; Wang, W.; Wang, C.; Zhang, L.; Zhang, X.; Liu, S.; Xu, Y.; Wang, H.; Dai, Q.; Liu, C.; et al. Discovery of Antibacterial Contezolid Acefosamil: Innovative O-Acyl Phosphoramidate Prodrug for IV and Oral Therapies. Acs Med. Chem. Lett. 2022, 13, 1030–1035. [Google Scholar] [CrossRef] [PubMed]
- Al-Halawa, D.A.; Seir, R.A.; Qasrawi, R. Antibiotic Resistance Knowledge, Attitudes, and Practices among Pharmacists: A Cross-Sectional Study in West Bank, Palestine. J. Environ. Public Health 2023, 2023, 2294048. [Google Scholar] [CrossRef] [PubMed]
- Wall, S. Prevention of antibiotic resistance - an epidemiological scoping review to identify research categories and knowledge gaps. Glob. Health Action 2019, 12, 1756191. [Google Scholar] [CrossRef]
- Owens, R.C., Jr. Antimicrobial stewardship: Concepts and strategies in the 21st century. Diagn. Microbiol. Infect. Dis. 2008, 61, 110–128. [Google Scholar] [CrossRef]
- Siegel, J.D.; Rhinehart, E.; Jackson, M.; Chiarello, L.; Healthcare Infection Control Practices Advisory, C. Management of multidrug-resistant organisms in health care settings, 2006. Am. J. Infect. Control 2007, 35, S165–S193. [Google Scholar] [CrossRef]
- Muto, C.A.; Jernigan, J.A.; Ostrowsky, B.E.; Richet, H.M.; Jarvis, W.R.; Boyce, J.M.; Farr, B.M. SHEA guideline for preventing nosocomial transmission of multidrug-resistant strains of Staphylococcus aureus and enterococcus. Infect. Control Hosp. Epidemiol. 2003, 24, 362–386. [Google Scholar] [CrossRef]
- Ventola, C.L. The antibiotic resistance crisis: Part 1: Causes and threats. P T A Peer-Rev. J. Formul. Manag. 2015, 40, 277–283. [Google Scholar]
- Lee, C.R.; Cho, I.H.; Jeong, B.C.; Lee, S.H. Strategies to minimize antibiotic resistance. Int. J. Environ. Res. Public Health 2013, 10, 4274–4305. [Google Scholar] [CrossRef]
- Ricciardi, W.; Giubbini, G.; Laurenti, P. Surveillance and Control of Antibiotic Resistance in the Mediterranean Region. Mediterr. J. Hematol. Infect. Dis. 2016, 8, e2016036. [Google Scholar] [CrossRef]































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Breijyeh, Z.; Karaman, R. Design and Synthesis of Novel Antimicrobial Agents. Antibiotics 2023, 12, 628. https://doi.org/10.3390/antibiotics12030628
Breijyeh Z, Karaman R. Design and Synthesis of Novel Antimicrobial Agents. Antibiotics. 2023; 12(3):628. https://doi.org/10.3390/antibiotics12030628
Chicago/Turabian StyleBreijyeh, Zeinab, and Rafik Karaman. 2023. "Design and Synthesis of Novel Antimicrobial Agents" Antibiotics 12, no. 3: 628. https://doi.org/10.3390/antibiotics12030628
APA StyleBreijyeh, Z., & Karaman, R. (2023). Design and Synthesis of Novel Antimicrobial Agents. Antibiotics, 12(3), 628. https://doi.org/10.3390/antibiotics12030628