2. Results and Discussion
Distribution of the ardA-H1 allele.ardA-H1 is carried by a copy of the Tn
5801 inserted into the bacterial chromosome in all the strains examined. This allele is conserved in different
S. aureus lineages found in nosocomial infections, as well as in other Gram-positive species, represented mainly by
E. faecium. For example, the first 250 genomes listed in the “bacteria” database included the HA-MRSA Mu50 (the archetype of the USA100 clone (ST5(CC5)-SCC
mecII), Acc: BA000017, 100% coverage, 100% identity); HA-MRSA JKD6009 (closely related to the BMB9393 strain (ST239(CC8)-SCC
mecIII), Acc: LR027876, 100% coverage, 100% identity);
E. faecium ISMMS_VRE_11 (Acc: CP016163, 100% coverage, 100% identity); and
Streptococcus agalactiae NGBS128 (Acc: CP012480, 98.00% coverage, 94.93% identity); in addition to other species of Gram-positive bacteria commonly found in human infections. Importantly, anti-restriction genes with a high nucleotide identity to
ardA-H1 were also found in Gram-positive cocci from animals, such as
Jeotgalibaca sp PTS2502 (Acc: CP019433, 100% coverage, 95.21% identity) isolated from pigs [
8,
9], as well as
Listeria monocytogenes L2624 (Acc: CP007686.1, 98.00% coverage, 94.93% identity), a human pathogen that can be isolated from various environmental sources [
10] (
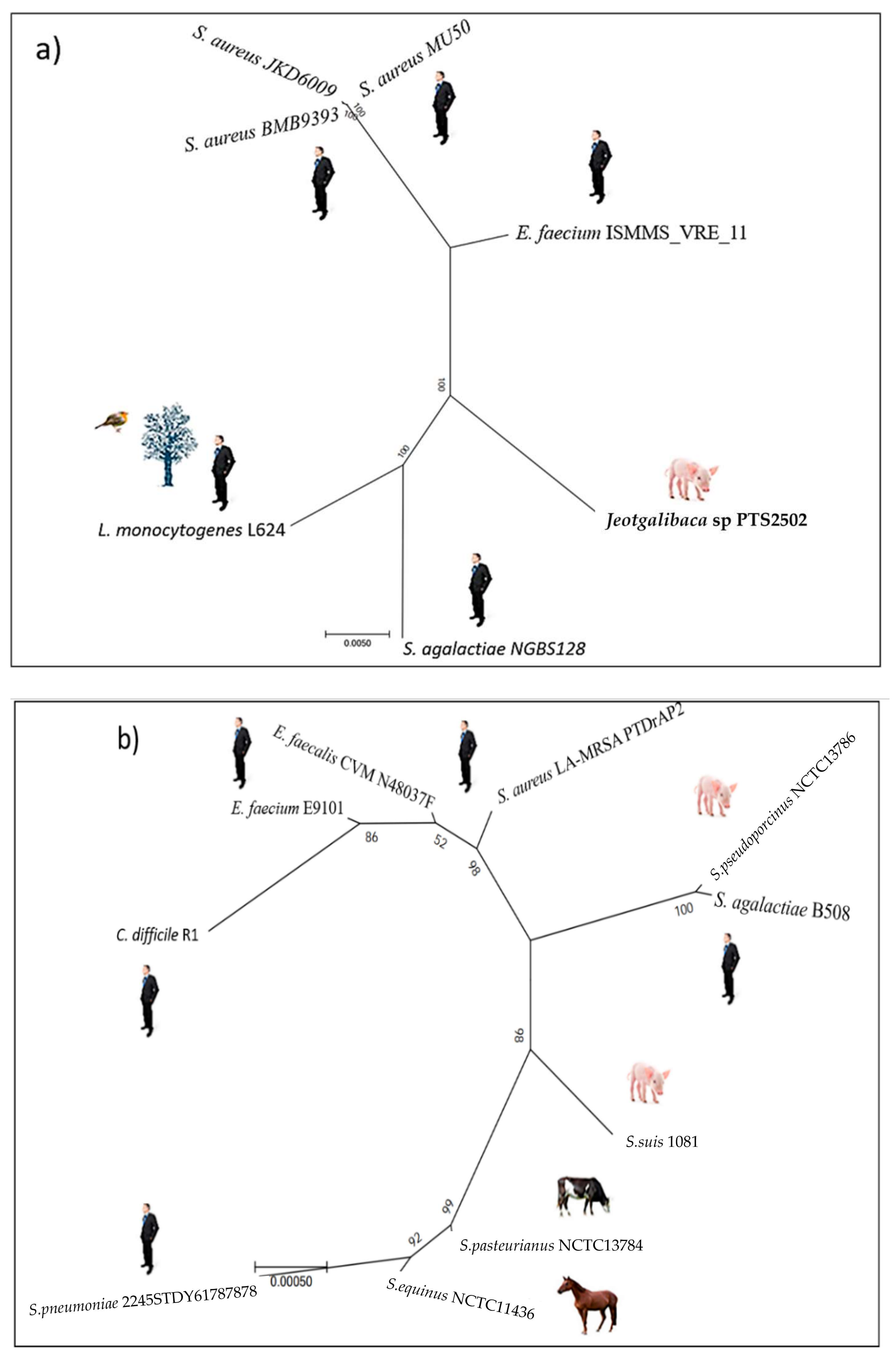
Figure 1a,
Supplementary File S1). As most of the MRSA analyzed are often collected from humans, and the
ardA-H1 allele is the main
ardA homolog among human isolates, this allele was first recognized among
S. aureus. However, toward the end of this study, a second
ardA-H allele was detected when MRSA isolated mainly from animals was included in this study. This new allele was called
ardA-H2.
ardA-H2 is widely spread in livestock MRSA (LA-MRSA). The use of Tn916 from E. faecalis DS16 as a reference led to the discovery of the allele ardA-H2, which was primarily found among LA-MRSA. Therefore, the reference sequence chosen to represent ArdA-H2 was the amino acid sequence (locus_tag: DD562_05595) carried by a Tn916 copy inserted into the chromosome sequence of the LA-MRSA strain PTDrAP2 (Acc: CP029172), which belongs to the ST398(CC398)-SCCmecV lineage.
In addition to its presence in LA-MRSA ST398-SCC
mecV,
ardA-H2 was conserved in a variety of Gram-positive bacteria (100% coverage, 100% nucleotide identity), including bacteria of clinical importance to humans, such as
Streptococcus pneumoniae 2245STDY6178787 (Acc: LR216060) and
S. agalactiae B508 (Acc: CP021770). Furthermore, Gram-positive species found in livestock animals besides LA-MRSA can carry an identical
ardA-H2, for example,
Streptococcus pseudoporcinus strain NCTC13786 (Acc: LR134341),
Streptococcus equinus strain CNU G6 (Acc: CP046629) and
Streptococcus suis strain 1081 (Acc: CP017667) (
Figure 1b,
Supplementary File S1). ArdA-H2 of the PTDrAP2 strain exhibited 63.35% amino acid identity (98% coverage, e-value: 3e-74) with ArdA-H1 of the HA-MRSA strain BMB9393. The alignment of the amino acid sequences of these two alleles in different Gram-positive bacteria, and also of
E. coli ArdA and its homologs in other Gram-negative species, showed conserved residues that were aligned unambiguously, as expected in homologous alignments (
Figure 2).
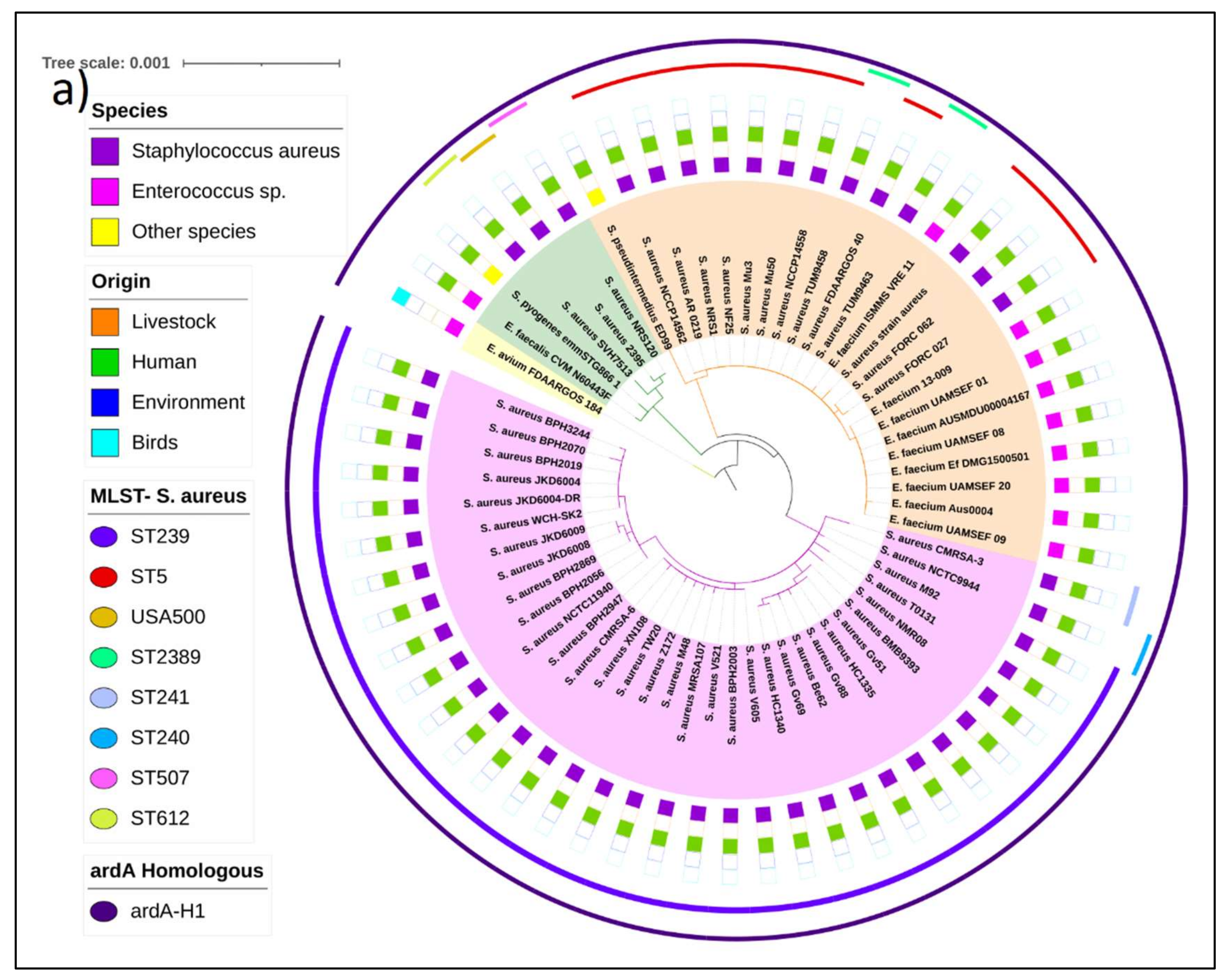
Phylogenetic trees of the Tn916 family of transposons carrying the ardA-H1 and ardA-H2 alleles. The estimated maximum likelihood (ML) tree topology for Tn
5801 transposon carrying
ardA-H1 revealed three main independent clades. The more basal genomes were from
Enterococcus avium,
E. faecalis, and
Streptococcus pyogenes, followed by three genomes of
S. aureus. It is possible that the origin of Tn
5801 from
S. aureus is in the group of catalase-negative, Gram-positive cocci. The Tn
5801 sequence of the HA-MRSA strain Mu50 and related genomes clustered in the orange clade (
Figure 3a). Intra-genus horizontal transfer of Tn
5801 from MU50-related genomes to some strains of
E. faecium is suggested by the topology of this tree (
Figure 3a, orange clade).
Another clade (pink) grouped the Tn
5801 sequence of the strain BMB9393 and other ST239 genomes from different countries, supporting the hypothesis that the acquisition of
ardA-H1 (carried by this transposon) by ST239 strains occurred before the adaptive radiation and spread of this lineage to different continents [
1].
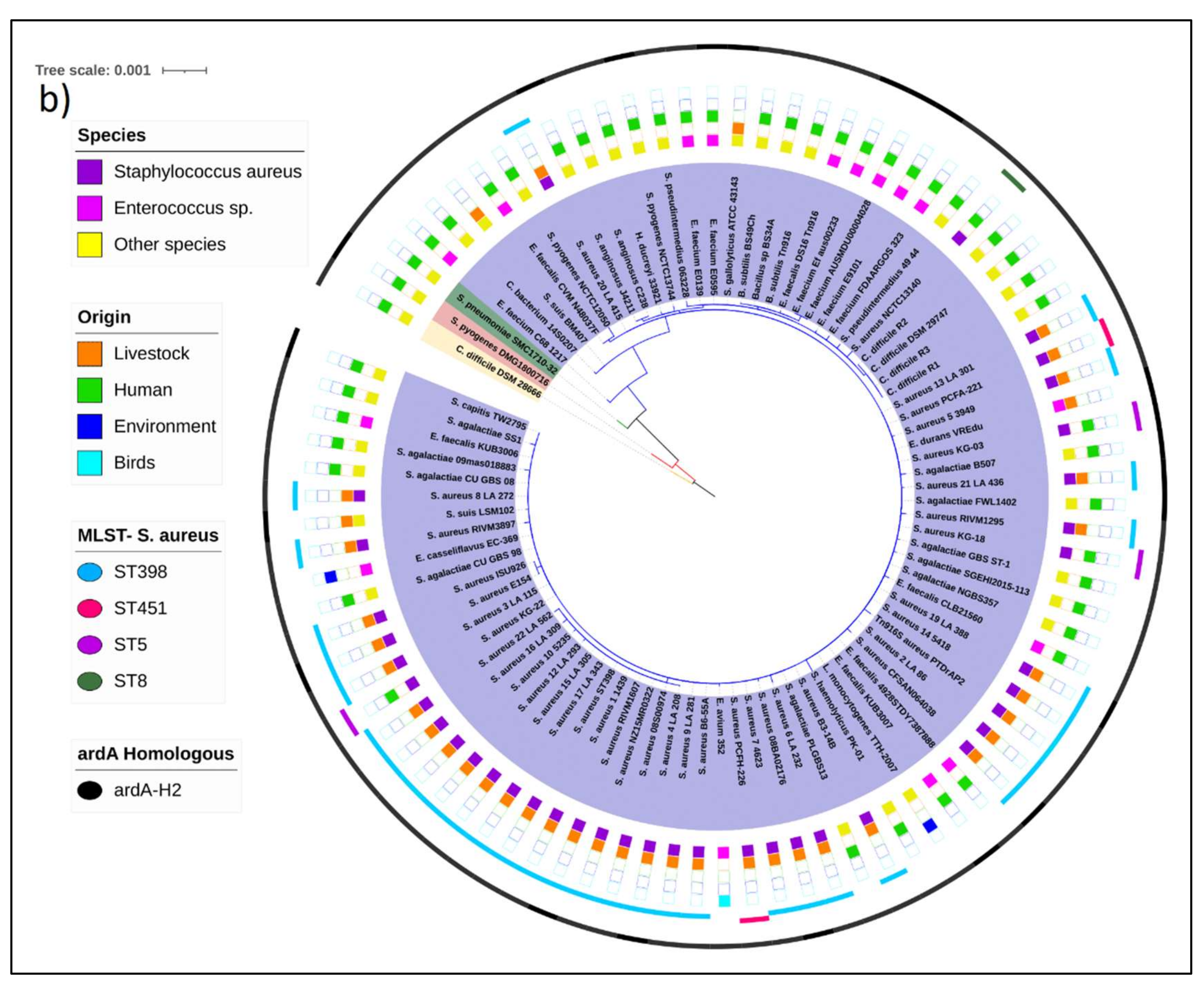
An ML tree was also constructed for the Tn
916, which carries the
ardA-H2 allele. The most basal sequences in this tree are from
Clostridioides difficile,
S. pyogenes, and
S. pneumoniae. It is notable that the Tn
916 from PTDrAP2 clustered with the transposon sequences carried by other LA-MRSA (ST398) and also by
S. agalactiae,
E. faecalis,
Staphylococcus pseudintermedius,
C. difficile, and
Bacillus subtillis, in addition to other Gram-positive species. León-Sampedro and colleagues [
11] also detected highly conserved Tn
916 elements integrated into the genomes of different Gram-positive bacteria. Taken together, these data suggest recent transmissions of
ardA-H2 between animal and human strains of a range of Gram-positive bacterial species, which is consistent with the unconstrained bacterial promiscuity promoted by
Tn916 [
11]. In contrast to Tn
5801, whose transfer appears to be more restrictive, the tree topology for Tn
916 suggests a more promiscuous transfer for the latter transposon (
Figure 3b). Despite this, our analysis suggested the occurrence of intergenus transfer of Tn
5801 from
S. aureus to
E. faecium (
Figure 3a). In fact, vancomycin resistance arose in a MU50-related strain of MRSA [
12], which was also found here as
ardA-H1 (+). In some reported cases, the vancomycin-resistant
S. aureus (VRSA) emerged during co-infection with vancomycin-resistant
Enterococcus (VRE), suggesting that gene transfers from
Enterococcus sp to
S. aureus can also occur in vivo during natural infections [
13,
14]. Thus, all these data suggest that the exchange of genetic material between
S. aureus and
Enterococcus sp can occur in both directions.
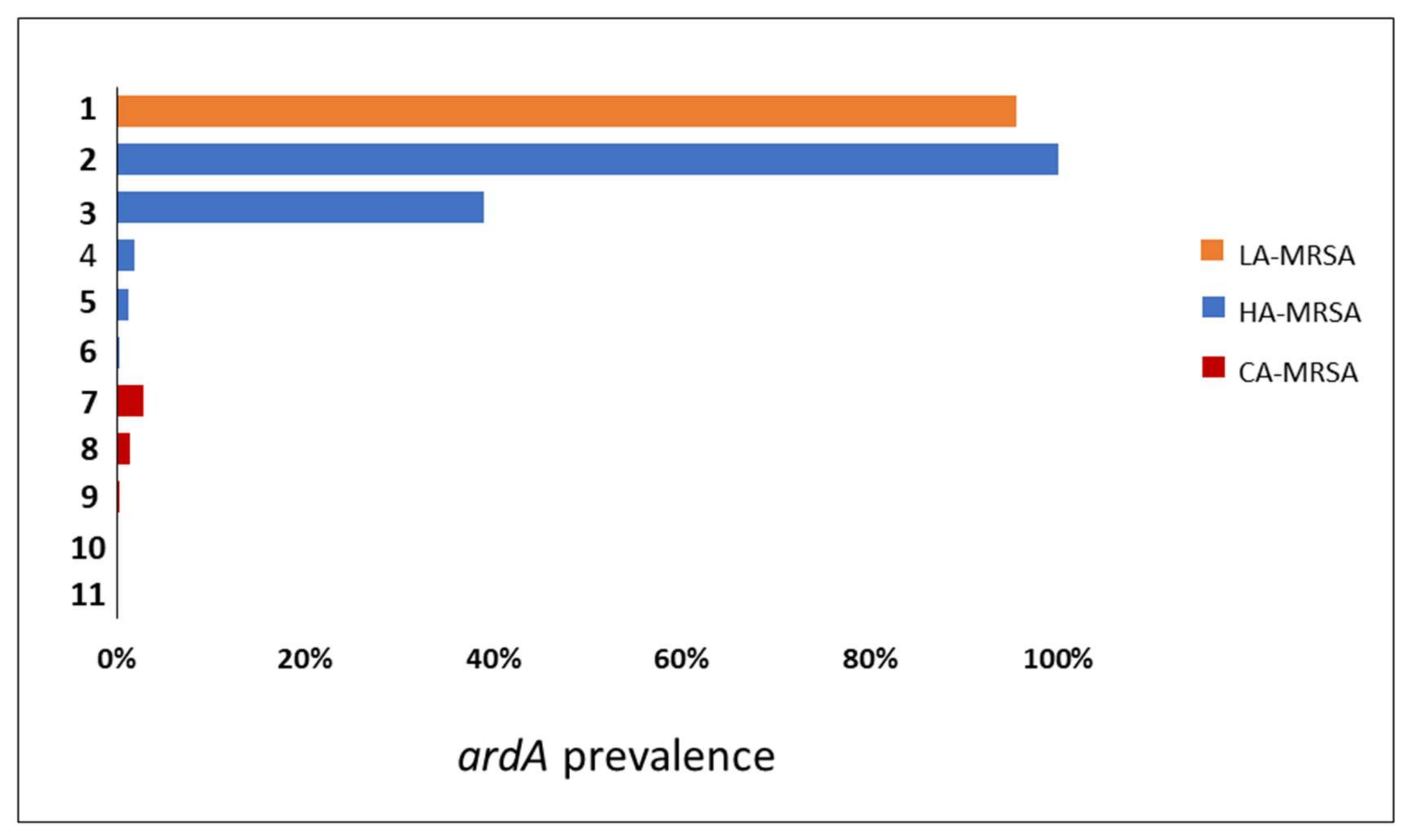
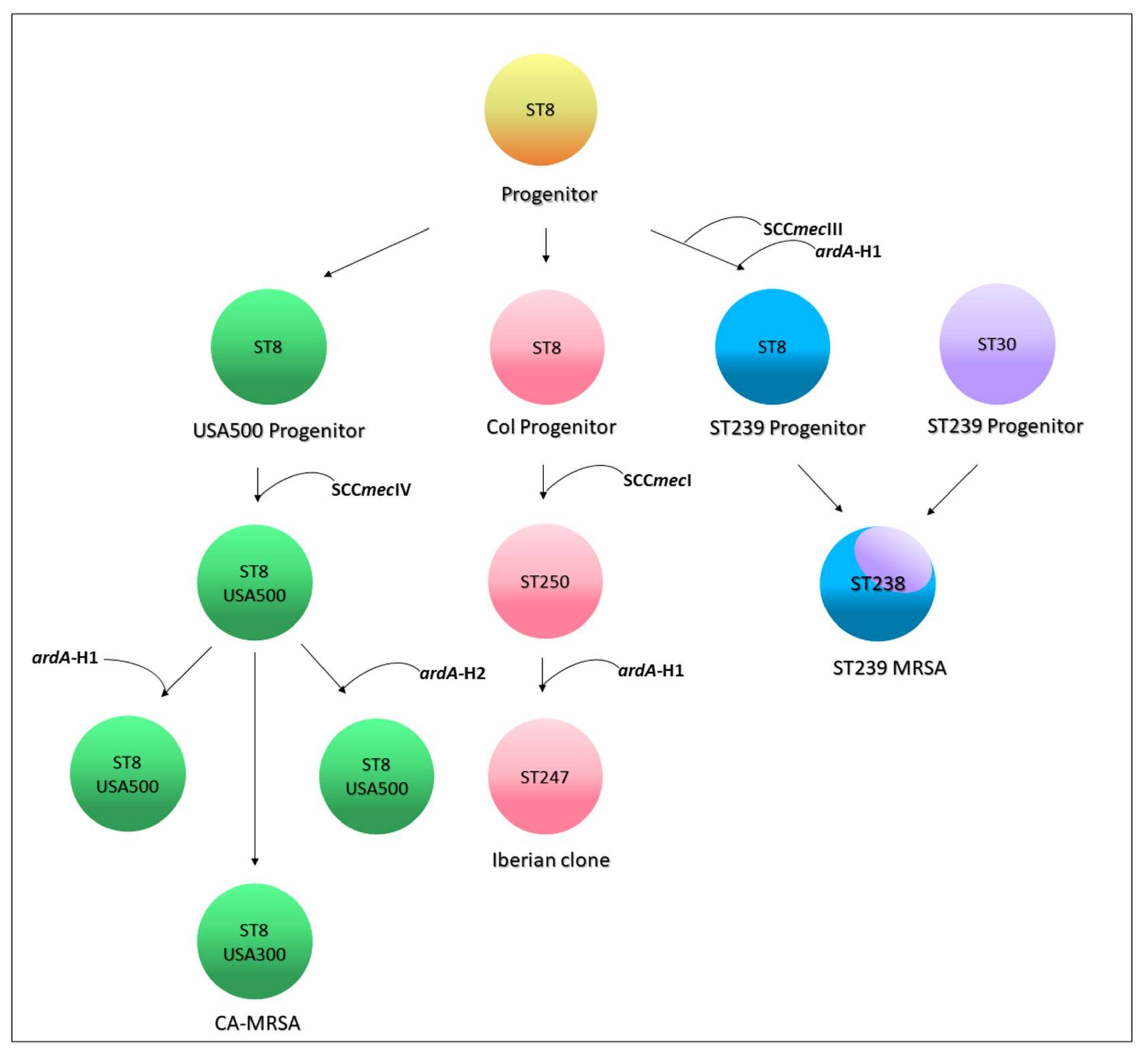
Searching for ardA-H in contemporary MRSA lineages. The search for
ardA-H1 in 6215
S. aureus genomes showed that this allele was most associated with CC8 genomes of HA-MRSA, including those related to multidrug-resistant strains of the ST239(CC8)-SCC
mecIII lineage (
n = 224/224; 100%) and to HA-MRSA strains named USA500 (
n = 132/339; 39%) (
Figure 4). Based on the evolution of ST8 [
1,
15], and the analyses performed here, we proposed that the
ardA-H1 was introduced into a CC8 genome ST239 progenitor probably before the ST239 diversification and worldwide spread of this HA-MRSA lineage [
1]. This model is also supported by the fact that all the ST239 genomes deposited in the GenBank carry
ardA-H1. However, in USA500(CC8)-related genomes,
ardA appears to have entered after USA500 diversification, because genomes that lack
ardA-H, contain
ardA-H1, and a few with
ardA-H2 were found in this study (
Figure 5). In addition to CC8,
ardA-H1 was detected at a lower frequency among CC5, CC22, and CC45 genomes. The
ardA-H2 allele, however, was almost universally found in strains related to LA-MRSA of the ST398 lineage (
n = 746/780; 95.64%) (
Figure 4). Likewise, this gene was present in the genomes of other ST analyzed related to ST398, including ST3783, ST3226, ST3709, ST752, ST3706 and ST541.
Notably,
ardA-H (mainly
ardA-H1) was found at a very low frequency in the 2384 genomes that belong to CCs commonly associated with CA-MRSA (
n = 16/2384; 0.34%), including those related to the clones USA300 (CC8;
n = 2/1447; 0.14%), USA400 (CC1;
n = 2/157; 1.27%), and USA1100 (CC30;
n = 12/438; 2.74%) (
Figure 4;
p < 0.0001). Among the genomes related to HA-MRSA, the prevalence of
ardA-H was 12.4% (
n = 379/3052). The presence of
ardA-H1 was particularly detected among CC8 MRSA-related genomes (ST239-SCC
mecIII and USA500), strains classically involved in nosocomial infections and known to present high-level multiresistance (356/563; 63.2%) (
Figure 4). It can be argued that the high number of resistance traits found in these hospital-associated CC8 genomes (ST239-SCC
mecIII and USA500) could be due to the intrinsic characteristics of the CC8 background and not to the presence of
ardA-H1 in the bacterial chromosome. However, contrary this assumption is the fact that among the genomes (
n = 1447) of the ST8 (CC8) strains related to the highly susceptible CA-MRSA, USA300 [
16], only two genomes carry
ardA-H2.
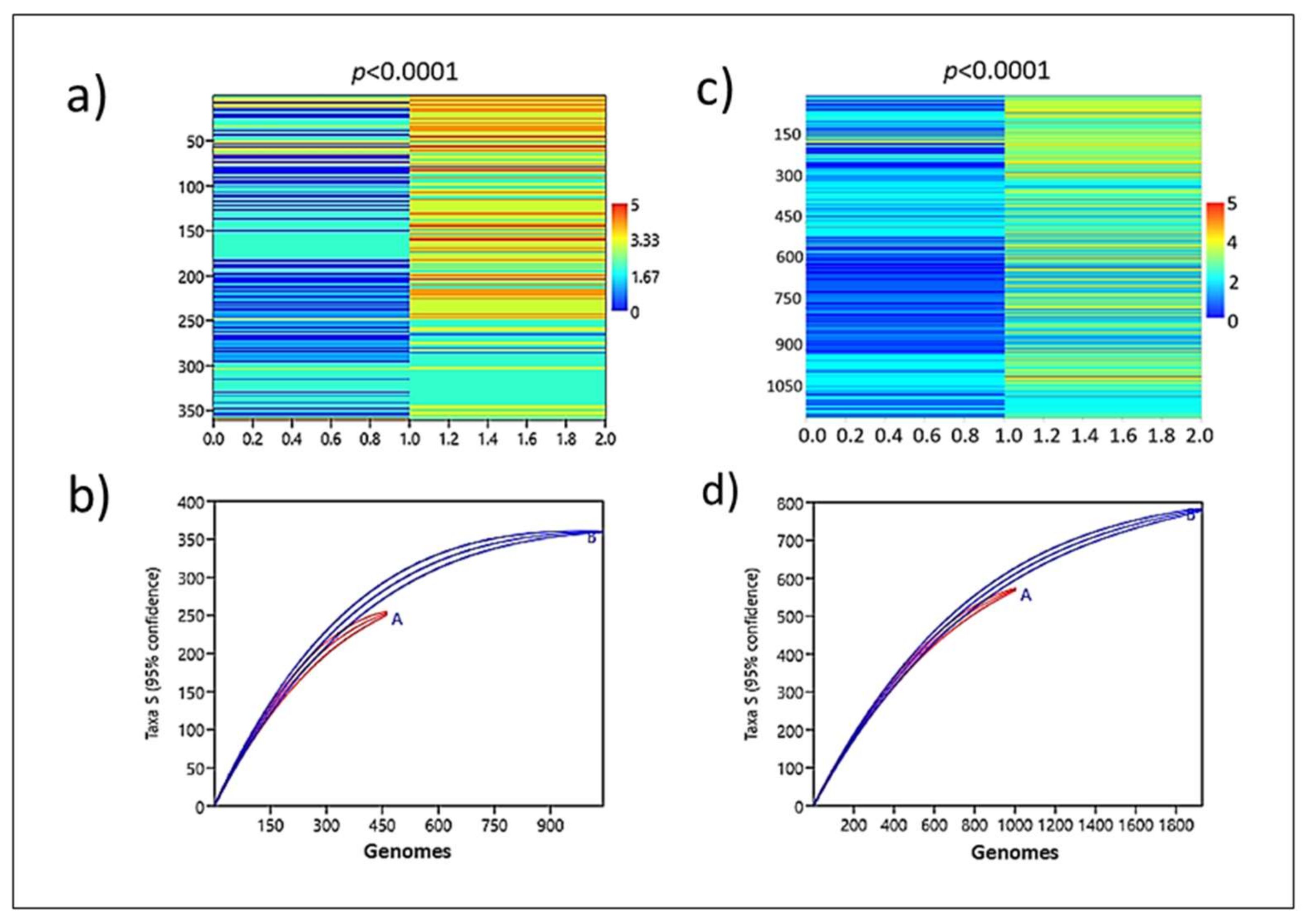
Analysis of the enrichment of the resistance gene. Corroborating a role played by
ardA-H in the acquisition of resistance genes, the genomic group formed by
ardA-H1 (+) accumulated significantly more resistance traits (
p < 0.0001) compared to that formed by
ardA-H (−) (
Figure 6a,b;
Supplementary File S2). Again, supporting these data, a strong association between multidrug resistance and the presence of
ardA-H2 in these genomes was also detected. Likewise, the enrichment analysis shows that resistant genes are clearly over-represented in
ardA-H2 (+) compared to
ardA-H (−) genomes (
Figure 6c,d). Thus, these data indicate an increased acquisition of antimicrobial-resistant traits in
ardA-H1 and
ardA-H2 populations of
S. aureus.
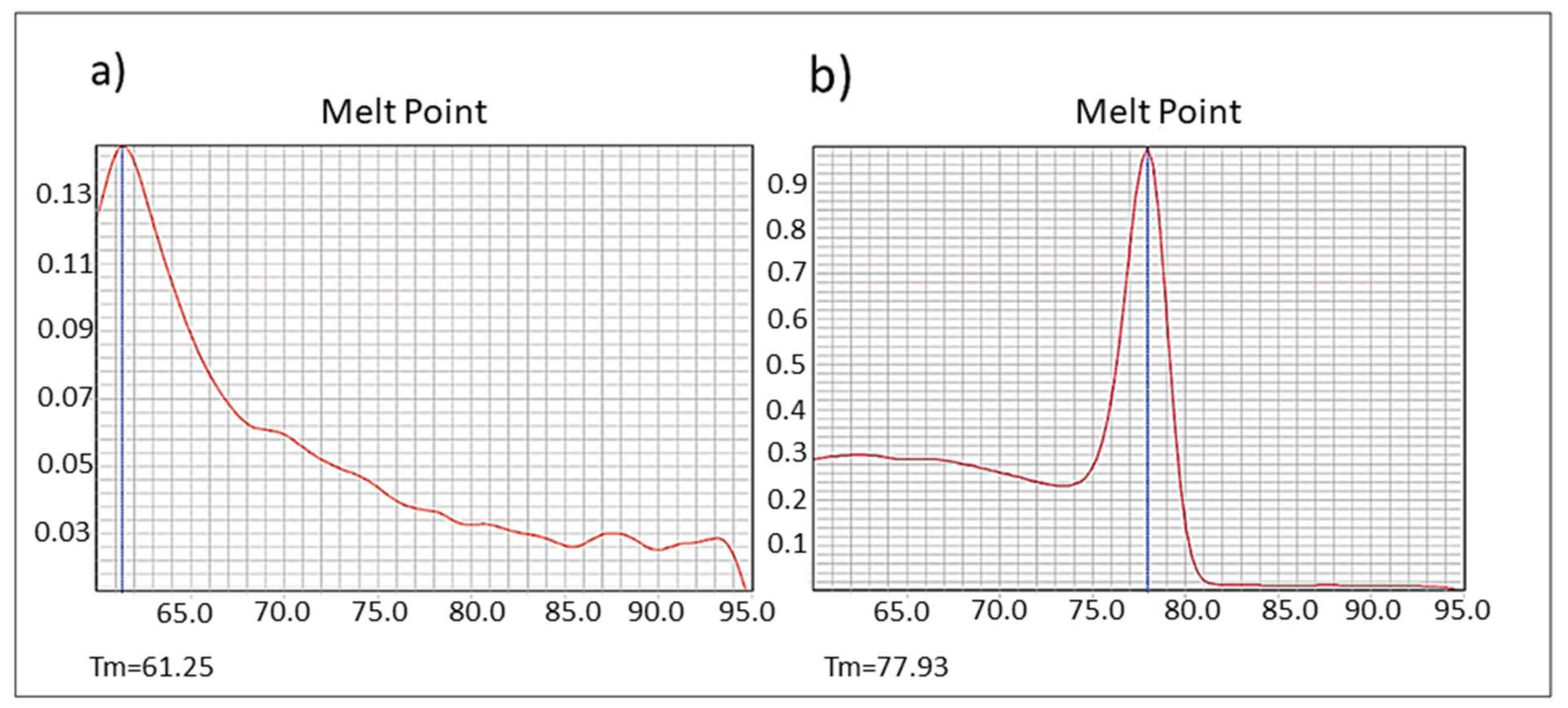
ardA-H1 expression as dsDNA. Some anti-restriction genes, such as the
E. coli ardC gene, are expressed in the recipient strain as dsDNA; also, the protein is not exported from the donor to the recipient cells, as demonstrated by González-Montes et al. 2020 [
17]. Therefore, this mechanism may increase gene acquisition in the bacterial hosts. Those authors also suggested that inhibition of ArdC activity could provide a new tool to hinder the transmission of multidrug resistance [
17]. However, other authors have hypothesized that
ardA could be transcribed from single-stranded (conjugative) DNA (ssDNA), prior to the synthesis of complementary DNA in the recipient strain, possibly due to the presence of specific secondary structures formed by ssDNA [
18]. As also demonstrated for
ardC [
17], here, we show that the
ardA-H1 naturally present in the genome of the BMB9393 was expressed in the form of dsDNA (
Figure 7a,b).
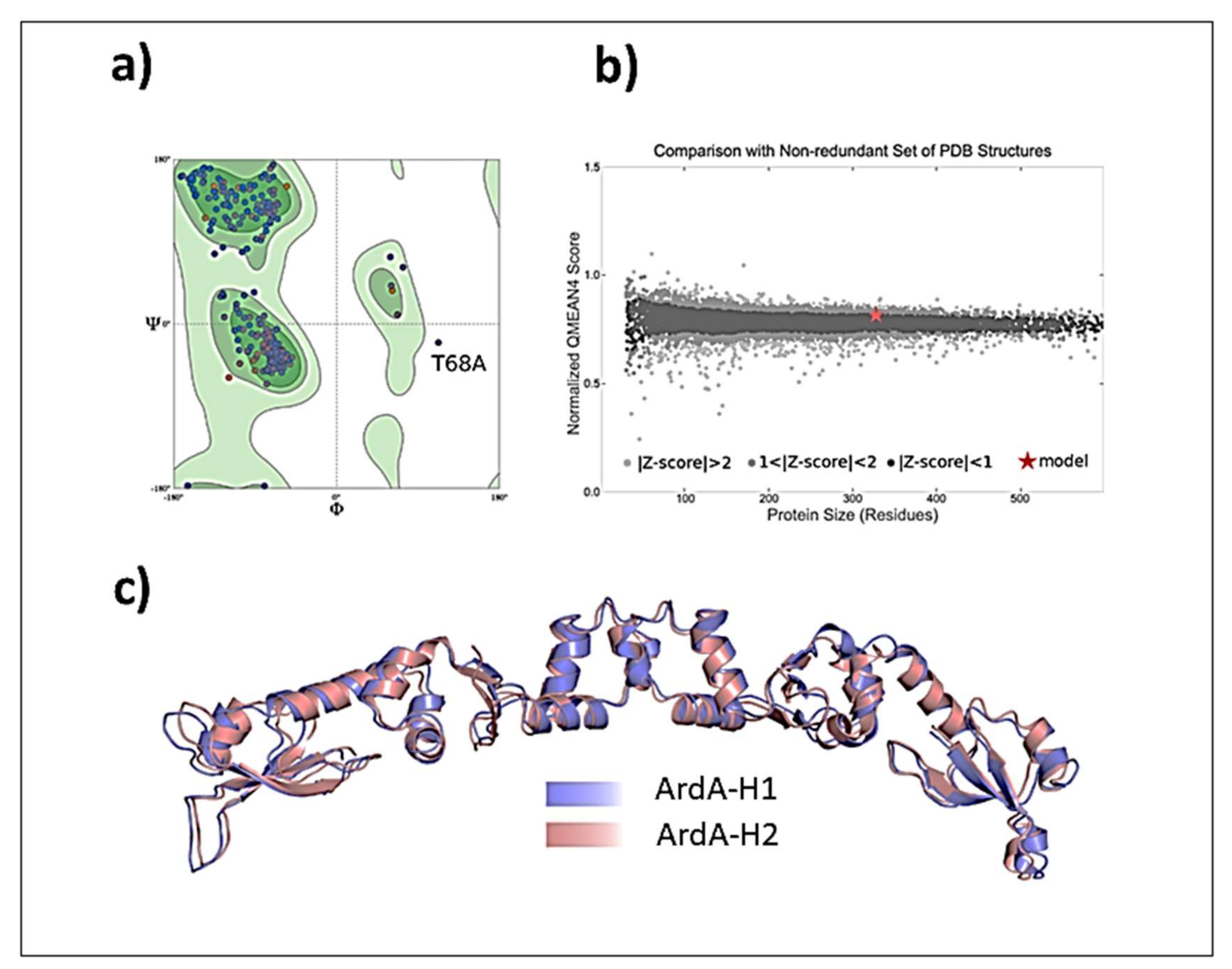
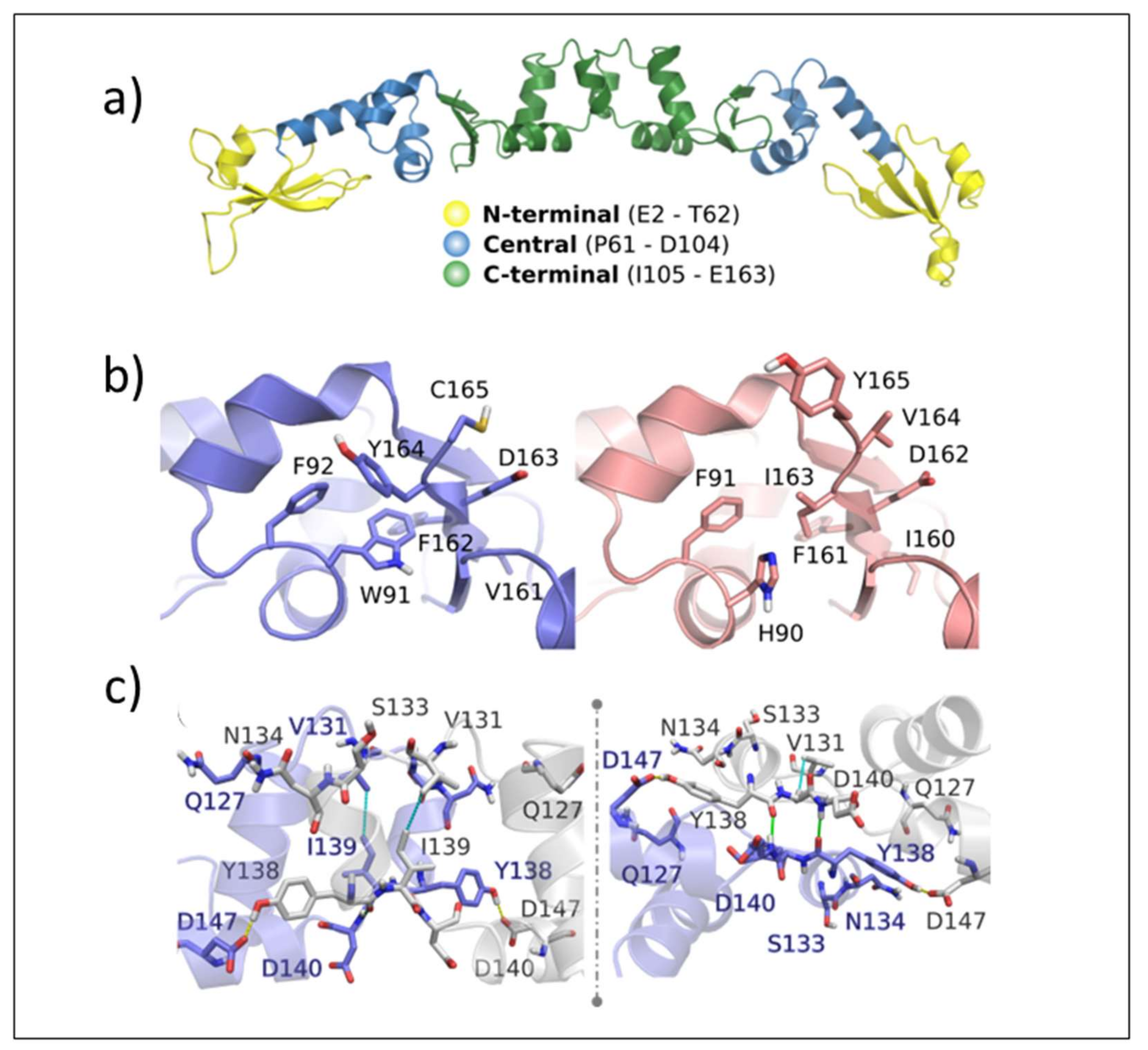
Validation of ArdA-H1 model. The analyses performed with MolProbity indicated that the model constructed for ArdA-H1 presented 96.91% of all residues in favored regions and only one outlier residue was located in the central domain (Thr78 from chain A) (
Figure 8a). The QMEANDisCo result shows good energetic and geometric quality, with an overall score of 0.79 ± 0.05 (
Figure 8b). Due to the high similarity between the protein sequences, ArdA-H1 shares the same folding of ArdA-H2 with preserved secondary structures (
Figure 8c). ArdA-H1 shares the same 3D arrangement of ArdA-H2 with each monomer of the dimeric structure containing the following three domains: (i) N-terminal (E2-T62), (ii) central (P61-D104), and (iii) C-terminal (I105-E165) (
Figure 8a). The most important amino acid residues for ArdA-H2 are listed in
Supplementary Table S1. These analyses focused on the central and C-terminal domains because previous work revealed that the N-terminal domain is not important for the anti-restriction activity, as it can be deleted without affecting the binding against the MTase core [
19]. The residues previously described as essential for the structural integrity of the ArdA-H2 protein and the anti-restriction activity [
6,
19] are highly conserved between the ArdA-H1 and ArdA-H2 structures (
Figure 9a,b). In the anti-restriction motif (
Figure 9c), ArdA-H1 contains residues Gln127, Ser133, and Asn134 instead of Ala126, Ala131, and Ser133, respectively, in ArdA-H2 (not shown). These residues are exposed to solvents in both structures, and their presence in ArdA-H1 might provide additional polar (e.g., hydrogen bonds) interactions during the molecular recognition process. Despite these differences, the high sequence and structural similarities of those proteins suggest that these alleles are likely to be active in vivo.
ardA-H1 effectively increases exogenous DNA incorporation into S. aureus cells. The amino acid sequence of the
E. faecalis ardA homolog (Tn
916, ORF 18) [
6] is identical to that of the ArdA-H2 allele found in LA-MRSA (
Figure 2). In fact, this homolog was effective in blocking restriction activities in vivo when ORF18 was cloned in
E. coli that expressed different type I RM families [
6]. Furthermore, ArdA-H2 has the same 3D arrangement and overlaps with the experimentally defined structure of ArdA ORF18. Thus, we chose to clone the
ardA-H1 gene from BMB9393, a hospital strain of the ST239(CC8)-SCC
mecIII lineage, one of most important MRSA in human infections, to test the effect of
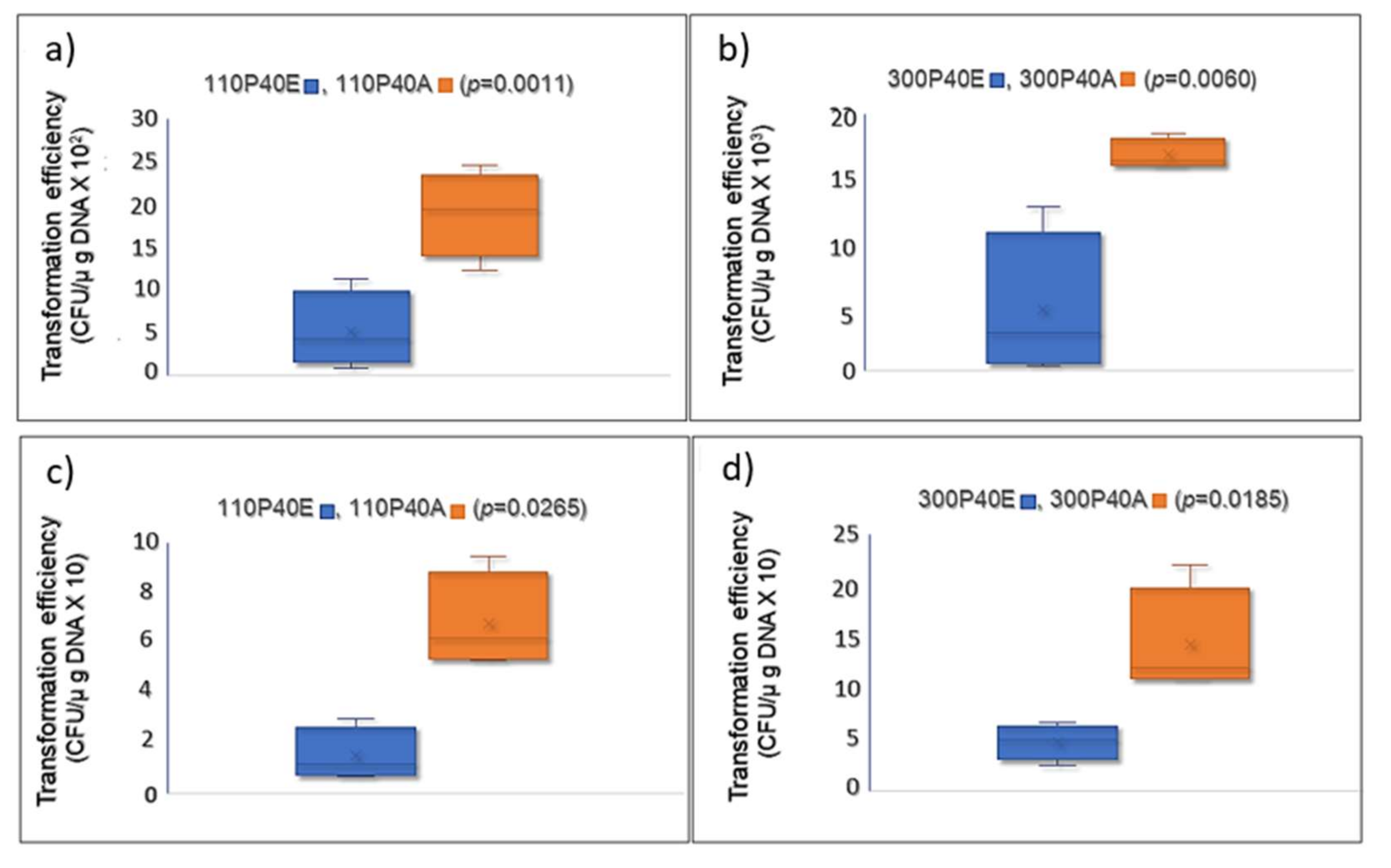
ardA-H1 on gene transfer using two clinical CA-MRSA strains of different genetic backgrounds. Our data clearly show increased acquisition of DNA for the clones that expressed
ardA-H1. The rates of
cat gene acquisition (carried by pBMB9393) by strains 110P40A and 300P40A, both expressing ArdA
-H1, were increased 3.67- and 3.53-fold, respectively, compared with 110P40E and 300P40E, the corresponding isogenic
ardA-H1 (−) strains (
Figure 10a,b). A similar increase (3.00–3.53 folds) in DNA transfer was observed when heterologous
E. coli DNA was transformed into isogenic strains (
Figure 10c,d).
The rates of
cat gene acquisition (carried by pBMB9393) by strains 110P40A and 300P40A, both expressing ArdA
-H1, were increased 3.67- and 3.53-fold, respectively, compared with 110P40E and 300P40E, the corresponding isogenic
ardA-H1 (−) strains (
Figure 10a,b). A similar increase (3.00–3.53 folds) in DNA transfer was observed when heterologous
E. coli DNA was transformed into isogenic strains (
Figure 10c,d). Consistent with these findings, McMahon and colleagues [
6] demonstrated the in vitro heterologous activity of ArdA homologs from different bacterial species against type I RM systems, including MRSA Mu50 (allele H1).
However, the
ardA-H1 gene from MU50 cloned in
E. coli failed to induce high levels of protein expression [
6], and those authors did not demonstrate its role in DNA transfer in
S. aureus.
Taken together, our study shows that the
ardA-H1 of the BMB9393 strain is transcribed in dsDNA format and indicates that the ArdA-H1 protein is expressed and active, as demonstrated by the increased transfer of exogenous DNA to pCN40::
ardA-H1 clones in comparison with isogenic strains
(ardA-negative), as shown previously for
ardA-H2 [
6,
7]. These data are further validated by the spatial sequence similarity between ArdA-H1 and ArdA-H2 proteins, as demonstrated here. Furthermore, we revealed that the genomes of strains ST239 (CC8; HA-MRSA) and ST398 (CC398; LA-MRSA) that carry
ardA-H alleles (
ardA-H1 and
ardA-H2; respectively) are enriched with antimicrobial resistance genes, while these genes were poorly detected in the genomes of
S. aureus lineages associated with the very susceptible CA-MRSA strains. Therefore, our data indicate that
ardA-H1 plays a role in promoting gene acquisition in MRSA and, consequently, might influence the dissemination of resistance genes in different environments.
3. Material and Methods
Searching for ardA gene in a local S. aureus database. A database, consisting of 6215 genomic sequences, was created in this study to encompass all genomic sequences available in the GeneBank of the most important MLST clonal complexes (CCs) of MRSA circulating in humans and animals (
Supplementary File S3,
http://data.mendeley.com/datasets/mkwvsp8rhg (published on 19 April 2022)). The two
ardA homolog alleles detected during this investigation in
S. aureus were named
ardA-H1 (501 bp) and
ardA-H2 (498 pb) and correspond to the nucleotide sequences found in strains BMB9393 (ST239 (CC8)) (Acc: CP005288; locus_tag: SABB_RSO2195) and PTDrAP2 (ST398(CC398)) (Acc: CP029172; locus_tag: DD562_05595), respectively. These sequences were used as references to search for
ardA homologs in the local
S. aureus database using basic local alignment search tool (BLAST) command line applications (
https://www.ncbi.nlm.nih.gov/books/NBK279690/ (accessed on 3 May 2019)). As we found
ardA-H1 located in the Tn
5801 transposon, which belongs to the Tn
916 family, the complete nucleotide sequence of Tn
916 of the
Enterococcus faecalis DS16 (Acc: U09422.1) was also used as a reference sequence for the search for
ardA homologs in the local database. In addition, BLASTn (
https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 3 May 2019)) was run using default parameters in the NCBI databases “bacteria (taxid:2)” and “
Enterococcus (taxid: 1350)”. The UniProtKB BLAST tool (
http://www.uniprot.org/blast/ (accessed on 20 May 2019)) with default parameters was also run to look for BMB9393 anti-restriction protein homologs (Uniprot accession number (UAcc): A0A2 × 2LRV4) in
S. aureus and other bacterial species. This same application was performed using the
ardA amino acid sequence of the
E. coli strain SE11 (UAcc: B6IC30). Subsequently, representative sequences of
ardA homologs from different Gram-positive and Gram-negative species were selected based on sequence differences. These sequences were aligned using Clustal Omega (1.2.4-
https://www.ebi.ac.uk/Tools/msa/clustalo (accessed on 20 May 2019)). The location of the
ardA homologs in
S. aureus genomes was examined using IslandViewer 4 with integrated prediction methods [
20] and the MicroScope genomic platform [
21].
Phylogenetic trees of transposable elements carrying homologs of ardA (ardA-H). To build a phylogenetic tree for Tn
5801, the sequences were selected by defining a cut-off value of 99.90% nucleotide identity and 91.00% coverage (
Supplementary File S4), using as a reference the Tn
5801 sequence of the strain ST239 (CC8) named BMB9393 (Acc: CP005288; coordinates 451.953 to 480.229). These sequences were aligned using REALPHY. The maximum likelihood tree was obtained by applying Mega7 algorithms with default parameters using the Tamura-Nei model with 1000 bootstrap replicates [
22]. The Tn
5801 sequence of the strain
E. avium FDAARGOS 184 (Acc: NZ_CP024590) was used to root the tree. For the phylogenetic tree of Tn
916, the reference was the nucleotide sequence of Tn
916 of the strain ST398 (CC398) named PTDrAP2 (Acc: CP029172; coordinates: 1,064,554 to 1,082,585), and the same cut-off value was applied for the selection of 41 Tn
916 sequences from
S. aureus and 48 from other Gram-positive species (
Supplementary File S4). The Tn
916 phylogenetic tree was constructed as described. The Tn
916 sequence of the strain
C. difficile DSM 28666 (ACC: NZ_CP012321) was used to root the tree. The Interactive Tree Of Life (iTOL) v.4 tool was used to visualize the trees [
23].
Gene set enrichment analysis for the resistance genes. Whole genomes carrying
ardA-H and an equal number of sequences that lacked these genes were randomly selected from the
S. aureus local database (
Supplementary File S2), using the Kutools for Excel 24.0 (Informer Technologies, Inc). The selected genomes were used to test for a statistical association between the absence/presence of
ardA-H and the acquisition of multidrug resistance in
S. aureus. Data on resistant genes were collected using the ResFinder 3.1 database composed only of genes acquired horizontally [
24], using threshold values of 90% and a coverage of 90%. This analysis was performed for all 360
ardA-H1 and 782
ardA-H2 genomes available on GenBank, balanced by the same number of genomes that lacked these genes (
ardA-H (−) genomes) (
Supplementary File S2). The Markov chain transition matrix was used to analyze the process of gene acquisitions in the state transition of
ardA-H (−) and
ardA-H (+) genomes. The χ
2 test was used to calculate the probability that data would be taken from a system with random proportions of transitions (i.e., no preferred state). This analysis was performed using Past software 4.03 [
25]. Multidrug resistance was defined as resistance to four antimicrobial drugs, including beta-lactams. High-level multidrug resistance was defined as resistance to more than four antimicrobials, including beta-lactams.
Three-dimensional (3D) structures of ArdA-H allele-1 and allele-2. Currently, there is a high-resolution 3D structure of ArdA-H2 in the Protein Data Bank (PDB), which has been solved experimentally (PDB code 2W82,
E. faecalis) by means of X-ray diffraction with a resolution of 2.8 Å. As no experimental structure of ArdA-H1 is available at PDB, we built the 3D homodimeric structure by comparative modelling with the SWISS-MODEL web server [
26,
27], available at
https://swissmodel.expasy.org/ (accessed on 21 October 2019), using the ArdA-H2 structure as a template found by HHblits (62.58% amino acid sequence identity and 49.00% similarity. The 3D ArdA-H1 and ArdA-H2 structures were further prepared using the Protein Preparation Wizard tool of the Maestro software [
28]. In this step, all hydrogen atoms were added using PROPKA [
29]. The ArdA-H1 model was further validated according to geometric and energetic properties in MolProbity [
30] and QMEAN (QMEANDisCo scoring function) [
27], which are available as structure assessment tools in the SWISS-MODEL web server.
The role of the ardA-H1 from strain BMB9393 in gene transfer. The
ardA-H1 sequence was amplified using DNA (kit Wizard Genomic DNA Purification; Promega, Madison, WI, EUA) from the HA-MRSA strain BMB9393 and the primers
ardAcompFwd/Rev (
Supplementary Table S2) to provide a 574 bp amplicon. The Platinum Taq DNA Polymerase High Fidelity was used, and the PCR protocol was performed as suggested by the manufacturer (Invitrogen; Waltham, MA, USA), at a melting temperature of 55°C. The PCR product was cloned into the
E. coli p-GEM vector-(Promega), and then transferred to the
S. aureus vector pCN40 [
31]. The pCN40::
ardA-H1 (pACN40) and the also empty pCN40 were transferred by electroporation to
E. coli DC10B [
32], and to the CA-MRSA clinical strains 07-040 (related to USA1100 clone) and USA300-114 (related to USA300 clone) to generate 110P40A (derived from 07-040) and 300P40A (derived from USA300-114). In addition, isogenic strains (110P40E and 300P40E) that naturally lack
ardA-H were obtained by transforming the empty pCN40 plasmid into wild type
S. aureus strains (07-040 and USA300-114, respectively). All procedures used for the transformation were previously described by Monk and colleagues [
32].
Table 1 and
Table 2 describe the strains and the plasmids used and/or constructed in this study, respectively.
Clones were confirmed by PCR and Sanger sequencing, which was performed at PSEQDNA (Rio de Janeiro, RJ, Brazil). Expression of
ardA-H1 was confirmed in the transformants by real-time qRT-PCR (Step One Real-Time PCR System (Applied Biosystem; Foster City, CA, USA)) using the Power SYBR Green RNA-to-Ct
TM 1-Step Kit (Applied Biosystem), as described by the manufacturer’s manual. RNA samples from the isogenic strains 07-040 and USA300-114 (
ardA-H negatives) were also included as negative controls. For these experiments, we tested three biological replicates with three technical replicates for each. The primers used are listed in
Supplementary Table S2.
Table 1 and
Table 2 list the strains and plasmids used in this work, respectively. The transformation experiments were performed using 1000 ng of the pLI50 shuttle vector (Addgene; Teddington, UK), recovered from
E. coli DC10B, or 30 ng of pBMB9393 (Acc: CP005289.1), a native
S. aureus plasmid of MRSA strain BMB9393 [
1].
Electrocompetent cells were prepared [
32] for the clones that expressed
ardA-H1 (
ardA-H1 (+)) and for the corresponding isogenic strains 110P40E and 300P40E, which naturally lack
ardA-H (
ardA-H (−)). The electroporation was performed exactly as described by Monk and colleagues [
32]. Transformants were selected using trypticase soy broth containing 10 µg/mL of chloramphenicol (Cm10), the selectable marker of the plasmids used. Three independent biological replicates were performed with three technical replicates (colony count) for each condition. Transformation efficiency (TfE) was determined using the following formula: TfE = CFU/µDNA. Unpaired, two-tailed, Student’s
t-test was used for statistical analysis for each pair of strains
ardA (+) and
ardA (−). Normality tests and
t-tests were performed using GraphPad Prisma 9.4.
To assess whether ardA-H1 is expressed as dsDNA, we performed real-time qRT-PCR as described above, using cDNA from the strain BMB9393 (Acc: CP005288) that carries the ardA-H1 gene in ICE Tn5801 and from the strain CR15-051, an ST30(CC30)-SCCmecIV MRSA, whose genome was previously sequenced by us (Acc: JAEAMC000000000) and does not carry the ardA-H gene.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}