


A Riboswitch-Driven Era of New Antibacterials
 , ,
, ,  ,
,  , and
, and 
Abstract
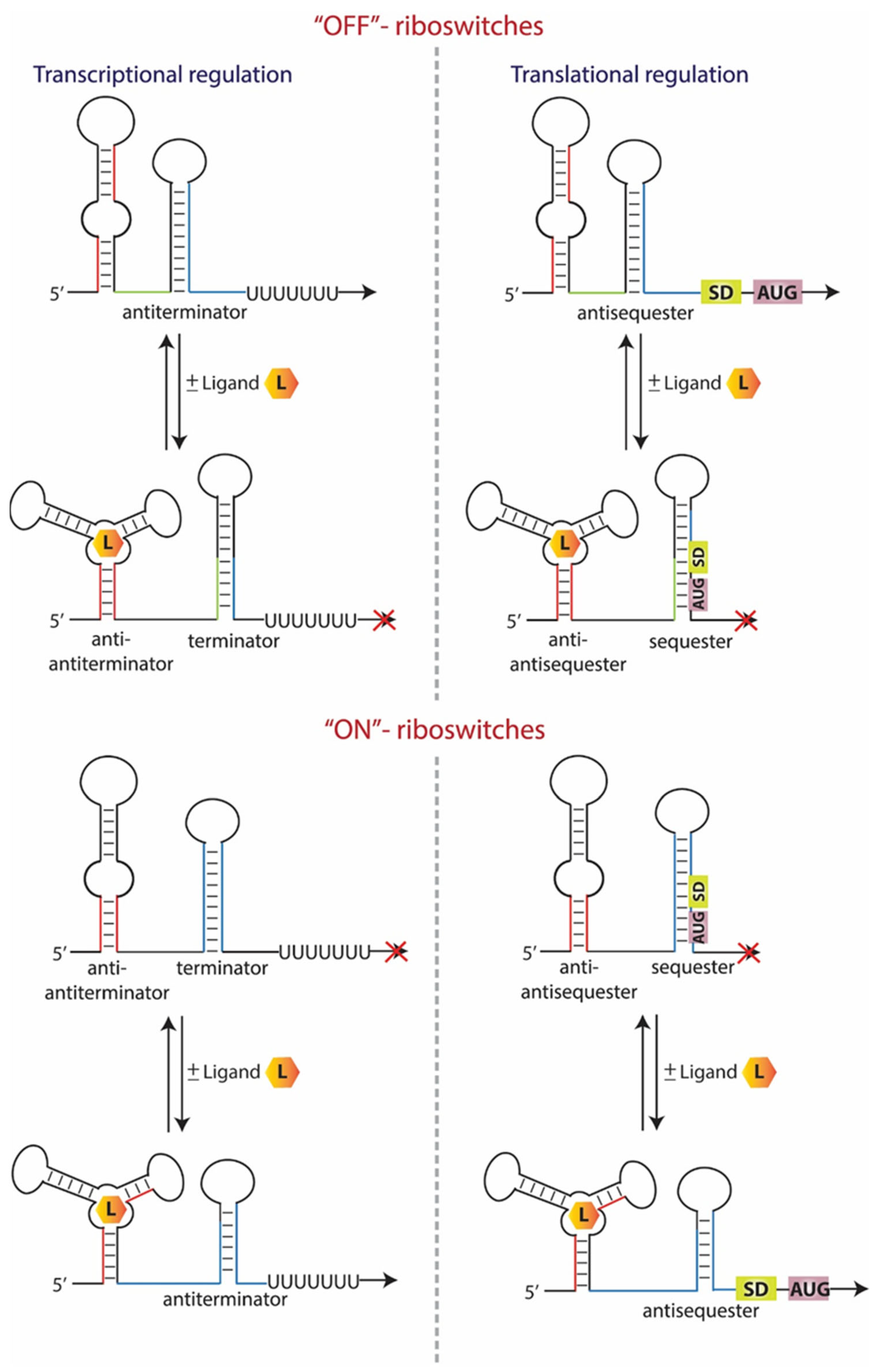
1. Introduction
2. TPP Riboswitches

{kind=link}
{kind=link}
{kind=link}
| Riboswitch | Antimicrobial Compound | Organism | Ref. | |
|---|---|---|---|---|
| TPP | Pyrithiamine |  | B. subtilis, A. oryzae | [49] |
| FMN | Roseoflavin |  | L. monocytogenes, B. subtilis, E. faecalis, S. pyogenes | [53,54,55] |
| 5FDQD |  | B. subtilis, C. difficile | [56,57] | |
| Ribocil |  | E. coli | [58] | |
| Ribocil-C |  | E. coli, S. aureus | [58,59] | |
| Ribocil C-PA |  | E. coli, K. pneumoniae | [60] | |
| 10-(2,2-dihydroxylethyl)-7,8-dimethylisoalloxazine (5a) |  | M. tuberculosis | [61] | |
| ASO-1 | T1T1C1T2C2C2C2A2T2C2C2A2G2A1C1T1 | S. aureus, L. monocytogenes, E. coli | [62] | |
| GlmS | ASO1 | C1T1T1T2A2A2C2T2G2T2A2C2T2G1C1C1 | S. aureus | [63] |
| carba-α-D-glucosamine |  | S. aureus | [64] | |
| carba-α-D-glucosamine-6-phosphate |  | S. aureus | [64] | |
| fluoro-carba-α-D-glucosamine-6-phosphate |  | B. subtilis, S. aureus | [65] | |
| Guanine | PC1 |  | S. aureus, C. difficile, MDR strains | [66,67,68] |
| T-box | Neomycin B |  | B. subtilis, S. aureus | [28,69] |
| PKZ18 |  | B. subtilis, S. aureus | [70] | |
| PKZ18-22 |  | B. subtilis, S. aureus, MRSA | [71,72] | |
3. FMN Riboswitches
4. GlmS Riboswitch-Ribozyme
5. Guanine Riboswitches
6. Cyclic-di GMP Riboswitches
7. Lysine Riboswitches
8. T-Box Riboswitches
9. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brown, E.D.; Wright, G.D. Antibacterial Drug Discovery in the Resistance Era. Nature 2016, 529, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.N. Ribosome-Targeting Antibiotics and Mechanisms of Bacterial Resistance. Nat. Rev. Microbiol. 2014, 12, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Deigan, K.E.; FerrÉ-D’AmarÉ, A.R. Riboswitches: Discovery of Drugs That Target Bacterial Gene-Regulatory RNAs. Acc. Chem. Res. 2011, 44, 1329–1338. [Google Scholar] [CrossRef]
- Winkler, W.C.; Breaker, R.R. Regulation of bacterial gene expression by riboswitches. Annu. Rev. Microbiol. 2005, 59, 487–517. [Google Scholar] [CrossRef]
- Sherwood, A.V.; Henkin, T.M. Riboswitch-Mediated Gene Regulation: Novel RNA Architectures Dictate Gene Expression Responses. Annu. Rev. Microbiol. 2016, 70, 361–374. [Google Scholar] [CrossRef]
- Breaker, R.R. Prospects for Riboswitch Discovery and Analysis. Mol. Cell 2011, 43, 867–879. [Google Scholar] [CrossRef]
- Henkin, T.M. Riboswitch RNAs: Using RNA to Sense Cellular Metabolism. Genes Dev. 2008, 22, 3383–3390. [Google Scholar] [CrossRef]
- Mandal, M.; Breaker, R.R. Gene Regulation by Riboswitches. Nat. Rev. Mol. Cell Biol. 2004, 5, 451–463. [Google Scholar] [CrossRef]
- Tucker, B.J.; Breaker, R.R. Riboswitches as Versatile Gene Control Elements. Curr. Opin. Struct. Biol. 2005, 15, 342–348. [Google Scholar] [CrossRef]
- Oliva, G.; Sahr, T.; Buchrieser, C. Small RNAs, 5′ UTR Elements and RNA-Binding Proteins in Intracellular Bacteria: Impact on Metabolism and Virulence. FEMS Microbiol. Rev. 2015, 39, 331–349. [Google Scholar] [CrossRef]
- Matsui, M.; Corey, D.R. Non-Coding RNAs as Drug Targets. Nat. Rev. Drug Discov. 2017, 16, 167–179. [Google Scholar] [CrossRef]
- Dersch, P.; Khan, M.A.; Mühlen, S.; Görke, B. Roles of Regulatory RNAs for Antibiotic Resistance in Bacteria and Their Potential Value as Novel Drug Targets. Front. Microbiol. 2017, 8, 803. [Google Scholar] [CrossRef] [PubMed]
- McCown, P.J.; Corbino, K.A.; Stav, S.; Sherlock, M.E.; Breaker, R.R. Riboswitch Diversity and Distribution. RNA 2017, 23, 995–1011. [Google Scholar] [CrossRef] [PubMed]
- Barrick, J.E.; Breaker, R.R. The Distributions, Mechanisms, and Structures of Metabolite-Binding Riboswitches. Genome Biol. 2007, 8, R239. [Google Scholar] [CrossRef]
- Sudarsan, N.; Lee, E.R.; Weinberg, Z.; Moy, R.H.; Kim, J.N.; Link, K.H.; Breaker, R.R. Riboswitches in Eubacteria Sense the Second Messenger Cyclic Di-GMP. Science 2008, 321, 411–413. [Google Scholar] [CrossRef]
- Cromie, M.J.; Shi, Y.; Latifi, T.; Groisman, E.A. An RNA Sensor for Intracellular Mg2+. Cell 2006, 125, 71–84. [Google Scholar] [CrossRef]
- Dann, C.E.; Wakeman, C.A.; Sieling, C.L.; Baker, S.C.; Irnov, I.; Winkler, W.C. Structure and Mechanism of a Metal-Sensing Regulatory RNA. Cell 2007, 130, 878–892. [Google Scholar] [CrossRef]
- Baker, J.L.; Sudarsan, N.; Weinberg, Z.; Roth, A.; Stockbridge, R.B.; Breaker, R.R. Widespread Genetic Switches and Toxicity Resistance Proteins for Fluoride. Science 2012, 335, 233–235. [Google Scholar] [CrossRef] [PubMed]
- Dambach, M.; Sandoval, M.; Updegrove, T.B.; Anantharaman, V.; Aravind, L.; Waters, L.S.; Storz, G. The Ubiquitous YybP-YkoY Riboswitch Is a Manganese-Responsive Regulatory Element. Mol. Cell 2015, 57, 1099–1109. [Google Scholar] [CrossRef]
- Price, I.R.; Gaballa, A.; Ding, F.; Helmann, J.D.; Ke, A. Mn2+-Sensing Mechanisms of YybP-YkoY Orphan Riboswitches. Mol. Cell 2015, 57, 1110–1123. [Google Scholar] [CrossRef]
- Furukawa, K.; Ramesh, A.; Zhou, Z.; Weinberg, Z.; Vallery, T.; Winkler, W.C.; Breaker, R.R. Bacterial Riboswitches Cooperatively Bind Ni2+ or Co2+ Ions and Control Expression of Heavy Metal Transporters. Mol. Cell 2015, 57, 1088–1098. [Google Scholar] [CrossRef] [PubMed]
- White, N.; Sadeeshkumar, H.; Sun, A.; Sudarsan, N.; Breaker, R.R. Na+ Riboswitches Regulate Genes for Diverse Physiological Processes in Bacteria. Nat. Chem. Biol. 2022, 18, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Henkin, T.M.; Yanofsky, C. Regulation by Transcription Attenuation in Bacteria: How RNA Provides Instructions for Transcription Termination/Antitermination Decisions. BioEssays 2002, 24, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Green, N.J.; Grundy, F.J.; Henkin, T.M. The T Box Mechanism: TRNA as a Regulatory Molecule. FEBS Lett. 2010, 584, 318–324. [Google Scholar] [CrossRef]
- Grundy, F.J.; Henkin, T.M. TRNA as a Positive Regulator of Transcription Antitermination in B. subtilis. Cell 1993, 74, 475–482. [Google Scholar] [CrossRef]
- Blount, K.F.; Breaker, R.R. Riboswitches as Antibacterial Drug Targets. Nat. Biotechnol. 2006, 24, 1558–1564. [Google Scholar] [CrossRef]
- Apostolidi, M.; Saad, N.Y.; Drainas, D.; Pournaras, S.; Becker, H.D.; Stathopoulos, C. A GlyS T-Box Riboswitch with Species-Specific Structural Features Responding to Both Proteinogenic and Nonproteinogenic TRNA Gly Isoacceptors. RNA 2015, 21, 1790–1806. [Google Scholar] [CrossRef]
- Stamatopoulou, V.; Apostolidi, M.; Li, S.; Lamprinou, K.; Papakyriakou, A.; Zhang, J.; Stathopoulos, C. Direct Modulation of T-Box Riboswitch-Controlled Transcription by Protein Synthesis Inhibitors. Nucleic Acids Res. 2017, 45, 10242–10258. [Google Scholar] [CrossRef]
- Wencker, F.D.R.; Marincola, G.; Schoenfelder, S.M.K.; Maaß, S.; Becher, D.; Ziebuhr, W. Another Layer of Complexity in Staphylococcus aureus Methionine Biosynthesis Control: Unusual RNase III-Driven T-Box Riboswitch Cleavage Determines Met Operon MRNA Stability and Decay. Nucleic Acids Res. 2021, 49, 2192–2212. [Google Scholar] [CrossRef]
- Giarimoglou, N.; Kouvela, A.; Patsi, I.; Zhang, J.; Stamatopoulou, V.; Stathopoulos, C. Lineage-Specific Insertions in T-Box Riboswitches Modulate Antibiotic Binding and Action. Nucleic Acids Res. 2022, 50, 5834–5849. [Google Scholar] [CrossRef]
- Nahvi, A. Coenzyme B12 Riboswitches Are Widespread Genetic Control Elements in Prokaryotes. Nucleic Acids Res. 2004, 32, 143–150. [Google Scholar] [CrossRef]
- Jia, X.; Zhang, J.; Sun, W.; He, W.; Jiang, H.; Chen, D.; Murchie, A.I.H. Riboswitch Regulation of Aminoglycoside Resistance Acetyl and Adenyl Transferases. Cell 2013, 153, 1419–1420. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Zhang, J.; Sun, W.; He, W.; Jiang, H.; Chen, D.; Murchie, A.I.H. Riboswitch Control of Aminoglycoside Antibiotic Resistance. Cell 2013, 152, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Croft, M.T.; Moulin, M.; Webb, M.E.; Smith, A.G. Thiamine Biosynthesis in Algae Is Regulated by Riboswitches. Proc. Natl. Acad. Sci. USA 2007, 104, 20770–20775. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, M.A.; Petrova, S.A.; Gelfand, M.S. Comparative Genomic Analysis of Fungal TPP-Riboswitches. Fungal Genet. Biol. 2018, 114, 34–41. [Google Scholar] [CrossRef]
- Bocobza, S.; Adato, A.; Mandel, T.; Shapira, M.; Nudler, E.; Aharoni, A. Riboswitch-Dependent Gene Regulation and Its Evolution in the Plant Kingdom. Genes Dev. 2007, 21, 2874–2879. [Google Scholar] [CrossRef]
- Sudarsan, N.; Wickiser, J.K.; Nakamura, S.; Ebert, M.S.; Breaker, R.R. An MRNA Structure in Bacteria That Controls Gene Expression by Binding Lysine. Genes Dev. 2003, 17, 2688–2697. [Google Scholar] [CrossRef]
- Winkler, W.; Nahvi, A.; Breaker, R.R. Thiamine Derivatives Bind Messenger RNAs Directly to Regulate Bacterial Gene Expression. Nature 2002, 419, 952–956. [Google Scholar] [CrossRef]
- Rodionov, D.A.; Vitreschak, A.G.; Mironov, A.A.; Gelfand, M.S. Comparative Genomics of Thiamin Biosynthesis in Procaryotes. J. Biol. Chem. 2002, 277, 48949–48959. [Google Scholar] [CrossRef]
- Gong, S.; Du, C.; Wang, Y. Regulation of the Thiamine Pyrophosphate (TPP)-sensing Riboswitch in NMT1 MRNA from Neurospora Crassa. FEBS Lett. 2020, 594, 625–635. [Google Scholar] [CrossRef]
- Miranda-Ríos, J. The THI-Box Riboswitch, or How RNA Binds Thiamin Pyrophosphate. Structure 2007, 15, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.E.; Ferré-D’Amaré, A.R. Crystal Structures of the Thi-Box Riboswitch Bound to Thiamine Pyrophosphate Analogs Reveal Adaptive RNA-Small Molecule Recognition. Structure 2006, 14, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Serganov, A.; Polonskaia, A.; Phan, A.T.; Breaker, R.R.; Patel, D.J. Structural Basis for Gene Regulation by a Thiamine Pyrophosphate-Sensing Riboswitch. Nature 2006, 441, 1167–1171. [Google Scholar] [CrossRef]
- Padhi, S.; Pradhan, M.; Bung, N.; Roy, A.; Bulusu, G. TPP Riboswitch Aptamer: Role of Mg2+ Ions, Ligand Unbinding, and Allostery. J. Mol. Graph. Model. 2019, 88, 282–291. [Google Scholar] [CrossRef]
- Thore, S.; Leibundgut, M.; Ban, N. Structure of the Eukaryotic Thiamine Pyrophosphate Riboswitch with Its Regulatory Ligand. Science 2006, 312, 1208–1211. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Breaker, R.R. Eukaryotic TPP Riboswitch Regulation of Alternative Splicing Involving Long-Distance Base Pairing. Nucleic Acids Res. 2013, 41, 3022–3031. [Google Scholar] [CrossRef] [PubMed]
- Panchal, V.; Brenk, R. Riboswitches as Drug Targets for Antibiotics. Antibiotics 2021, 10, 45. [Google Scholar] [CrossRef] [PubMed]
- Tylicki, A.; Łempicka, A.; Romaniuk-Demonchaux, K.; Czerniecki, J.; Dobrzyń, P.; Strumiło, S. Effect of Oxythiamin on Growth Rate, Survival Ability and Pyruvate Decarboxylase Activity InSaccharomyces Cerevisiae. J. Basic Microbiol. 2003, 43, 522–529. [Google Scholar] [CrossRef]
- Sudarsan, N.; Cohen-Chalamish, S.; Nakamura, S.; Emilsson, G.M.; Breaker, R.R. Thiamine Pyrophosphate Riboswitches Are Targets for the Antimicrobial Compound Pyrithiamine. Chem. Biol. 2005, 12, 1325–1335. [Google Scholar] [CrossRef]
- Chen, L.; Cressina, E.; Dixon, N.; Erixon, K.; Agyei-Owusu, K.; Micklefield, J.; Smith, A.G.; Abell, C.; Leeper, F.J. Probing Riboswitch–Ligand Interactions Using Thiamine Pyrophosphate Analogues. Org. Biomol. Chem. 2012, 10, 5924. [Google Scholar] [CrossRef]
- Lünse, C.E.; Schüller, A.; Mayer, G. The Promise of Riboswitches as Potential Antibacterial Drug Targets. Int. J. Med. Microbiol. 2014, 304, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Wakchaure, P.D.; Ganguly, B. Molecular Level Insights into the Inhibition of Gene Expression by Thiamine Pyrophosphate (TPP) Analogs for TPP Riboswitch: A Well-Tempered Metadynamics Simulations Study. J. Mol. Graph. Model. 2021, 104, 107849. [Google Scholar] [CrossRef] [PubMed]
- Ott, E.; Stolz, J.; Mack, M. The RFN Riboswitch of Bacillus Subtilis Is a Target for the Antibiotic Roseoflavin Produced by Streptomyces Davawensis. RNA Biol. 2009, 6, 276–280. [Google Scholar] [CrossRef]
- Mansjö, M.; Johansson, J. The Riboflavin Analog Roseoflavin Targets an FMN-Riboswitch and Blocks Listeria Monocytogene s Growth, but Also Stimulates Virulence Gene-Expression and Infection. RNA Biol. 2011, 8, 674–680. [Google Scholar] [CrossRef]
- Wang, H.; Mann, P.A.; Xiao, L.; Gill, C.; Galgoci, A.M.; Howe, J.A.; Villafania, A.; Barbieri, C.M.; Malinverni, J.C.; Sher, X.; et al. Dual-Targeting Small-Molecule Inhibitors of the Staphylococcus aureus FMN Riboswitch Disrupt Riboflavin Homeostasis in an Infectious Setting. Cell Chem. Biol. 2017, 24, 576–588.e6. [Google Scholar] [CrossRef]
- Blount, K.F.; Megyola, C.; Plummer, M.; Osterman, D.; O’Connell, T.; Aristoff, P.; Quinn, C.; Chrusciel, R.A.; Poel, T.J.; Schostarez, H.J.; et al. Novel Riboswitch-Binding Flavin Analog That Protects Mice against Clostridium difficile Infection without Inhibiting Cecal Flora. Antimicrob. Agents Chemother. 2015, 59, 5736–5746. [Google Scholar] [CrossRef]
- Vicens, Q.; Mondragón, E.; Reyes, F.E.; Coish, P.; Aristoff, P.; Berman, J.; Kaur, H.; Kells, K.W.; Wickens, P.; Wilson, J.; et al. Structure–Activity Relationship of Flavin Analogues That Target the Flavin Mononucleotide Riboswitch. ACS Chem. Biol. 2018, 13, 2908–2919. [Google Scholar] [CrossRef] [PubMed]
- Howe, J.A.; Wang, H.; Fischmann, T.O.; Balibar, C.J.; Xiao, L.; Galgoci, A.M.; Malinverni, J.C.; Mayhood, T.; Villafania, A.; Nahvi, A.; et al. Selective Small-Molecule Inhibition of an RNA Structural Element. Nature 2015, 526, 672–677. [Google Scholar] [CrossRef]
- Howe, J.A.; Xiao, L.; Fischmann, T.O.; Wang, H.; Tang, H.; Villafania, A.; Zhang, R.; Barbieri, C.M.; Roemer, T. Atomic Resolution Mechanistic Studies of Ribocil: A Highly Selective Unnatural Ligand Mimic of the E. coli FMN Riboswitch. RNA Biol. 2016, 13, 946–954. [Google Scholar] [CrossRef]
- Motika, S.E.; Ulrich, R.J.; Geddes, E.J.; Lee, H.Y.; Lau, G.W.; Hergenrother, P.J. Gram-Negative Antibiotic Active through Inhibition of an Essential Riboswitch. J. Am. Chem. Soc. 2020, 142, 10856–10862. [Google Scholar] [CrossRef]
- Harale, B.; Kidwai, S.; Ojha, D.; Singh, M.; Chouhan, D.K.; Singh, R.; Khedkar, V.; Rode, A.B. Synthesis and Evaluation of Antimycobacterial Activity of Riboflavin Derivatives. Bioorg. Med. Chem. Lett. 2021, 48, 128236. [Google Scholar] [CrossRef] [PubMed]
- Traykovska, M.; Penchovsky, R. Engineering Antisense Oligonucleotides as Antibacterial Agents That Target FMN Riboswitches and Inhibit the Growth of Staphylococcus aureus, Listeria monocytogenes, and Escherichia coli. ACS Synth. Biol. 2022, 11, 1845–1855. [Google Scholar] [CrossRef]
- Traykovska, M.; Popova, K.B.; Penchovsky, R. Targeting GlmS Ribozyme with Chimeric Antisense Oligonucleotides for Antibacterial Drug Development. ACS Synth. Biol. 2021, 10, 3167–3176. [Google Scholar] [CrossRef] [PubMed]
- Schüller, A.; Matzner, D.; Lünse, C.E.; Wittmann, V.; Schumacher, C.; Unsleber, S.; Brötz-Oesterhelt, H.; Mayer, C.; Bierbaum, G.; Mayer, G. Activation of the GlmS Ribozyme Confers Bacterial Growth Inhibition. ChemBioChem 2017, 18, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Matzner, D.; Schüller, A.; Seitz, T.; Wittmann, V.; Mayer, G. Fluoro-Carba-Sugars Are Glycomimetic Activators of the GlmS Ribozyme. Chem.-A Eur. J. 2017, 23, 12604–12612. [Google Scholar] [CrossRef]
- Mulhbacher, J.; Brouillette, E.; Allard, M.; Fortier, L.-C.; Malouin, F.; Lafontaine, D.A. Novel Riboswitch Ligand Analogs as Selective Inhibitors of Guanine-Related Metabolic Pathways. PLoS Pathog. 2010, 6, e1000865. [Google Scholar] [CrossRef]
- Ster, C.; Allard, M.; Boulanger, S.; Lamontagne Boulet, M.; Mulhbacher, J.; Lafontaine, D.A.; Marsault, É.; Lacasse, P.; Malouin, F. Experimental Treatment of Staphylococcus aureus Bovine Intramammary Infection Using a Guanine Riboswitch Ligand Analog. J. Dairy Sci. 2013, 96, 1000–1008. [Google Scholar] [CrossRef]
- Kofoed, E.M.; Yan, D.; Katakam, A.K.; Reichelt, M.; Lin, B.; Kim, J.; Park, S.; Date, S.V.; Monk, I.R.; Xu, M.; et al. De Novo Guanine Biosynthesis but Not the Riboswitch-Regulated Purine Salvage Pathway Is Required for Staphylococcus aureus Infection In Vivo. J. Bacteriol. 2016, 198, 2001–2015. [Google Scholar] [CrossRef]
- Anupam, R.; Denapoli, L.; Muchenditsi, A.; Hines, J.V. Identification of Neomycin B-Binding Site in T Box Antiterminator Model RNA. Bioorg. Med. Chem. 2008, 16, 4466–4470. [Google Scholar] [CrossRef][Green Version]
- Frohlich, K.M.; Weintraub, S.F.; Bell, J.T.; Todd, G.C.; Väre, V.Y.P.; Schneider, R.; Kloos, Z.A.; Tabe, E.S.; Cantara, W.A.; Stark, C.J.; et al. Discovery of Small-Molecule Antibiotics against a Unique TRNA-Mediated Regulation of Transcription in Gram-Positive Bacteria. ChemMedChem 2019, 14, 758–769. [Google Scholar] [CrossRef]
- Väre, V.Y.P.; Schneider, R.F.; Kim, H.; Lasek-Nesselquist, E.; McDonough, K.A.; Agris, P.F. Small-Molecule Antibiotics Inhibiting TRNA-Regulated Gene Expression Is a Viable Strategy for Targeting Gram-Positive Bacteria. Antimicrob. Agents Chemother. 2020, 65, e01247-20. [Google Scholar] [CrossRef] [PubMed]
- Seyler, T.M.; Moore, C.; Kim, H.; Ramachandran, S.; Agris, P.F. A New Promising Anti-Infective Agent Inhibits Biofilm Growth by Targeting Simultaneously a Conserved RNA Function That Controls Multiple Genes. Antibiotics 2021, 10, 41. [Google Scholar] [CrossRef] [PubMed]
- Serganov, A.; Huang, L.; Patel, D.J. Coenzyme Recognition and Gene Regulation by a Flavin Mononucleotide Riboswitch. Nature 2009, 458, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Winkler, W.C.; Cohen-Chalamish, S.; Breaker, R.R. An MRNA Structure That Controls Gene Expression by Binding FMN. Proc. Natl. Acad. Sci. USA 2002, 99, 15908–15913. [Google Scholar] [CrossRef] [PubMed]
- Vicens, Q.; Mondragón, E.; Batey, R.T. Molecular Sensing by the Aptamer Domain of the FMN Riboswitch: A General Model for Ligand Binding by Conformational Selection. Nucleic Acids Res. 2011, 39, 8586–8598. [Google Scholar] [CrossRef] [PubMed]
- Crielaard, S.; Maassen, R.; Vosman, T.; Rempkens, I.; Velema, W.A. Affinity-Based Profiling of the Flavin Mononucleotide Riboswitch. J. Am. Chem. Soc. 2022, 144, 10462–10470. [Google Scholar] [CrossRef]
- Gelfand, M. A Conserved RNA Structure Element Involved in the Regulation of Bacterial Riboflavin Synthesis Genes. Trends Genet. 1999, 15, 439–442. [Google Scholar] [CrossRef]
- Wickiser, J.K.; Winkler, W.C.; Breaker, R.R.; Crothers, D.M. The Speed of RNA Transcription and Metabolite Binding Kinetics Operate an FMN Riboswitch. Mol. Cell 2005, 18, 49–60. [Google Scholar] [CrossRef]
- Vitreschak, A.G. Regulation of Riboflavin Biosynthesis and Transport Genes in Bacteria by Transcriptional and Translational Attenuation. Nucleic Acids Res. 2002, 30, 3141–3151. [Google Scholar] [CrossRef]
- Long, Q.; Ji, L.; Wang, H.; Xie, J. Riboflavin Biosynthetic and Regulatory Factors as Potential Novel Anti-Infective Drug Targets. Chem. Biol. Drug Des. 2010, 75, 339–347. [Google Scholar] [CrossRef]
- Otani, S.; Takatsu, M.; Nakano, M.; Kasai, S.; Miura, R. Letter: Roseoflavin, a New Antimicrobial Pigment from Streptomyces. J. Antibiot. 1974, 27, 86–87. [Google Scholar] [CrossRef]
- Mack, M.; van Loon, A.P.G.M.; Hohmann, H.-P. Regulation of Riboflavin Biosynthesis in Bacillus Subtilis Is Affected by the Activity of the Flavokinase/Flavin Adenine Dinucleotide Synthetase Encoded by RibC. J. Bacteriol. 1998, 180, 950–955. [Google Scholar] [CrossRef] [PubMed]
- Grill, S.; Busenbender, S.; Pfeiffer, M.; Köhler, U.; Mack, M. The Bifunctional Flavokinase/Flavin Adenine Dinucleotide Synthetase from Streptomyces Davawensis Produces Inactive Flavin Cofactors and Is Not Involved in Resistance to the Antibiotic Roseoflavin. J. Bacteriol. 2008, 190, 1546–1553. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.R.; Blount, K.F.; Breaker, R.R. Roseoflavin Is a Natural Antibacterial Compound That Binds to FMN Riboswitches and Regulates Gene Expression. RNA Biol. 2009, 6, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Langer, S.; Hashimoto, M.; Hobl, B.; Mathes, T.; Mack, M. Flavoproteins Are Potential Targets for the Antibiotic Roseoflavin in Escherichia coli. J. Bacteriol. 2013, 195, 4037–4045. [Google Scholar] [CrossRef]
- Langer, S.; Nakanishi, S.; Mathes, T.; Knaus, T.; Binter, A.; Macheroux, P.; Mase, T.; Miyakawa, T.; Tanokura, M.; Mack, M. The Flavoenzyme Azobenzene Reductase AzoR from Escherichia coli Binds Roseoflavin Mononucleotide (RoFMN) with High Affinity and Is Less Active in Its RoFMN Form. Biochemistry 2013, 52, 4288–4295. [Google Scholar] [CrossRef]
- Wong, S.S.-Y.; Woo, P.C.-Y.; Luk, W.-K.; Yuen, K.-Y. Susceptibility Testing of Clostridium Difficile against Metronidazole and Vancomycin by Disk Diffusion and Etest. Diagn. Microbiol. Infect. Dis. 1999, 34, 1–6. [Google Scholar] [CrossRef]
- Aspevall, O.; Lundberg, A.; Burman, L.G.; Åkerlund, T.; Svenungsson, B. Antimicrobial Susceptibility Pattern of Clostridium Difficile and Its Relation to PCR Ribotypes in a Swedish University Hospital. Antimicrob. Agents Chemother. 2006, 50, 1890–1892. [Google Scholar] [CrossRef]
- Rizvi, N.F.; Howe, J.A.; Nahvi, A.; Klein, D.J.; Fischmann, T.O.; Kim, H.-Y.; McCoy, M.A.; Walker, S.S.; Hruza, A.; Richards, M.P.; et al. Discovery of Selective RNA-Binding Small Molecules by Affinity-Selection Mass Spectrometry. ACS Chem. Biol. 2018, 13, 820–831. [Google Scholar] [CrossRef]
- Chen, Q.; Li, Y.; Lin, C.; Chen, L.; Luo, H.; Xia, S.; Liu, C.; Cheng, X.; Liu, C.; Li, J.; et al. Expanding the DNA-Encoded Library Toolbox: Identifying Small Molecules Targeting RNA. Nucleic Acids Res. 2022, 50, e67. [Google Scholar] [CrossRef]
- McCown, P.J.; Roth, A.; Breaker, R.R. An Expanded Collection and Refined Consensus Model of GlmS Ribozymes. RNA 2011, 17, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Komatsuzawa, H.; Fujiwara, T.; Nishi, H.; Yamada, S.; Ohara, M.; McCallum, N.; Berger-Bächi, B.; Sugai, M. The Gate Controlling Cell Wall Synthesis in Staphylococcus aureus. Mol. Microbiol. 2004, 53, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Milewski, S. Glucosamine-6-Phosphate Synthase—The Multi-Facets Enzyme. Biochim. Biophys. Acta-Protein Struct. Mol. Enzymol. 2002, 1597, 173–192. [Google Scholar] [CrossRef]
- Xin, Y.; Hamelberg, D. Deciphering the Role of Glucosamine-6-Phosphate in the Riboswitch Action of GlmS Ribozyme. RNA 2010, 16, 2455–2463. [Google Scholar] [CrossRef]
- Collins, J.A.; Irnov, I.; Baker, S.; Winkler, W.C. Mechanism of MRNA Destabilization by the GlmS Ribozyme. Genes Dev. 2007, 21, 3356–3368. [Google Scholar] [CrossRef]
- Khan, M.A.; Göpel, Y.; Milewski, S.; Görke, B. Two Small RNAs Conserved in Enterobacteriaceae Provide Intrinsic Resistance to Antibiotics Targeting the Cell Wall Biosynthesis Enzyme Glucosamine-6-Phosphate Synthase. Front. Microbiol. 2016, 7, 908. [Google Scholar] [CrossRef]
- McCarthy, T.J.; Plog, M.A.; Floy, S.A.; Jansen, J.A.; Soukup, J.K.; Soukup, G.A. Ligand Requirements for GlmS Ribozyme Self-Cleavage. Chem. Biol. 2005, 12, 1221–1226. [Google Scholar] [CrossRef]
- Klein, D.J.; Ferré-D’Amaré, A.R. Structural Basis of GlmS Ribozyme Activation by Glucosamine-6-Phosphate. Science 2006, 313, 1752–1756. [Google Scholar] [CrossRef]
- Lim, J.; Grove, B.C.; Roth, A.; Breaker, R.R. Characteristics of Ligand Recognition by AglmS Self-Cleaving Ribozyme. Angew. Chem. Int. Ed. 2006, 45, 6689–6693. [Google Scholar] [CrossRef]
- Mayer, G.; Famulok, M. High-Throughput-Compatible Assay for GlmS Riboswitch Metabolite Dependence. ChemBioChem 2006, 7, 602–604. [Google Scholar] [CrossRef]
- Lünse, C.E.; Schmidt, M.S.; Wittmann, V.; Mayer, G. Carba-Sugars Activate the GlmS-Riboswitch of Staphylococcus aureus. ACS Chem. Biol. 2011, 6, 675–678. [Google Scholar] [CrossRef]
- Fei, X.; Holmes, T.; Diddle, J.; Hintz, L.; Delaney, D.; Stock, A.; Renner, D.; McDevitt, M.; Berkowitz, D.B.; Soukup, J.K. Phosphatase-Inert Glucosamine 6-Phosphate Mimics Serve as Actuators of the GlmS Riboswitch. ACS Chem. Biol. 2014, 9, 2875–2882. [Google Scholar] [CrossRef] [PubMed]
- Mandal, M.; Boese, B.; Barrick, J.E.; Winkler, W.C.; Breaker, R.R. Riboswitches Control Fundamental Biochemical Pathways in Bacillus Subtilis and Other Bacteria. Cell 2003, 113, 577–586. [Google Scholar] [CrossRef]
- Mulhbacher, J.; Lafontaine, D.A. Ligand Recognition Determinants of Guanine Riboswitches. Nucleic Acids Res. 2007, 35, 5568–5580. [Google Scholar] [CrossRef] [PubMed]
- Noeske, J.; Richter, C.; Grundl, M.A.; Nasiri, H.R.; Schwalbe, H.; Wöhnert, J. An Intermolecular Base Triple as the Basis of Ligand Specificity and Affinity in the Guanine- and Adenine-Sensing Riboswitch RNAs. Proc. Natl. Acad. Sci. USA 2005, 102, 1372–1377. [Google Scholar] [CrossRef]
- Edwards, A.L.; Batey, R.T. A Structural Basis for the Recognition of 2′-Deoxyguanosine by the Purine Riboswitch. J. Mol. Biol. 2009, 385, 938–948. [Google Scholar] [CrossRef]
- Hamal Dhakal, S.; Panchapakesan, S.S.S.; Slattery, P.; Roth, A.; Breaker, R.R. Variants of the Guanine Riboswitch Class Exhibit Altered Ligand Specificities for Xanthine, Guanine, or 2′-Deoxyguanosine. Proc. Natl. Acad. Sci. USA 2022, 119, e2120246119. [Google Scholar] [CrossRef]
- Johansen, L.E.; Nygaard, P.; Lassen, C.; Agersø, Y.; Saxild, H.H. Definition of a Second Bacillus Subtilis Pur Regulon Comprising the Pur and Xpt-PbuX Operons plus PbuG, NupG (YxjA), and PbuE (YdhL). J. Bacteriol. 2003, 185, 5200–5209. [Google Scholar] [CrossRef]
- Kim, J.N.; Blount, K.F.; Puskarz, I.; Lim, J.; Link, K.H.; Breaker, R.R. Design and Antimicrobial Action of Purine Analogues That Bind Guanine Riboswitches. ACS Chem. Biol. 2009, 4, 915–927. [Google Scholar] [CrossRef]
- Yan, L.-H.; Le Roux, A.; Boyapelly, K.; Lamontagne, A.-M.; Archambault, M.-A.; Picard-Jean, F.; Lalonde-Seguin, D.; St-Pierre, E.; Najmanovich, R.J.; Fortier, L.-C.; et al. Purine Analogs Targeting the Guanine Riboswitch as Potential Antibiotics against Clostridioides Difficile. Eur. J. Med. Chem. 2018, 143, 755–768. [Google Scholar] [CrossRef]
- Krajewski, S.S.; Isoz, I.; Johansson, J. Antibacterial and Antivirulence Effect of 6-N-Hydroxylaminopurine in Listeria monocytogenes. Nucleic Acids Res. 2017, 45, gkw1308. [Google Scholar] [CrossRef] [PubMed]
- Matyjasik, M.M.; Hall, S.D.; Batey, R.T. High Affinity Binding of N2-Modified Guanine Derivatives Significantly Disrupts the Ligand Binding Pocket of the Guanine Riboswitch. Molecules 2020, 25, 2295. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.R.; Baker, J.L.; Weinberg, Z.; Sudarsan, N.; Breaker, R.R. An Allosteric Self-Splicing Ribozyme Triggered by a Bacterial Second Messenger. Science 2010, 329, 845–848. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.D.; Shanahan, C.A.; Moore, E.L.; Simon, A.C.; Strobel, S.A. Structural Basis of Differential Ligand Recognition by Two Classes of Bis-(3′-5′)-Cyclic Dimeric Guanosine Monophosphate-Binding Riboswitches. Proc. Natl. Acad. Sci. USA 2011, 108, 7757–7762. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.G.Y.; Sudarsan, N.; Breaker, R.R. Mechanism for Gene Control by a Natural Allosteric Group I Ribozyme. RNA 2011, 17, 1967–1972. [Google Scholar] [CrossRef]
- Smith, K.D.; Lipchock, S.V.; Ames, T.D.; Wang, J.; Breaker, R.R.; Strobel, S.A. Structural Basis of Ligand Binding by a C-Di-GMP Riboswitch. Nat. Struct. Mol. Biol. 2009, 16, 1218–1223. [Google Scholar] [CrossRef]
- Kulshina, N.; Baird, N.J.; Ferré-D’Amaré, A.R. Recognition of the Bacterial Second Messenger Cyclic Diguanylate by Its Cognate Riboswitch. Nat. Struct. Mol. Biol. 2009, 16, 1212–1217. [Google Scholar] [CrossRef]
- Smith, K.D.; Lipchock, S.V.; Livingston, A.L.; Shanahan, C.A.; Strobel, S.A. Structural and Biochemical Determinants of Ligand Binding by the C-Di-GMP Riboswitch. Biochemistry 2010, 49, 7351–7359. [Google Scholar] [CrossRef]
- Furukawa, K.; Gu, H.; Sudarsan, N.; Hayakawa, Y.; Hyodo, M.; Breaker, R.R. Identification of Ligand Analogues That Control C-Di-GMP Riboswitches. ACS Chem. Biol. 2012, 7, 1436–1443. [Google Scholar] [CrossRef]
- Karaolis, D.K.R.; Rashid, M.H.; Chythanya, R.; Luo, W.; Hyodo, M.; Hayakawa, Y. C-Di-GMP (3′-5′-Cyclic Diguanylic Acid) Inhibits Staphylococcus aureus Cell-Cell Interactions and Biofilm Formation. Antimicrob. Agents Chemother. 2005, 49, 1029–1038. [Google Scholar] [CrossRef]
- Ishihara, Y.; Hyodo, M.; Hayakawa, Y.; Kamegaya, T.; Yamada, K.; Okamoto, A.; Hasegawa, T.; Ohta, M. Effect of Cyclic Bis(3′-5′)Diguanylic Acid and Its Analogs on Bacterial Biofilm Formation. FEMS Microbiol. Lett. 2009, 301, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Mano, E.; Hyodo, M.; Sato, Y.; Ishihara, Y.; Ohta, M.; Hayakawa, Y. Synthesis of Cyclic Bis(3′-5′)-2′-Deoxyguanylic/Guanylic Acid (c-DGpGp) and Its Biological Activities to Microbes. ChemMedChem 2007, 2, 1410–1413. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Qu, T.; Zhao, H.; Su, L.; Yu, Q.; Gao, J.; Wu, B. The Effect of C-Di-GMP (3′–5′-Cyclic Diguanylic Acid) on the Biofilm Formation and Adherence of Streptococcus Mutans. Microbiol. Res. 2010, 165, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Hutton, C.; Southwood, T.; Turner, J. Inhibitors of Lysine Biosynthesis as Antibacterial Agents. Mini-Rev. Med. Chem. 2003, 3, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Bugg, T.D.H.; Brandish, P.E. From Peptidoglycan to Glycoproteins: Common Features of Lipid-Linked Oligosaccharide Biosynthesis. FEMS Microbiol. Lett. 1994, 119, 255–262. [Google Scholar] [CrossRef]
- Grundy, F.J.; Lehman, S.C.; Henkin, T.M. The L Box Regulon: Lysine Sensing by Leader RNAs of Bacterial Lysine Biosynthesis Genes. Proc. Natl. Acad. Sci. USA 2003, 100, 12057–12062. [Google Scholar] [CrossRef]
- Rodionov, D.A.; Vitreschak, A.G.; Mironov, A.A.; Gelfand, M.S. Regulation of Lysine Biosynthesis and Transport Genes in Bacteria: Yet Another RNA Riboswitch? Nucleic Acids Res. 2003, 31, 6748–6757. [Google Scholar] [CrossRef]
- Klein, D.J. The Kink-Turn: A New RNA Secondary Structure Motif. EMBO J. 2001, 20, 4214–4221. [Google Scholar] [CrossRef]
- Blount, K.F.; Wang, J.X.; Lim, J.; Sudarsan, N.; Breaker, R.R. Antibacterial Lysine Analogs That Target Lysine Riboswitches. Nat. Chem. Biol. 2007, 3, 44–49. [Google Scholar] [CrossRef]
- Shiota, T.; Folk, J.E.; Tietze, F. Inhibition of Lysine Utilization in Bacteria by S-(β-Aminoethyl)Cysteine and Its Reversal by Lysine Peptides. Arch. Biochem. Biophys. 1958, 77, 372–377. [Google Scholar] [CrossRef]
- Hirshfield, I.N.; Tomford, J.W.; Zamecnik, P.C. Thiosine-Resistant Mutants of Escherichia coli K-12 with Growth-Medium-Dependent Lysyl-TRNA Synthetase Activity. Biochim. Biophys. Acta-Nucleic Acids Protein Synth. 1972, 259, 344–356. [Google Scholar] [CrossRef]
- Di Girolamo, M.; Busiello, V.; Coccia, R.; Foppoli, C. Aspartokinase III Repression and Lysine Analogs Utilization for Protein Synthesis. Physiol. Chem. Phys. Med. NMR 1990, 22, 241–245. [Google Scholar] [PubMed]
- Lu, Y.; Chen, N.-Y.; Paulus, H. Identification of AecA Mutations in Bacillus Subtilis as Nucleotide Substitutions in the Untranslated Leader Region of the Aspartokinase II Operon. J. Gen. Microbiol. 1991, 137, 1135–1143. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Patte, J.-C.; Akrim, M.; Méjean, V. The Leader Sequence of the Escherichia coli LysC Gene Is Involved in the Regulation of LysC Synthesis. FEMS Microbiol. Lett. 1998, 169, 165–170. [Google Scholar] [CrossRef]
- Ataide, S.F.; Wilson, S.N.; Dang, S.; Rogers, T.E.; Roy, B.; Banerjee, R.; Henkin, T.M.; Ibba, M. Mechanisms of Resistance to an Amino Acid Antibiotic That Targets Translation. ACS Chem. Biol. 2007, 2, 819–827. [Google Scholar] [CrossRef]
- Garst, A.D.; Héroux, A.; Rambo, R.P.; Batey, R.T. Crystal Structure of the Lysine Riboswitch Regulatory MRNA Element. J. Biol. Chem. 2008, 283, 22347–22351. [Google Scholar] [CrossRef] [PubMed]
- Serganov, A.; Huang, L.; Patel, D.J. Structural Insights into Amino Acid Binding and Gene Control by a Lysine Riboswitch. Nature 2008, 455, 1263–1267. [Google Scholar] [CrossRef] [PubMed]
- Henkin, T.M. The T Box Riboswitch: A Novel Regulatory RNA That Utilizes TRNA as Its Ligand. Biochim. Biophys. Acta-Gene Regul. Mech. 2014, 1839, 959–963. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ferré-D’Amaré, A.R. Structure and Mechanism of the T-Box Riboswitches. Wiley Interdiscip. Rev. RNA 2015, 6, 419–433. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ferré-D’Amaré, A.R. Co-Crystal Structure of a T-Box Riboswitch Stem I Domain in Complex with Its Cognate TRNA. Nature 2013, 500, 363–366. [Google Scholar] [CrossRef]
- Zhang, J.; Ferré-D’Amaré, A. The TRNA Elbow in Structure, Recognition and Evolution. Life 2016, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- Grigg, J.C.; Ke, A. Sequence, Structure, and Stacking. RNA Biol. 2013, 10, 1761–1764. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rollins, S.M.; Grundy, F.J.; Henkin, T.M. Analysis of Cis -Acting Sequence and Structural Elements Required for Antitermination of the Bacillus Subtilis TyrS Gene. Mol. Microbiol. 1997, 25, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Su, Z.; Lehmann, J.; Stamatopoulou, V.; Giarimoglou, N.; Henderson, F.E.; Fan, L.; Pintilie, G.D.; Zhang, K.; Chen, M.; et al. Structural Basis of Amino Acid Surveillance by Higher-Order TRNA-MRNA Interactions. Nat. Struct. Mol. Biol. 2019, 26, 1094–1105. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ferré-D’Amaré, A.R. Direct Evaluation of TRNA Aminoacylation Status by the T-Box Riboswitch Using TRNA-MRNA Stacking and Steric Readout. Mol. Cell 2014, 55, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Suddala, K.C.; Zhang, J. High-Affinity Recognition of Specific TRNAs by an MRNA Anticodon-Binding Groove. Nat. Struct. Mol. Biol. 2019, 26, 1114–1122. [Google Scholar] [CrossRef]
- Raina, M.; Ibba, M. TRNAs as Regulators of Biological Processes. Front. Genet. 2014, 5, 171. [Google Scholar] [CrossRef]
- Vitreschak, A.G.; Mironov, A.A.; Lyubetsky, V.A.; Gelfand, M.S. Comparative Genomic Analysis of T-Box Regulatory Systems in Bacteria. RNA 2008, 14, 717–735. [Google Scholar] [CrossRef]
- Gerdeman, M.S. In Vitro Structure-Function Studies of the Bacillus Subtilis TyrS MRNA Antiterminator: Evidence for Factor-Independent TRNA Acceptor Stem Binding Specificity. Nucleic Acids Res. 2002, 30, 1065–1072. [Google Scholar] [CrossRef]
- Means, J.A.; Hines, J.V. Fluorescence Resonance Energy Transfer Studies of Aminoglycoside Binding to a T Box Antiterminator RNA. Bioorg. Med. Chem. Lett. 2005, 15, 2169–2172. [Google Scholar] [CrossRef]
- Zapp, M.L.; Stern, S.; Green, M.R. Small Molecules That Selectively Block RNA Binding of HIV-1 Rev Protein Inhibit Rev Function and Viral Production. Cell 1993, 74, 969–978. [Google Scholar] [CrossRef]
- Wong, C.-H.; Hendrix, M.; Scott Priestley, E.; Greenberg, W.A. Specificity of Aminoglycoside Antibiotics for the A-Site of the Decoding Region of Ribosomal RNA. Chem. Biol. 1998, 5, 397–406. [Google Scholar] [CrossRef]
- Kirk, S.R.; Tor, Y. TRNA Phe Binds Aminoglycoside Antibiotics. Bioorg. Med. Chem. 1999, 7, 1979–1991. [Google Scholar] [CrossRef]
- Mikkelsen, N.E.; Brännvall, M.; Virtanen, A.; Kirsebom, L.A. Inhibition of RNase P RNA Cleavage by Aminoglycosides. Proc. Natl. Acad. Sci. USA 1999, 96, 6155–6160. [Google Scholar] [CrossRef] [PubMed]
- McPike, M.P.; Goodisman, J.; Dabrowiak, J.C. Specificity of Neomycin Analogues Bound to the Packaging Region of Human Immunodeficiency Virus Type 1 RNA. Bioorg. Med. Chem. 2004, 12, 1835–1843. [Google Scholar] [CrossRef] [PubMed]
- Blount, K.F. Using Pyrene-Labeled HIV-1 TAR to Measure RNA-Small Molecule Binding. Nucleic Acids Res. 2003, 31, 5490–5500. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hendrix, M.; Priestley, E.S.; Joyce, G.F.; Wong, C.-H. Direct Observation of Aminoglycoside−RNA Interactions by Surface Plasmon Resonance. J. Am. Chem. Soc. 1997, 119, 3641–3648. [Google Scholar] [CrossRef]
- Anupam, R.; Nayek, A.; Green, N.J.; Grundy, F.J.; Henkin, T.M.; Means, J.A.; Bergmeier, S.C.; Hines, J.V. 4,5-Disubstituted Oxazolidinones: High Affinity Molecular Effectors of RNA Function. Bioorg. Med. Chem. Lett. 2008, 18, 3541–3544. [Google Scholar] [CrossRef]
- Orac, C.M.; Zhou, S.; Means, J.A.; Boehm, D.; Bergmeier, S.C.; Hines, J.V. Synthesis and Stereospecificity of 4,5-Disubstituted Oxazolidinone Ligands Binding to T-Box Riboswitch RNA. J. Med. Chem. 2011, 54, 6786–6795. [Google Scholar] [CrossRef]
- Armstrong, I.; Aldhumani, A.H.; Schopis, J.L.; Fang, F.; Parsons, E.; Zeng, C.; Hossain, M.I.; Bergmeier, S.C.; Hines, J.V. RNA Drug Discovery: Conformational Restriction Enhances Specific Modulation of the T-Box Riboswitch Function. Bioorg. Med. Chem. 2020, 28, 115696. [Google Scholar] [CrossRef]
- Nahvi, A.; Sudarsan, N.; Ebert, M.S.; Zou, X.; Brown, K.L.; Breaker, R.R. Genetic Control by a Metabolite Binding MRNA. Chem. Biol. 2002, 9, 1043–1049. [Google Scholar] [CrossRef]
- Mironov, A.S.; Gusarov, I.; Rafikov, R.; Lopez, L.E.; Shatalin, K.; Kreneva, R.A.; Perumov, D.A.; Nudler, E. Sensing Small Molecules by Nascent RNA. Cell 2002, 111, 747–756. [Google Scholar] [CrossRef]
- Machtel, P.; Bąkowska-Żywicka, K.; Żywicki, M. Emerging Applications of Riboswitches—From Antibacterial Targets to Molecular Tools. J. Appl. Genet. 2016, 57, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Etzel, M.; Mörl, M. Synthetic Riboswitches: From Plug and Pray toward Plug and Play. Biochemistry 2017, 56, 1181–1198. [Google Scholar] [CrossRef] [PubMed]
- Roßmanith, J.; Narberhaus, F. Modular Arrangement of Regulatory RNA Elements. RNA Biol. 2017, 14, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Sinumvayo, J.P.; Zhao, C.; Tuyishime, P. Recent Advances and Future Trends of Riboswitches: Attractive Regulatory Tools. World J. Microbiol. Biotechnol. 2018, 34, 171. [Google Scholar] [CrossRef]
- Groher, F.; Suess, B. Synthetic Riboswitches—A Tool Comes of Age. Biochim. Biophys. Acta-Gene Regul. Mech. 2014, 1839, 964–973. [Google Scholar] [CrossRef]
- Seeliger, J.C.; Topp, S.; Sogi, K.M.; Previti, M.L.; Gallivan, J.P.; Bertozzi, C.R. A Riboswitch-Based Inducible Gene Expression System for Mycobacteria. PLoS ONE 2012, 7, e29266. [Google Scholar] [CrossRef]
- Han, K.; Liang, Z.; Zhou, N. Design Strategies for Aptamer-Based Biosensors. Sensors 2010, 10, 4541–4557. [Google Scholar] [CrossRef]
- Ketterer, S.; Gladis, L.; Kozica, A.; Meier, M. Engineering and Characterization of Fluorogenic Glycine Riboswitches. Nucleic Acids Res. 2016, 44, 5983–5992. [Google Scholar] [CrossRef]
- Kellenberger, C.A.; Chen, C.; Whiteley, A.T.; Portnoy, D.A.; Hammond, M.C. RNA-Based Fluorescent Biosensors for Live Cell Imaging of Second Messenger Cyclic Di-AMP. J. Am. Chem. Soc. 2015, 137, 6432–6435. [Google Scholar] [CrossRef] [PubMed]
- Jo, J.-J.; Shin, J.-S. Construction of Intragenic Synthetic Riboswitches for Detection of a Small Molecule. Biotechnol. Lett. 2009, 31, 1577–1581. [Google Scholar] [CrossRef] [PubMed]
- Pernites, R.; Ponnapati, R.; Felipe, M.J.; Advincula, R. Electropolymerization Molecularly Imprinted Polymer (E-MIP) SPR Sensing of Drug Molecules: Pre-Polymerization Complexed Terthiophene and Carbazole Electroactive Monomers. Biosens. Bioelectron. 2011, 26, 2766–2771. [Google Scholar] [CrossRef]
- Connelly, C.M.; Numata, T.; Boer, R.E.; Moon, M.H.; Sinniah, R.S.; Barchi, J.J.; Ferré-D’Amaré, A.R.; Schneekloth, J.S. Synthetic Ligands for PreQ1 Riboswitches Provide Structural and Mechanistic Insights into Targeting RNA Tertiary Structure. Nat. Commun. 2019, 10, 1501. [Google Scholar] [CrossRef]
- Hickey, S.F.; Hammond, M.C. Structure-Guided Design of Fluorescent S-Adenosylmethionine Analogs for a High-Throughput Screen to Target SAM-I Riboswitch RNAs. Chem. Biol. 2014, 21, 345–356. [Google Scholar] [CrossRef]
- Budhathoki, P.; Bernal-Perez, L.F.; Annunziata, O.; Ryu, Y. Rationally-Designed Fluorescent Lysine Riboswitch Probes. Org. Biomol. Chem. 2012, 10, 7872. [Google Scholar] [CrossRef]
- Porter, E.B.; Marcano-Velázquez, J.G.; Batey, R.T. The Purine Riboswitch as a Model System for Exploring RNA Biology and Chemistry. Biochim. Biophys. Acta-Gene Regul. Mech. 2014, 1839, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Chinnappan, R.; Dubé, A.; Lemay, J.-F.; Lafontaine, D.A. Fluorescence Monitoring of Riboswitch Transcription Regulation Using a Dual Molecular Beacon Assay. Nucleic Acids Res. 2013, 41, e106. [Google Scholar] [CrossRef]
- Pavlova, N.; Penchovsky, R. Genome-Wide Bioinformatics Analysis of FMN, SAM-I, GlmS, TPP, Lysine, Purine, Cobalamin, and SAH Riboswitches for Their Applications as Allosteric Antibacterial Drug Targets in Human Pathogenic Bacteria. Expert Opin. Ther. Targets 2019, 23, 631–643. [Google Scholar] [CrossRef]
- Antunes, D.; Santos, L.H.S.; Caffarena, E.R.; Guimarães, A.C.R. Bacterial 2′-Deoxyguanosine Riboswitch Classes as Potential Targets for Antibiotics: A Structure and Dynamics Study. Int. J. Mol. Sci. 2022, 23, 1925. [Google Scholar] [CrossRef]
- Childs-Disney, J.L.; Yang, X.; Gibaut, Q.M.R.; Tong, Y.; Batey, R.T.; Disney, M.D. Targeting RNA Structures with Small Molecules. Nat. Rev. Drug Discov. 2022, 8, 1–27. [Google Scholar] [CrossRef] [PubMed]



| Natural Riboswitches | Synthetic Riboswitches | |
|---|---|---|
| Signals | Metabolites, ions, synthetic ligands | |
| Regulated mechanism | Transcription, translation | Transcription, translation, RNAi pathway, RNA self-cleavage |
| Structural modules | Aptamer domain and expression platform | Aptamer–aptamer domain and expression platform, two riboswitches, aptamer domain and ribozyme, aptamer domain and expression platform from different riboswitches |
| Applications | Gene expression regulation | Small molecule reporters, conditional gene regulation, metabolic flux engineering, fluorescent biosensors |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giarimoglou, N.; Kouvela, A.; Maniatis, A.; Papakyriakou, A.; Zhang, J.; Stamatopoulou, V.; Stathopoulos, C. A Riboswitch-Driven Era of New Antibacterials. Antibiotics 2022, 11, 1243. https://doi.org/10.3390/antibiotics11091243
Giarimoglou N, Kouvela A, Maniatis A, Papakyriakou A, Zhang J, Stamatopoulou V, Stathopoulos C. A Riboswitch-Driven Era of New Antibacterials. Antibiotics. 2022; 11(9):1243. https://doi.org/10.3390/antibiotics11091243
Chicago/Turabian StyleGiarimoglou, Nikoleta, Adamantia Kouvela, Alexandros Maniatis, Athanasios Papakyriakou, Jinwei Zhang, Vassiliki Stamatopoulou, and Constantinos Stathopoulos. 2022. "A Riboswitch-Driven Era of New Antibacterials" Antibiotics 11, no. 9: 1243. https://doi.org/10.3390/antibiotics11091243
APA StyleGiarimoglou, N., Kouvela, A., Maniatis, A., Papakyriakou, A., Zhang, J., Stamatopoulou, V., & Stathopoulos, C. (2022). A Riboswitch-Driven Era of New Antibacterials. Antibiotics, 11(9), 1243. https://doi.org/10.3390/antibiotics11091243