3.1. Chemical Synthesis
The reactions were carried out under an argon atmosphere, if required, and were monitored by thin-layer chromatography. All microwave-mediated reactions were carried out in G10 or G20 Anton Paar vials, using an Anton Paar Monowave 300 Extra microwave synthesizer. Flash chromatography was performed with silica gel 60 (40–63 μm). Spectroscopic
1H and
13C NMR, MS and/or analytical data were obtained using chromatographically homogeneous samples.
1H NMR (500 MHz) and
13C NMR (125 MHz) spectra were recorded in CDCl
3 unless otherwise indicated. Chemical shifts (δ) are reported in ppm, and coupling constants are given in Hz. For each compound, detailed peak assignments have been made according to COSY, HSQC and HMBC experiments. The numbering of molecules is indicated in the
Supplementary Material. Optical rotations were measured with a sodium (589 nm) lamp at 20 °C. IR spectra were recorded on an FT–IR spectrophotometer, and the wavelengths are reported in cm
−1. High-resolution mass spectra (HRMS) were recorded with a TOF mass analyzer under electrospray ionization (ESI) in positive ionization mode detection [
42].
3.1.1. Protected Nitrile 1
To a solution of E (100 mg 0.19 mmol, 1 equiv.) and D (192 mg, 78 mmol, 4 equiv.) in freshly distillated DCM (7 mL) was added 4 Å molecular sieves (1.4 g). After stirring at r.t. for 1 h, boron trifluoride diethyl etherate (96 µL, 0.78 mmol, 4 equiv.) was added at –80 °C. After 15 min at –80 °C, the cooling bath was removed, then the mixture was allowed to warm to r.t. for 16 h. The mixture was filtrated over a pad of celite, then washed with EtOAc. The filtrate was treated with a saturated aqueous solution of sodium bicarbonate. The organic layer was washed with water and brine, then concentrated in vacuo. Flash chromatographic purification (cyclohexane/EtOAc 75:25) afforded the product as a white solid (96 mg, 67%): Rf 0.17 (cyclohexane/EtOAc 7/3); [α]D + 94.1 (c 1.0, CH2Cl2); IR (film) 3675, 2971, 2900, 1696, 1684, 1394, 1229, 1076, 1047, 891, 869; 1H NMR (500 MHz, CDCl3) δ 8.58 (br s, 1H, NH), 7.73 (d, JH6-H5 = 8.2 Hz, 1H, H6), 5.74–5.53 (m, 2H, H5 + H1′), 5.11 (s, 1H, H1″), 4.53 (d, JH2″-H3″ = 6.2 Hz, H2″), 4.43 (d, JH3″-H2″ = 6.2 Hz, 1H, H3″), 4.31 (t, JH4″-H5″b = JH4″-H5″a = 5.4 Hz, 1H, H4″), 4.17–4.03 (m, 2H, H4′ + H2′), 3.98–3.84 (m, 2H, H3′ + H5′), 3.45–3.40 (m, 2H, H5″a, H5″b), 2.93–2.80 (m, 2H, H6′b, H6′a), 1.60 (q, J = 7.4 Hz, 2H, H7″), 1.46 (q, J = 7.4 Hz, 2H, H7″), 0.87 – 0.74 (m, 24H, -C(CH3)3, H8″), 0.16–0.04 (m, 12H, -Si(CH3)2); 13C NMR (125 MHz, CDCl3) δ 162.9 (C4), 149.95 (C2), 140.15 (C6), 118.12 (C6″), 116.63 (C7′), 112.34 (C1″), 101.56 (C5), 90.20 (C1′), 86.00 (C3″), 85.49 (C4″), 84.21 (C4′), 81.56 (C2″), 75.23 (C5′), 75.05 (C2′), 71.05 (C3′), 53.45 (C5″), 29.18 (C7″), 28.85 (C7″), 25.80 (-C(CH3)3), 21.72 (C6′), 18.0, 17.9 (-C(CH3)3), 8.36 (C8″), −4.0, −4.3, −4.9, −4.9 (-Si(CH3)2); HRMS ESI+ Calcd. for C33H57N6O9Si2+ (M + H)+ 737.3720, found 737.3731.
3.1.2. Protected Amidoxime 2
The mixture of nitrile 1 (363 mg, 0.49 mmol, 1 equiv.) and 50% aqueous hydroxylamine (0.23 mL, 3.94 mmol, 8 equiv) in MeOH (8 mL) was heated at reflux for 6 h, then concentrated in vacuo. The oily residue was diluted with EtOAc (30 mL), then washed with water and brine (2 × 10mL). The organic layer was dried (MgSO4), then concentrated in vacuo. Flash chromatographic purification (cyclohexane/EtOAc 75:25) afforded product 2 as a white solid (329 mg, 87%): Rf 0.25 (cyclohexane/EtOAc = 1/1); [α]D +13 (c 1.0, CH2Cl2); IR (film): 1260, 1165, 1091, 911, 834, 735. 1H NMR (500 MHz, CDCl3) δ 7.75 (d, J H6-H5 = 8.1 Hz, 1H, H6), 5.88 (d, JH1′-H2′ = 5.5 Hz, 1H, H1′), 5.73 (d, JH5-H6 = 8.2 Hz, 1H, H5), 5.31 (s, 1H, H1″), 4.62 (dd, JH3″-H2″ = 6.3 Hz, JH3″-H4″ = 1.7 Hz, 1H, H3″), 4.53 (d, JH2″-H3″ = 6.3 Hz, 1H, H2″), 4.34 (td, JH4″-H5″ = 5.6 Hz, JH4″-H3″ = 1.7 Hz, 1H, H4″), 4.25 (t, JH5′-H4′ = 3.0 Hz, 1H, H5′), 4.16-4.09 (m, 2H, H2′, H4′), 4.05 (t, JH3′-H2′ = 3.8 Hz, 1H, H3′), 3.49 (dd, JH6a′-H5′ = 12.8 Hz, JH6a′-H6b′ = 5.6 Hz, 1H, H6a′), 3.45 (dd, JH6b′-H5′ = 12.8 Hz, JH6b′-H6a′ = 5.6 Hz, 1H, H6b′), 2.80 (dd, JH5a′-H6′ = 14.3 Hz, JH5a′-H5b′ = 5.5 Hz, 1H, H5a′), 2.52 (dd, JH5b′-H6′ = 14.3 Hz, JH5b′-H5a′ = 8.8 Hz, 1H, H5b′), 1.69 (q, JH7a″-H8″ = 7.4 Hz, 2H, H7a″), 1.55 (q, JH7b″-H8″ = 7.5 Hz, 2H, H7b″), 0.91-0.83 ( m, 24 H, H8”, SiCH3), 0.10-0.02 (m, 12 H, Si(CH3)2). 13C NMR (125 MHz, CDCl3) δ 171.8 (C7′), 162.8 (C4), 150.1 (C2), 140.5 (C6), 111.8 (C1″), 102.1 (C5), 89.0 (C1′), 86.2 (C2″), 86.1 (C4″), 84.8 (C4′), 81.5 (C5′), 75.3 (C2′), 72.3 (C3′), 53.6 (C5″), 38.7 (C6′ab), 29.4 (C7″), 29.0 (C7″), 25.93, 25.86 (-C(CH3)3), 18.1 (-C(CH3)3), 8.5, 7.7 (C8″), −4.1, −4.6, −4.8, (-SiC); HRMS ESI+ calcd. for C33H60N7O10Si2+ (M + H)+ 770.3935 found 770.3914.
3.1.3. General Procedure for the Synthesis of Protected Oxadiazoles
In a 10 mL microwave reaction vial, to a solution of amidoxime 2 (1 equiv.) in toluene (3 mL) was added acyl chloride 3a–3i (1.1 equiv.) and DBU (2.5 equiv.). The reaction was stirred at r.t. for 30 min. Then the reaction mixture was irradiated under microwave irradiation at 150 °C for 30 min to 1 h. The reaction mixture was then concentrated in vacuo. Flash chromatographic purifications (cyclohexane/EtOAc 9:1) afforded the products 11a–11i in 42 to 83% yield.
3.1.4. Protected Oxadiazole 11a
Compound 11a was obtained as a white solid (40 mg, 67% yield): Rf 0.25 (cyclohexane/EtOAc = 7/3); [α]D +14 (c 1.0, CH2Cl2); IR (film): 3006, 2873, 1558, 1427, 1368, 1275, 1146, 1016, 988, 935, 887, 820, 764, 750; 1H NMR (500 MHz, CDCl3) δ 7.89 (d, JH6-H5 = 8.2 Hz, 1H, H6), 5.85 (d, JH1′-H2′ = 4.7 Hz, 1H, H1′), 5.73 (dd, JH5-H6 = 8.2 Hz, 1H, H5), 5.24 (s, 1H, H1″), 4.64 (dd, JH3″-H2″ = 6.2 Hz, JH3″-H4″ = 1.5 Hz, 1H, H3″), 4.52 (d, JH2″-H3″ = 6.2 Hz, 1H, H2″), 4.32 (t, JH5′-H4′ = 5.8 Hz, 1H, H5′), 4.24–4.21 (m, 1H, H4″), 4.16 (t, JH2′-H3′ = 4.7 Hz, 1H, H2′), 4.05 (dd, JH4′-H3′ = 4.4 Hz, JH4′-H5′ = 1.6 Hz, 1H, H4′), 3.99 (t, JH3′-H4′ = 4.4 Hz, 1H, H3′), 3.50 (dd, JH6′a-H5′ = 12.7 Hz, JH6′a-H6′b = 5.2 Hz, 1H, H6′a), 3.43 (dd, JH6′b-H5′ = 12.7 Hz, JH6′b-H5′ = 5.2 Hz, 1H, H6′b), 3.38 (dd, JH5″a-H4″ = 15.0 Hz, JH5″a-H5″b = 9.3 Hz, 1H, H5″a), 3.19 (dd, JH5″b-H4″ = 15.0 Hz, JH5″b-H5″a = 9.3 Hz, 1H, H5″b), 2.83 (t, JH2′-H3′ = 7.6 Hz, 1H, H1*), 1.80–1.74 (m, 2H, H2*), 1.69 (q, JH7″-H8″ = 7.5 Hz, 2 H, H7″), 1.56 (q, JH7″-H8″ = 7.5 Hz, 2 H, H7″), 1.30–1.24 (m, 24H, H3*-H14*), 0.92–0.82 (m, 27H, -SiC(CH3)3, H8″, H15*), 0.08–0.05 (12H, Si(CH3)2); 13C NMR (125 MHz, CDCl3) δ 180.2 (C8′), 166.9 (C7′), 163.0 (C4), 150.1 (C2), 140.2 (C6), 118.00 (C6″), 112.2 (C1″), 101.8 (C5), 88.7 (C1′), 86.1 (C2″), 85.0 (C4″), 84.6 (C4′), 81.7 (C3″), 78.2 (C5′), 75.5 (C2′), 72.0 (C3′), 53.4 (C5″), 31.9, 29.6, 29.4, 29.3, 29.2, 29.1, 28.9, 25.8 (C2*-C9*), 26.6, 26.5 (C6′, C1*), 25.76 (SiCCH3), 22.69 (s), 18.0 (SiCCH3), 14.1 (C10*), 8.4, 7.5 (C8″), –4.2, –4.5, –4.8 (SiCH3); HRMS ESI+ calcd. for C44H78N7O10Si2+ (M + H)+ 920.5343 found 920.5326.
3.1.5. Protected Oxadiazole 11b
Compound 11b was obtained as a white soli (53 mg, 55% yield): Rf 0.25 (cyclohexane/EtOAc = 7/3); [α]D +10 (c 1.0, CH2Cl2); IR (film): 3006, 2873, 1558, 1427, 1368, 1275, 1146, 1016, 988, 935, 887, 820, 764, 750; 1H NMR (500 MHz, CDCl3) δ 8.61 (s, 1H, NH), 7.88 (d, JH5-H6 = 8.1Hz, 1H, H6), 5.84 (d, JH1′-H2′ = 4.3Hz, 1H, H1′), 5.72 (d, JH5-H6 = 8.1Hz, 1H, H5), 5.24 (s, 1H, H1″), 4.63 (d, JH2″-H3″ = 6.1Hz, 1H, H3″), 4.52 (d, JH2″-H3″ = 6.0 Hz, 1H, H2″), 4.33–4.29 (m, 1H, H4″), 4.26–4.19 (m, 1H, H5′), 4.18–4.14 (m, 1H, H2′), 4.05 (d, JH3′-H4′ = 3.0 Hz, 1H, H4′), 4.00 (d, JH3′-H4′ = 3.0 Hz, 1H, H3′), 3.50 (dd, JH5″a-H5″b = 12.9 Hz, JH4″-H5″a = 5.1Hz, 1H, H5″a), 3.42 (dd, JH5″a-H5″b = 12.9 Hz, JH4″-H5″b = 5.8 Hz, 1H, H5″b), 3.37 (dd, JH6′a-H6′b = 14.9 Hz, JH6′a-H5′ = 5.0 Hz, 1H, H6′a), 3.19 (dd, JH6′a-H6′b = 14.9 Hz, JH6′b-H5′ = 9.2 Hz, 1H, H6′b), 2.82 (t, JH1*-H2* = 7.6 Hz, 2H, H1*), 1.84–1.62 (m, 4H, H2*, H7″), 1.56 (q, JH7″-H8″ = 7.2 Hz, 2H, H7″), 1.40–1.17 (m, 16H, H2*-H9*), 0.97–0.75 (m, 21H, H10*, H8″, SiC(CH3)3), 0.07, 0.07, 0.06 (s, 12H, SiCH3); 13C NMR (125 MHz, CDCl3) δ 180.3 (C8′), 166.9 (C7′), 162.9 (C4), 150.0 (C2), 140.3 (C6), 118.1 (C6″), 112.3 (C1″), 101.8 (C5), 88.8 (C1′), 86.1 (C2″), 85.0 (C4″), 84.7 (C4′), 81.7 (C3”), 78.3 (C5′), 75.5 (C2′), 72.1 (C3′), 53.4 (C6′ab), 32.0 (C4*), 29.8, 29.7, 29.5(C5”b), 29.3, 29.3, 29.2 (C5”a), 29.1 (C7”), 28.9 (C7”), 26.7 (C1*), 26.6 (C2*), 25.9 (SiC(CH3)3), 25.8 (SiC(CH3)3), 22.8 (C3*), 18.0 (C14*), 14.2 (C15*), 8.5 (C8″), 7.6 (C8″), −4.0 (SiCH3), −4.4 (SiCH3), −4.6 (SiC), −4.7 (SiC). HRMS ESI+ calcd. for C49H88N7O10Si2+ (M + H)+ 990.6125 found 990.6105.
3.1.6. Protected Oxadiazole 11c
Compound 11c was obtained as a colorless oil (50 mg, 75% yield): Rf 0.25 (cyclohexane/EtOAc = 7/3); [α]D +16 (c 1.0, CH2Cl2); IR (film): 2926, 2855, 2105, 1695, 1462, 1378, 1275, 1167, 1100, 925, 867, 839, 764, 750; 1H NMR (500 MHz, CDCl3) δ 7.89 (d, JH6-H5 = 8.0 Hz, 1H, H6), 5.85 (d, JH1′-H2′ = 4.4 Hz, 1H, H1′), 5.73 (dd, JH5-H6 = 8.0 Hz, JH5-H1′ = 2.2 Hz, 1H, H5), 5.24 (s, 1H, H1″), 4.64 (dd, JH3″-H2″ = 6.6 Hz, JH3″-H4″ = 1.4 Hz, 1H, H3″), 4.51 (d, JH2″-H3″ = 6.5 Hz, 1H, H2″), 4.32 (t, JH5′-H4′ = 5.7 Hz, 1H, H5′), 4.24–4.21 (m, 1H, H4″), 4.16 (t, JH2′-H3′ = 4.4 Hz, 1H, H2′), 4.05 (dd, JH4′-H3′ = 4.2 Hz, JH4′-H5′ = 1.5 Hz, 1H, H4′), 3.98 (t, JH3′-H4′ = 4.2 Hz, 1H, H3′), 3.50 (dd, JH6′a-H5′ = 12.8 Hz, JH6′a-H6′b = 5.8 Hz, 1H, H6′a), 3.43 (dd, JH6′b-H5′ = 12.8 Hz, JH6′b-H5′ = 5.8 Hz, 1H, H6′b), 3.38 (dd, JH5″a-H4″ = 15.0 Hz, JH5″a-H5″b = 9.3 Hz, 1H, H5″a), 3.19 (dd, JH5″b-H4″ = 15.0 Hz, JH5″b-H5″a = 9.3 Hz, 1H, H5″b), 2.83 (t, JH2′-H3′ = 7.6 Hz, 1H, H1*), 1.79–1.73 (m, 2H, H2*), 1.69 (q, JH7″-H8″ = 7.4 Hz, 2H, H7″), 1.56 (q, JH7″-H8″ = 7.4 Hz, 2 H, H7″), 1.38–1.24 ( m, 30H, H3*-H17*), 0.92–0.84 (m, 27H, SiC(CH3)3, H8″, H18*), 0.07–0.05 (12 H, Si(CH3)2); 13C NMR (125 MHz, CDCl3) δ 180.1 (C8′), 166.8 (C7′), 162.8 (C4), 149.9 (C2), 140.1 (C6), 117.9 (C6″), 112.1 (C1″), 101.6 (C5), 88.6 (C1′), 85.9 (C2″), 84.9 (C4″), 84.5 (C4′), 81.6 (C3”), 78.1 (C5′), 75.4 (C2′), 71.9 (C3′), 53.2 (C6′ab), 31.9 (C4*), 29.6, 29.3, 29.3(C5”b), 29.2, 29.1, 29.1 (C5”a), 29.0 (C7”), 28.7 (C7”), 26.6 (C1*), 26.4 (C2*), 25.7 (SiC(CH3)3), 25.6 (SiC(CH3)3), 22.6 (C3*), 17.9 (C17*), 14.1 (C18*), 8.3 (C8″), 7.4 (C8″), −4.2 (SiCH3), −4.5 (SiCH3), −4.8 (SiC), −4.8 (SiC); HRMS ESI+ calcd. for C52H94N7O10Si2+ (M + H)+ 1032.6595 found 1032.6576.
3.1.7. Protected Oxadiazole 11d
Compound 11d was obtained as a colorless oil (47.5 mg, 72% yield): Rf 0.25 (cyclohexane/EtOAc = 7/3); [α]D +33 (c 1.0, CH2Cl2); IR (film): 2928, 2856, 2105, 1695, 1462, 1378, 1275, 1167, 1099, 924, 903, 867, 838, 764, 750; 1H NMR (500 MHz, CDCl3) δ 7.89 (d, JH6-H5 = 8.2 Hz, 1H, H6), 5.85 (d, JH1′-H2′ = 4.7 Hz, 1H, H1′), 5.73 (d, JH5-H6 = 8.2 Hz, 1H, H5), 5.35–5.30 (m, 2 H, H8*, H9*), 5.24 (s, 1H, H1″), 4.64 (dd, JH3″-H2″ = 6.2 Hz, JH3″-H4″ = 1.5 Hz, 1H, H3″), 4.52 (d, JH2″-H3″ = 6.5 Hz, 1H, H2″), 4.31 (t, JH5′-H4′ = 5.8 Hz, 1H, H5′), 4.24–4.21 (m, 1H, H4″), 4.15 (t, JH2′-H3′ = 4.5 Hz, 1H, H2′), 4.05 (dd, JH4′-H3′ = 4.2 Hz, JH4′-H5′ = 1.6 Hz, 1H, H4′), 3.98 (t, JH3′-H4′ = 4.3 Hz, 1H, H3′), 3.50 (dd, JH6′a-H5′ = 13.0 Hz, JH6′a-H6′b = 5.2 Hz, 1H, H6′a), 3.43 (dd, JH6′b-H5′ = 13.0 Hz, JH6′b-H5′ = 5.8 Hz, 1H, H6′b), 3.38 (dd, JH5″a-H4″ = 15.0 Hz, JH5″a-H5″b = 9.2 Hz, 1H, H5″a), 3.19 (dd, JH5″b-H4″ = 15.0 Hz, JH5″b-H5″a = 9.2 Hz, 1H, H5″b), 2.84 (t, JH2′-H3′ = 7.8 Hz, 1H, H1*), 2.02–1.98 (m, 4 H, H7*, H10*), 1.80–1.74 (m, 2 H, H2*), 1.69 (q, JH7″-H8″ = 7.4 Hz, 2 H, H7″), 1.56 (q, JH7″-H8″ = 7.4 Hz, 2 H, H7″), 1.42 –1.24 (m, 20 H, H3*-H6*, H9*-H16*), 0.92–0.84 ( m, 27H, -SiC(CH3)3, H8″, H17*), 0.07–0.05 (12 H, Si(CH3)2); 13C NMR (125 MHz, CDCl3) δ 180.2 (C8′), 166.9 (C7′), 163.0 (C4), 150.0 (C2), 140.2 (C6), 130.2 (C8*), 129.7 (C9*), 118.0 (C6″), 112.3 (C1″), 101.8 (C5), 88.8 (C1′), 86.1 (C2″), 85.0 (C4″), 84.7 (C4′), 81.7 (C3”), 78.3 (C5′), 75.5 (C2′), 72.0 (C3′), 53.4 (C6′ab), 32.0 (C4*), 29.9, 29.8(C5”b), 29.6, 29.4, 29.3 (C5”a), 29.1 (C7”), 28.9 (C7”), 27.3 (C7*), 27.2 (C10*), 26.7 (C1*), 26.6 (C2*), 25.9 (SiC(CH3)3), 25.8 (SiC(CH3)3), 22.8 (C3*), 18.1 (C16* ), 14.2 (C17*), 8.5 (C8″), 7.6 (C8″), −4.0 (SiCH3), −4.4 (SiCH3), −4.6 (SiC), −4.7 (SiC); HRMS ESI+ calcd. for C51H90N7O10Si2+ (M + H)+ 1016.6282 found 1016.6258.
3.1.8. Protected Oxadiazole 11e
Compound 11e was obtained as a colorless oil (23 mg, 42% yield): Rf 0.25 (cyclohexane/EtOAc = 7/3); [α]D +33 (c 1.0, CH2Cl2); IR (film): 2930, 2106, 1697, 1487, 1368, 1275, 1275, 1168, 1100, 924, 870, 839, 764, 750; 1H NMR (500 MHz, CDCl3) δ 8.06 (d, JHa-Hb = 8.8 Hz, 2 H, Ha), 7.91 (dd, JH6-H5 = 8.3 Hz, JH6-H1′ = 3.3 Hz, 1H, H6), 7.41 (t, JHd-He = 7.9 Hz, 2 H, Hd), 7.22 (t, JHe-Hd = 7.4 Hz, 1H, He), 7.09 (d, JHd-Hc = 8.8 Hz, 2 H, Hc), 7.07 (d, JHb-Ha = 8.8 Hz, 2 H, Hb), 5.85 (d, JH1′-H2′ = 4.4 Hz, 1H, H1′), 5.74 (dd, JH5-H6 = 8.3 Hz, JH5-H1′ = 2.0 Hz, 1H, H5), 5.29 (s, 1H, H1″), 4.65 (dd, JH3″-H2″ = 6.2 Hz, JH3″-H4″ = 1.5 Hz, 1H, H3″), 4.54 (d, JH2″-H3″ = 6.2 Hz, 1H, H2″), 4.32–4.21 (m, 2 H, H5′, H4″), 4.18–4.14 (m, 2 H, H2′, H4′), 4.01 (t, JH3′-H4′ = 4.2 Hz, 1H, H3′), 3.52 (dd, JH6′a-H5′ = 12.8 Hz, JH6′a-H6′b = 5.5 Hz, 1H, H6′a), 3.47–3.42 (m, 2 H, H6′b, H5″a), 3.27 (dd, JH5″b-H4″ = 15.0 Hz, JH5″b-H5″a = 9.2 Hz, 1H, H5″b), 1.70 (q, JH7″-H8″ = 7.4 Hz, 2 H, H7″), 1.56 (q, JH7″-H8″ = 7.4 Hz, 2 H, H7″), 0.92–0.82 (m, 24H, -SiC(CH3)3, H8″), 0.07–0.05 (12 H, Si(CH3)2); 13C NMR (125 MHz, CDCl3) δ 175.4 (C8′), 167.6 (C7′), 161.9 (C4), 155.4 (C10′), 150.0 (C2), 130.2 (Ca, Cd), 124.9 (Ce), 120.3 (Cc), 118.3 (Cb), 118.1 (C6″), 112.2 (C1″), 101.8 (C5), 88.8 (C1′), 86.1 (C2″), 85.0 (C4″), 84.8 (C4′), 81.7 (C3”), 78.2 (C5′), 75.5 (C2′), 72.1 (C3′), 53.4 (C6′ab), 32.0 (C4*), 29.9(C5”ab), 29.3 (C7”), 28.9 (C7”), 25.8 (SiC(CH3)3), 25.8 (SiC(CH3)3, 18.1, 18.0 (C9′), 8.5 (C8″), 7.6 (C8″), −4.0 (SiCH3), −4.4 (SiCH3), −4.6 (SiC), −4.7 (SiC). HRMS ESI+ calcd. for C46H66N7O11Si2+ (M + H)+ 948.4353 found 948.4329.
3.1.9. Protected Oxadiazole 11f
Compound 11f was obtained as a colorless oil (55 mg, 83% yield): Rf 0.25 (cyclohexane/EtOAc = 7/3); [α]D +15 (c 1.0, CH2Cl2); IR (film): 2928, 2856, 2105, 1696, 1580, 1462, 1378, 1274, 1167, 1099, 967, 910, 867, 838, 764, 748; 1H NMR (500 MHz, CDCl3) δ 7.89 (d, JH6-H5 = 8.2 Hz, 1H, H6), 5.84 (d, JH1′-H2′ = 4.4 Hz, 1H, H1′), 5.73 (dd, JH5-H6 = 8.2 Hz, JH5-H1′ = 2.0 Hz, 1H, H5), 5.42–5.33 (m, 2 H, H8*, H9*), 5.24 (s, 1H, H1″), 4.63 (dd, JH3″-H2″ = 6.2 Hz, JH3″-H4″ = 1.6 Hz, 1H, H3″), 4.51 (d, JH2″-H3″ = 6.0 Hz, 1H, H2″), 4.31 (t, JH5′-H4′ = 5.7 Hz, 1H, H5′), 4.24–4.20 (m, 1H, H4″), 4.15 (t, JH2′-H3′ = 4.5 Hz, 1H, H2′), 4.05 (dd, JH4′-H3′ = 4.1Hz, JH4′-H5′ = 1.7 Hz, 1H, H4′), 3.98 (t, JH3′-H4′ = 4.4 Hz, 1H, H3′), 3.50 (dd, JH6′a-H5′ = 12.6 Hz, JH6′a-H6′b = 5.1Hz, 1H, H6′a), 3.43 (dd, JH6′b-H5′ = 12.6 Hz, JH6′b-H5′ = 6.1Hz, 1H, H6′b), 3.38 (dd, JH5″a-H4″ = 14.7 Hz, JH5″a-H5″b = 9.0 Hz, 1H, H5″a), 3.19 (dd, JH5″b-H4″ = 15.0 Hz, JH5″b-H5″a = 9.0 Hz, 1H, H5″b), 2.83 (t, JH2′-H3′ = 7.7 Hz, 1H, H1*), 1.97–1.94 (m, 4 H, H7*, H10*), 1.80–1.73 (m, 2 H, H2*), 1.70 (q, JH7″-H8″ = 7.6 Hz, 2 H, H7″), 1.56 (q, JH7″-H8″ = 7.6 Hz, 2 H, H7″), 1.32–1.25 ( m, 20 H, H3*-H6*, H9*-H16*), 0.92–0.84 (m, 27H, -SiC(CH3)3, H8″, H17*), 0.07–0.05 (12 H, Si(CH3)2). 13C NMR (125 MHz, CDCl3) δ 180.2 (C8′), 166.9 (C7′), 162.9 (C4), 150.0 (C2), 140.2 (C6), 130.6 (C8*), 130.2 (C9*), 118.0 (C6″), 112.3 (C1″), 101.8 (C5), 88.8 (C1′), 86.1 (C2″), 85.0 (C4″), 84.7 (C4′), 81.7 (C3”), 78.3 (C5′), 75.5 (C2′), 72.0 (C3′), 53.4 (C6′ab), 32.7 (C7*), 32.6 (C10*), 32.0 (C4*), 29.7, 29.6 (C5”b), 29.5, 29.4, 29.3 (C5”a), 29.1 (C7”), 28.9 (C7”), 26.7 (C1*), 26.6 (C2*), 25.9 (SiC(CH3)3), 25.8 (SiC(CH3)3), 22.8 (C3*), 18.1 (C16*), 14.2 (C17*), 8.5 (C8″), 7.6 (C8″), −4.0 (SiCH3), −4.4 (SiCH3), −4.6 (SiC), −4.7 (SiC); HRMS ESI+ calcd. for C51H90N7O10Si2+ (M + H)+ 1016.6282 found 1016.6274.
3.1.10. Protected Oxadiazole 11g
Compound 11g was obtained as a colorless oil (38 mg, 58% yield): Rf 0.25 (cyclohexane/EtOAc = 7/3); [α]D +6 (c 1.0, CH2Cl2); IR (film): 2929, 2856, 2105, 1695, 1580, 1462, 1377, 1274, 1167, 1100, 924, 865, 839, 764, 750; 1H NMR (500 MHz, CDCl3) δ 7.89 (d, JH6-H5 = 8.1Hz, 1H, H6), 7.28–7.16 (m, 5 H, Habc), 5.85 (d, JH1′-H2′ = 4.7 Hz, 1H, H1′), 5.73 (dd, JH5-H6 = 8.1Hz, JH5-H1′ = 2.0 Hz, 1H, H5), 5.24 (s, 1H, H1″), 4.64 (dd, JH3″-H2″ = 6.1Hz, JH3″-H4″ = 1.4 Hz, 1H, H3″), 4.52 (d, JH2″-H3″ = 6.1Hz, 1H, H2″), 4.32 (t, JH5′-H4′ = 5.7 Hz, 1H, H5′), 4.24–4.21 (m, 1H, H4″), 4.16 (t, JH2′-H3′ = 4.4 Hz, 1H, H2′), 4.05 (dd, JH4′-H3′ = 4.3 Hz, JH4′-H5′ = 1.7 Hz, 1H, H4′), 3.98 (t, JH3′-H4′ = 4.5 Hz, 1H, H3′), 3.50 (dd, JH6′a-H5′ = 12.8 Hz, JH6′a-H6′b = 5.8 Hz, 1H, H6′a), 3.43 (dd, JH6′b-H5′ = 12.8 Hz, JH6′b-H5′ = 5.8 Hz, 1H, H6′b), 3.38 (dd, JH5″a-H4″ = 15.0 Hz, JH5″a-H5″b = 9.3 Hz, 1H, H5″a), 3.19 (dd, JH5″b-H4″ = 15.0 Hz, JH5″b-H5″a = 9.3 Hz, 1H, H5″b), 2.82 (t, JH2′-H3′ = 7.6 Hz, 1H, H1*), 2.59 (t, JH9′-H10′ = 7.6 Hz, 1H, H9*), 1.79–1.73 (m, 2H, H2*), 1.70 (q, JH7″-H8″ = 7.6 Hz, 2 H, H7″), 1.63–1.58 (m, 2H, H8*), 1.56 (q, JH7″-H8″ = 7.6 Hz, 2H, H7″), 1.36–1.26 ( m, 10 H, H3*-H7*), 0.92–0.84 ( m, 24H, -SiC(CH3)3, H8″, H18*), 0.07–0.05 (12 H, Si(CH3)2); 13C NMR (125 MHz, CDCl3) δ 180.2 (C8′), 166.9 (C7′), 162.9 (C4), 150.0 (C2), 142.9 (C10*), 140.2 (C6), 128.5 (Ca), 128.3 (Cb), 125.7 (Cc), 118.0 (C6″), 112.3 (C1″), 101.8 (C5), 88.8 (C1′), 86.1 (C2″), 85.0 (C4″), 84.7 (C4′), 81.7 (C3”), 78.3 (C5′), 75.5 (C2′), 72.0 (C3′), 53.4 (C6′ab), 36.1 (C9*), 31.6 (C4*), 29.5, 29.4, 29.4 (C5”b), 29.3, 29.3, 29.2 (C5”a), 29.1 (C7”), 28.9 (C7”), 26.7 (C1*), 26.6 (C2*), 25.9 (SiC(CH3)3), 25.8 (SiC(CH3)3), 17.9 (C8*), 8.5 (C8″), 7.6 (C8″), −4.0 (SiCH3), −4.4 (SiCH3), −4.6 (SiC), −4.7 (SiC). HRMS ESI− calcd. for C49H78N7O10Si2− (M + H)− 980.5354 found 980.5297.
3.1.11. Protected Oxadiazole 11h
Compound 11h was obtained as a colorless oil and as a 1/1 mixture of stereoisomers (38 mg, 58% yield): Rf 0.25 (cyclohexane/EtOAc = 7/3); [α]D +6 (c 1.0, CH2Cl2); IR (film): 2929, 2856, 2105, 1695, 1580, 1462, 1377, 1274, 1167, 1100, 924, 865, 839, 764, 750; 1H NMR (500 MHz, CDCl3) δ 7.90 (d, JH5-H6 = 8.1 Hz, 1H, H6), 5.85 (d, JH1′-H2′ = 3.1 Hz, 0.5H, H1′), 5.84 (d, JH1′-H2′ = 3.4 Hz, 0.5H, H1′), 5.74 (d, JH5-H6 = 8.1 Hz, 1H, H5), 5.22 (s, 0.5H, H1″), 5.22 (s, 0.5 H, H1″), 4.63 (d, JH2″-H3″ = 6.1 Hz, 1H, H3″), 4.51 (d, JH2″-H3″ = 6.1 Hz, 1H, H2″), 4.35–4.23 (m, 2H, H4″, H5′), 4.23–4.14 (m, 3 H, H3**, H2′), 4.08 (dd, JH4′-H5′ = 8.5 Hz, JH3′-H4′ = 4.4 Hz, 0.5H, H4′), 4.07 (dd, JH4′-H5′ = 8.5 Hz, JH3′-H4′ = 4.4 Hz, 0.5 H, H4′), 4.00 (t, JH3′-H4′ = JH2′-H3′ = 4.4 Hz, 1H, H3′), 3.95 (t, J = 7.6 Hz, 1H, H1*), 3.50 (dd, JH5″a-H5″b = 12.7, JH4″-H5″a = 4.9 Hz, 0.5H, H5″a), 3.49 (dd, JH5″a-H5″b = 12.7, JH4″-H5″a = 4.9 Hz, 0.5H, H5″a), 3.46–3.34 (m, 2 H, H6′a, H5″b), 3.26–3.16 (m, 1H, H6′b), 2.18–1.96 (m, 2 H, H2*), 1.75–1.65 (m, 2H, H7″), 1.59–1.51 (m, 2H, H7″), 1.40–1.5 (m, 17H, H3*-H9*, H4**), 0.94–0.78 (m, 21 H, H10*, H8″, SiC(CH3)3), 0.07, 0.07, 0.06 (s, 12H, SiCH3); 13C NMR (125 MHz, CDCl3) δ 176.74, 176.67 (C2**), 168.66, 168.63 (C8′), 167.23, 167.20 (C7′), 162.9, 162.8 (C4), 150.00, 149.95 (C2), 140.2 (C6), 117.96, 117.93 (C6″), 112.34, 112.29 (C1″), 101.76 (C5), 88.81 (C1′), 86.03 (C2″), 84.98, 84.86 (C4′, C4″), 81.7 (C3″), 78.2, 78.1 (C5′), 75.5 (C2′), 71.9 (C3′), 62.02 (C3**), 53.27, 53.25 (C5″), 44.60, 44.55 (C1*), 31.89 (C2*), 30.2, 29.7, 29.5, 29.4, 29.3, 29.2, 29.1, 28.8, 27.20, 22.7 (C3*-C9*, C7″), 25.81, 25.76 (SiCCH3), 18.0 (SiCCH3), 14.15, 14.12 (C4**), 8.4, 7.5 (C8″), −4.2, −4.5, −4.8 (SiCH3); HRMS ESI− calcd. for C47H80N7O12Si2− (M + H)− 990,5409 found 990.5455.
3.1.12. Protected Oxadiazole 11i
Compound 11i was obtained as a colorless oil (57 mg, 74% yield): Rf 0.25 (cyclohexane/EtOAc = 7/3); [α]D +10 (c 1.0, CH2Cl2); IR (film): 2928, 2856, 1697, 1463, 1260, 1168, 1102, 840, 779, 740; 1H NMR (500 MHz, CDCl3) δ 7.93 (d, JH6-H5 = 8.5 Hz, 1H, H6), 5.83 (d, JH1′-H2′ = 4.3 Hz, 1H, H1′), 5.73 (dd, JH5-H6 = 8.3 Hz, JH5-H1′ = 2.2 Hz, 1H, H5), 5.23 (s, 1H, H1″), 4.63 (dd, JH3″-H2″ = 6.3 Hz, JH3″-H4″ = 1.2 Hz, 1H, H3″), 4.51 (d, JH2″-H3″ = 6.4 Hz, 1H, H2″), 4.31-4.24 (m, 2 H, H5′,H4″), 4.16 (d, JH2′-H3′ = 3.9 Hz, 1H, H2′), 4.17 (q, JH3***-H4*** = 7.1Hz, 1H, H3***), 4.09 (dd, JH4′-H3′ = 4.9 Hz, JH4′-H5′ = 1.4 Hz, 1H, H4′), 4.01 (t, JH3′-H4′ = 4.6 Hz, 1H, H3′), 3.52 (dd, JH5″a-H5″b = 13 Hz, JH5″a-H6′ = 4.9 Hz, 1H, H5″a), 3.43 (dd, JH5″b-H5″a = 13 Hz, JH5″b-H4″ = 5.8 Hz, 1H, H5″b), 3.40 (dd, JH6″a-H6″b = 15.4 Hz, JH6″a-H5′ = 5.5 Hz, 1H, H6″a), 3.21 (dd, JH6″b-H6″a = 15.4 Hz, JH6″b-H5″ = 8.4 Hz, 1H, H6″b), 2.12-2.00 (m, 4 H, H2*,H2**), 1.68 (q, JH7″a-H8″ = 7.4 Hz, 2 H, H7″a), 1.55 (q, JH7″b-H8″ = 7.4 Hz, 2 H, H7″b), 1.28–1.19 ( m, 33 H, H3**-H10**, H4*-H10*, H4***), 1.14–1.03 ( m, 3H, H3*), 0.91–0.83 ( m, 30H, -SiC(CH3)3, H8″, H11*, H11**), 0.09–0.07 (12 H, Si(CH3)2). 13C NMR (125 MHz, CDCl3) δ 183.4 (C8′), 166.8 (C7′), 163.0 (C4), 150.0 (C2), 140.3 (C6), 118.0 (C6″), 112.3 (C1″), 101.7 (C5), 89.0 (C1′), 86.1 (C2″), 85.1 (C4″), 84.7 (C4′), 81.7 (C3”), 78.3 (C5′), 75.6 (C2′), 71.8 (C3′), 53.4 (C5”), 38.5 (C1*), 33.5 (C2*), 33.4 (C2**), 32.0, 29.7, 29.7, 29.5, 29.4, 29.3, 28.9 (C7”), 27.3 (C7”), 26.0 (SiC(CH3)3), 25.9 (SiC(CH3)3), 22.8 (C3*), 18.0 (C3** ), 14.2 (C4***,C11**,C11*), 8.5 (C8″), 7.6 (C8″), −4.0 (SiCH3), −4.3(SiCH3), −4.6(SiC), −4.7(SiC). HRMS ESI+ calcd. for C55H100N7O10Si2+ (M + H)+ 1074.7064 found 1074.7060.
3.1.13. General Procedure for Compounds Deprotection
The deprotection of compounds
11a–
11i was performed according to reference [
42] and afforded the fully deprotected compounds
12a–
12i in 22 to 85% yield over two steps.
3.1.14. Oxadiazole 12a
Compound 12a was obtained from the protected oxadiazole 11a (30 mg, 32 μmol, 1 equiv.) as a white solid (16.7 mg, 85% yield): Rf 0.15 (DCM/MeOH/NH4OH 14% 80/18/2); [α]D + 15 (c 1.0, CH2Cl2); IR (film): 2912, 2888, 2240, 1750, 1700, 1650, 1400, 1275, 1260, 1000, 764, 750.; 1H NMR (500 MHz, MeOD) δ 7.80 (d, JH6-H5 = 8.1Hz, 1H, H6), 5.77 (d, JH1′-H2′ = 3.1Hz, 1H, H1′), 5.73 (d, JH5-H6 = 8.1Hz, 1H, H5), 5.14 (s, 1H, H1″), 4.33–4.27 (m, 1H, H2′), 4.18–4.12 (m, 2 H, H3′, H4′), 4.09 (t, JH4″-H5″ = 7.1Hz, 1H, H4″), 4.04 (m, 2 H, H2″, H3″), 3.98 (m, 1H, H5′), 3.37–3.32 (m, 1H, H6′a) 3.29–3.26 (m,1H, H5″a), 3.20 (dd, JH6′b-H6′a = 15.0, JH6′b-H5′ = 5.7 Hz, 1H, H6′b), 3.11 (dd, JH5″b-H5″a = 13.0, JH5″b-H4″ = 7.1Hz,1H, H5″b), 2.91 (t, JH2*-H3* = 7.5 Hz, 2 H, H2*), 1.79 (dt, JH3*-H2* = JH3*-H4* = 7.4 Hz, 2 H, H3*), 1.44–1.20 (m,14 H), 0.89 (t, JH11*-H10* = 6.8 Hz, 3 H, H11*).; 13C NMR (125 MHz, MeOD) δ 181.9 (C1*), 168.7 (C7′), 166.1(C4), 152.1 (C2), 142.2 (C6), 111.0 (C1″), 102.5 (C5), 91.6 (C1′), 85.7 (C3′), 80.0 (C4″), 78.1 (C5′), 76.3 (C3″), 75.5 (C2′), 73.8 (C2″), 71.3 (C4′), 44.3 (C5″), 33.0, 30.6 (C6′), 30.39, 30.20, 30.02, 27.6 (C3*), 27.1 (C2*),23.7, 14.4; HRMS ESI+ calcd. for C27H43N5O10 (M + H)+ 598.3083, found 598.3082.
3.1.15. Oxadiazole 12b
Compound 12b was obtained from the protected oxadiazole 11b (27 mg, 27 μmol, 1 equiv.) as a white solid (10.7 mg, 59% yield): Rf 0.15 (DCM/MeOH/NH4OH 14% 80/18/2); [α]D + 1 (c 1.0, CH3OH); IR (film): 2924, 2853, 1673, 1466, 1272, 1203, 1136, 800, 724; 1H NMR (500 MHz, MeOD) δ 7.81 (d, JH6-H5 = 8.1Hz, 1H, H6), 5.77 (d, JH1′-H2′ = 3.2 Hz, 1H, H1′), 5.72 (d, JH5-H6 = 8.1Hz, 1H, H5), 5.14 (s, 1H, H1″), 4.30 (td, JH5′-H6′ = 6.5 Hz, JH5′-H4′ = 2.5 Hz, 1H, H5′), 4.19-4.01 (m, 5 H, H2′, H3′, H4′, H3″, H2″), 3.98 (d, JH4″-H5″ = 4.3 Hz, 1H, H4″), 3.30–3.27 (m, 2 H, H6′ab), 3.21 (dd, JH5″a-H4″ = 14.9 Hz, JH5″a-H5″b = 5.7 Hz, 1H, H5″a), 3.12 (dd, JH5″b-H4″ = 13.0 Hz, JH5″b-H5″a = 9.0 Hz, 1H, H5″b), 2.91 (t, JH1*-H2* = 7.5 Hz, 2H, H1*), 1.82–1.76 (m, 2H, H2*), 1.38–1.29 ( m, 24 H, H3*-H14*), 0.90 (t, JH15*-H14* = 7.0 Hz, 3H, H15*); 13C NMR (125 MHz, MeOD) δ 181.1 (C8′), 168.7 (C7′), 166.1 (C4), 152.0 (C2), 142.2 (C6), 110.9 (C1″), 102.4 (C5), 91.5 (C1′), 85.6 (C2″), 79.9 (C4′), 78.0 (C5′), 76.2 (C4”), 75.5 (C2′), 73.8 (C3″), 71.2 (C3′), 44.3 (C5”), 33.0 (C6′), 30.8, 30.7, 30.7, 30.5, 30.4, 30.3, 30.2, 30.0 (C4*-C14*), 27.6 (C2*), 27.1 (C1*), 23.7 (C3*), 14.4 (C15*). HRMS ESI+ calcd. for C32H54N5O10+ (M + H)+ 668.3865 found 668.3860.
3.1.16. Oxadiazole 12c
Compound 12c was obtained from the protected oxadiazole 11c (28 mg, 27 μmol, 1 equiv.) as a colorless oil (9.2 mg, 40% yield): Rf 0.15 (DCM/MeOH/NH4OH 14% 80/18/2); [α]D + 0.8 (c 1.0, CH3OH); IR (film): 2921, 2851, 1672, 1432, 1275, 1204, 1138, 961, 800, 764.; 1H NMR (500 MHz, MeOD) δ 7.81 (d, JH6-H5 = 8.0 Hz, 1H, H6), 5.76 (d, JH1′-H2′ = 3.2 Hz, 1H, H1′), 5.72 (d, JH5-H6 = 8.0 Hz, 1H, H5), 5.14 (s, 1H, H1″), 4.30 (td, JH5′-H6′ = 6.5 Hz, JH5′-H4′ = 2.5 Hz, 1H, H5′), 4.17–4.01 (m, 5 H, H2′, H3′, H4′, H3″, H2″), 3.98 (d, JH4″-H5″ = 4.1Hz, 1H, H4″), 3.30–3.25 (m, 2 H, H6′ab), 3.21 (dd, JH5″a-H4″ = 15.0 Hz, JH5″a-H5″b = 5.6 Hz, 1H, H5″a), 3.12 (dd, JH5″b-H4″ = 13.0 Hz, JH5″b-H5″a = 9.0 Hz, 1H, H5″b), 2.91 (t, JH1*-H2* = 7.5 Hz, 2 H, H1*), 1.80–1.76 (m, 2 H, H2*), 1.36–1.29 ( m, 30 H, H3*-H17*), 0.90 (t, JH15*-H14* = 6.9 Hz, 3H, H18*); 13C NMR (125 MHz, MeOD) δ 181.8 (C8′), 168.7 (C7′), 166.1 (C4), 152.0 (C2), 142.2 (C6), 110.9 (C1″), 102.4 (C5), 91.6 (C1′), 85.6 (C2″), 79.9 (C4′), 78.1 (C5′), 76.2 (C4”), 75.5 (C2′), 73.8 (C3″), 71.2 (C3′), 44.3 (C5”), 33.0 (C6′), 30.8, 30.5, 30.4, 30.3, 30.2, 30.0 (C4*-C17*), 27.6 (C2*), 27.1 (C1*), 23.7 (C3*), 14.4 (C18*); HRMS ESI+ calcd. for C35H60N5O10+ (M + H)+ 710.4335 found 710.4325.
3.1.17. Oxadiazole 12d
Compound 12d was obtained from the protected oxadiazole 11d (22 mg, 21 μmol, 1 equiv.) as a colorless oil (12 mg, 66% yield): Rf 0.15 (DCM/MeOH/NH4OH 14% 80/18/2); [α]D + 0.9 (c 1.0, CH3OH); IR (film): 3007, 2920, 2850, 1670, 1435, 1275, 1204, 1138, 960, 801, 764, 750; 1H NMR (500 MHz, MeOD) δ 7.81 (d, JH6-H5 = 8.1Hz, 1H, H6), 5.76 (d, JH1′-H2′ = 3.3 Hz, 1H, H1′), 5.73 (d, JH5-H6 = 8.1Hz, 1H, H5), 5.34 (t, JH8*-H9* = 4.6 Hz, 2 H, H8*, H9*), 5.14 (s, 1H, H1″), 4.30 (td, JH5′-H6′ = 6.5 Hz, JH5′-H4′ = 2.5 Hz, 1H, H5′), 4.15–4.01 (m, 5 H, H2′, H3′, H4′, H3″, H2″), 3.98 (d, JH4″-H5″ = 4.3 Hz, 1H, H4″), 3.29–3.25 (m, 2 H, H6′ab), 3.21 (dd, JH5″a-H4″ = 14.6 Hz, JH5″a-H5″b = 5.8 Hz, 1H, H5″a), 3.12 (dd, JH5″b-H4″ = 12.3 Hz, JH5″b-H5″a = 9.2 Hz, 1H, H5″b), 2.91 (t, JH1*-H2* = 7.3 Hz, 2 H, H1*), 2.06–2.01 (m, 4 H, H7*, H10*), 1.81–1.76 (m, 2 H, H2*), 1.35–1.29 ( m, 20 H, H3*-H6*, H11*-H16*), 0.91 (t, JH15*-H14* = 6.7 Hz, 3H, H17*); 13C NMR (125 MHz, MeOD) δ 181.8 (C8′), 168.7 (C7′), 166.1 (C4), 152.0 (C2), 142.2 (C6), 130.9 (C8*), 130.7 (C9*), 110.9 (C1″), 102.3 (C5), 91.6 (C1′), 85.5 (C2″), 80.0 (C4′), 78.1 (C5′), 76.2 (C4”), 75.5 (C2′), 73.8 (C3″), 71.2 (C3′), 44.3 (C5”), 33.0 (C6′), 30.8, 30.7, 30.6, 30.4, 30.3, 30.3, 30.1, 30.0 (C4*-C6*, C11*-C16*), 28.1 (C7*), 28.0 (C10*), 27.6 (C2*), 27.1 (C1*), 23.7 (C3*), 14.4 (C17*). HRMS ESI− calcd. for C34H54N5O10− (M + H)− 692.3876 found 692.3890.
3.1.18. Oxadiazole 12e
Compound 12e was obtained from the protected oxadiazole 11e (20 mg, 21 μmol, 1 equiv.) as a colorless oil (8 mg, 60% yield): Rf 0.15 (DCM/MeOH/NH4OH 14% 80/18/2); [α]D + 1 (c 1.0, CH3OH); IR (film): 3058, 2912, 2840, 1658, 1435, 1275, 1244, 1198, 993, 799, 751; 1H NMR (500 MHz, MeOD) δ 8.10 (d, JHa-Hb = 8.8 Hz, 2 H, Ha), 7.81 (d, JH6-H5 = 8.4 Hz, 1H, H6), 7.45 (t, JHd-He = 8.0 Hz, 2H, Hd), 7.25 (t, JHe-Hd = 7.5 Hz, 1H, He), 7.13–7.10 (m, 4 H, Hc, Hb), 5.78 (d, JH1′-H2′ = 3.0 Hz, 1H, H1′), 5.70 (d, JH5-H6 = 8.4 Hz, 1H, H5), 5.18 (s, 1H, H1″), 4.38 (td, JH5′-H6′ = 5.7 Hz, JH5′-H4′ = 3.1Hz, 1H, H5′), 4.17–4.14 (m, 2 H, H2′, H3′), 4.09–4.05 (m, 3 H, H4′, H3″, H2″), 3.99 (d, JH4″-H5″ = 3.5 Hz, 1H, H4″), 3.38–3.32 (m, 2H, H6′ab), 3.29–3.27 (m, JH5″a-H5″b = 5.8 Hz, 1H, H5″a), 3.17–3.11 (m, 1H, H5″b); 13C NMR (125 MHz, MeOD) δ 176.6 (C8′), 169.5 (C7′), 166.0 (C4), 163.6 (C9*), 156.6 (C10*), 152.1 (C2), 142.2 (C6), 131.3 (Cd), 131.2 (Ca), 126.0 (Ce), 121.3 (Cc), 119.0 (Cb), 110.8 (C1″), 102.4 (C5), 91.5 (C1′), 85.7 (C2″), 79.9 (C4′), 78.0 (C5′), 76.2 (C4”), 75.4 (C2′), 73.9 (C3″), 71.3 (C3′), 44.4 (C5”), 30.4 (C11*); HRMS ESI+ calcd. for C29H32N5O11+ (M + H)+ 626.2092 found 626.2078.
3.1.19. Oxadiazole 12f
Compound 12f was obtained from the protected oxadiazole 11f (44 mg, 41 μmol, 1 equiv.) as a colorless oil (22.8 mg, 63% yield): Rf 0.15 (DCM/MeOH/NH4OH 14% 80/18/2); [α]D + 8 (c 1.0, CH3OH); IR (film): 3052, 2926, 2854, 1698, 1410, 1275, 1203, 1132, 965, 799, 764, 750; 1H NMR (500 MHz, MeOD) δ 7.81 (d, JH6-H5 = 8.1Hz, 1H, H6), 5.77 (d, JH1′-H2′ = 3.2 Hz, 1H, H1′), 5.73 (d, JH5-H6 = 8.1Hz, 1H, H5), 5.41–5.36 (m, 2 H, H8*, H9*), 5.14 (s, 1H, H1″), 4.30 (td, JH5′-H6′ = 6.5 Hz, JH5′-H4′ = 2.5 Hz, 1H, H5′), 4.16–4.02 (m, 5 H, H2′, H3′, H4′, H3″, H2″), 3.98 (d, JH4″-H5″ = 4.8 Hz, 1H, H4″), 3.30–3.25 (m, 2 H, H6′ab), 3.21 (dd, JH5″a-H4″ = 14.9 Hz, JH5″a-H5″b = 5.7 Hz, 1H, H5″a), 3.12 (dd, JH5″b-H4″ = 13.1Hz, JH5″b-H5″a = 9.0 Hz, 1H, H5″b), 2.91 (t, JH1*-H2* = 7.6 Hz, 2H, H1*), 2.00–1.95 (m, 4H, H7*, H10*), 1.81–1.76 (m, 2H, H2*), 1.37–1.29 ( m, 20H, H3*-H6*, H11*-H16*), 0.90 (t, JH15*-H14* = 6.9 Hz, 3H, H17*); 13C NMR (125 MHz, MeOD) δ 181.8 (C8′), 168.7 (C7′), 166.1 (C4), 152.1 (C2), 142.2 (C6), 131.6 (C8*), 131.4 (C9*), 110.9 (C1″), 102.4 (C5), 91.5 (C1′), 85.6 (C2″), 80.0 (C4′), 78.0 (C5′), 76.2 (C4”), 75.5 (C2′), 73.8 (C3″), 71.3 (C3′), 44.3 (C5”), 33.6 (C7*), 33.5 (C10*), 33.0 (C6′), 30.7, 30.6, 30.5, 30.4, 30.3, 30.2, 30.0, 29.9 (C4*-C6*, C11*-C16*), 27.6 (C2*), 27.1 (C1*), 23.7 (C3*), 14.4 (C17*); HRMS ESI+ calcd. for C34H56N5O10+ (M + H)+ 694.4022 found 694.4012.
3.1.20. Oxadiazole 12g
Compound 12g was obtained from the protected oxadiazole 11g (20 mg, 20 μmol, 1 equiv.) as a colorless oil (12 mg, 63% yield): Rf 0.15 (DCM/MeOH/NH4OH 14% 80/18/2); [α]D + 1 (c 1.0, CH3OH); IR (film): 3028, 2913, 2841, 1660, 1435, 1275, 1133, 993, 800, 764, 750; 1H NMR (500 MHz, MeOD) δ 7.81 (d, JH6-H5 = 8.3 Hz, 1H, H6), 7.23 (d, JHa-Hb = 7.1Hz, 2 H, Ha), 7.16–7.11 (m, 3 H, Hb, Hc), 5.76 (d, JH1′-H2′ = 3.1Hz, 1H, H1′), 5.72 (d, JH5-H6 = 8.2 Hz, 1H, H5), 5.14 (s, 1H, H1″), 4.29 (td, JH5′-H6′ = 6.5 Hz, JH5′-H4′ = 2.7 Hz, 1H, H5′), 4.15–4.01 (m, 5 H, H2′, H3′, H4′, H3″, H2″), 3.98 (d, JH4″-H5″ = 3.9 Hz, 1H, H4″), 3.30–3.26 (m, 2H, H6′ab), 3.20 (dd, JH5″a-H4″ = 15.3 Hz, JH5″a-H5″b = 5.3 Hz, 1H, H5″a), 3.11 (dd, JH5″b-H4″ = 13.1Hz, JH5″b-H5″a = 9.0 Hz, 1H, H5″b), 2.90 (t, JH1*-H2* = 7.4 Hz, 2 H, H1*), 2.59 (t, JH9*-H10* = 7.7 Hz, 2 H, H9*), 1.80–1.74 (m, 2 H, H2*), 1.63–1.57 (m, 2 H, H8*), 1.37–1.28 ( m, 10 H, H3*-H7*); 13C NMR (125 MHz, MeOD) δ 181.8 (C8′), 168.7 (C7′), 166.1 (C4), 152.1 (C2), 143.9 (C10*), 142.2 (C6), 129.4 (Ca), 129.2 (Cb), 126.6 (Cc), 110.9 (C1″), 102.4 (C5), 91.5 (C1′), 85.6 (C2″), 79.9 (C4′), 78.1 (C5′), 76.2 (C4”), 75.5 (C2′), 73.8 (C3″), 71.2 (C3′), 44.3 (C5”), 36.9 (C9*), 32.7 (C6′), 30.5, 30.4, 30.3, 30.2, 30.1, 29.9 (C3*-C8*), 27.6 (C2*), 27.1 (C1*); HRMS ESI+ calcd. for C32H46N5O10+ (M + H)+ 660.3239 found 660.3226.
3.1.21. Oxadiazole 12h
Compound 12h was obtained from the protected oxadiazole 11h (42 mg, 42 μmol, 1 equiv.) as a white solid (19 mg, 68% yield) and as a 1/1 mixture of stereoisomers: Rf 0.15 (DCM/MeOH/NH4OH 14% 80/18/2); [α]D + 1.4 (c 1.0, CH3OH); IR (film): 2925, 2855, 1688, 1579, 1464, 1274, 1126, 748; 1H NMR (500 MHz, MeOD) δ 7.80 (d, JH5-H6 = 8.1 Hz, 0.5H, H6), 7.79 (d, JH5-H6 = 8.1 Hz, 0.5H, H6), 5.75 (d, JH1′-H2′ = 3.1 Hz, 1H, H1′), 5.72 (d, JH5-H6 = 8.1 Hz, 1H, H5), 5.14 (s, 0.5H, H1″), 5.12 (s, 0.5H, H1″), 4.31 (td, JH5′-H6′a = JH5′-H6′b = 6.0, JH4′-H5′ = 3.0 Hz, 1H, H5′), 4.26–4.16 (m, 2H, H3**), 4.16–4.09 (m, 2H, H1*, H2′), 4.09–4.01 (m, 3H, H4″, H4′, H3″), 3.97 (d, JH2″-H3″ = 3.5 Hz, 1H, H2″), 3.38–3.32 (m, 1H, H6′a), 3.30–3.26 (m, 1H, H5″a), 3.23 (dd, JH6′a-H6′b = 15.0, JH5′-H6′b = 6.0 Hz, 1H, H6′b), 3.14 (dd, JH5″a-H5″b = 13.0, JH4″-H5″b = 8.2 Hz, 1H, H5″b), 2.17–1.95 (m, 2H, H2*), 1.43–1.27 (m, 14H, H3*-H9*), 1.25 (t, JH3**-H4** = 7.1 Hz, 3H, H4**), 0.90 (t, JH9*-H10* = 6.9 Hz, 3H, H10*); 13C NMR (125 MHz, MeOD) δ 178.28, 178.22 (C2**), 170.5, 170.4 (C8′), 169.22, 169.18 (C7′), 166.16 (C4), 152.1 (C2), 142.2 (C6), 111.1, 111.0 (C1″), 102.4 (C5), 91.60, 91.57 (C1′), 85.9 (C4′), 80.0 (C4″), 78.3, 78.1 (C5′), 76.3 (C2″), 75.5 (C1*), 73.9 (C3′), 71.34, 71.30 (C2′), 63.2 (C3**), 44.3 (C5″), 33.1, 31.2, 31.0, 30.6, 30.5, 30.2, 28.1, 23.7 (C2*-C9*), 30.4 (C6′), 14.5, 14.4 (C4**, C10*); HRMS ESI+ calcd. for C30H48N5O12+ (M + H)+ 670.3294 found 670.3320.
3.1.22. Oxadiazole 12i
Compound 12i was obtained from the protected oxadiazole 11i (34 mg, 32 μmol, 1 equiv.) as a colorless oil (5.3 mg, 22% yield): Rf 0.15 (DCM/MeOH/NH4OH 14% 80/18/2); [α]D + 1 (c 1.0, CH3OH); IR (film): 2923, 1690, 1275, 1260, 764, 750; 1H NMR (500 MHz, MeOD) δ 7.89 (d, JH6-H5 = 8.1Hz, 1H, H6), 5.81 (d, JH1′-H2′ = 2.7 Hz, 1H, H1′), 5.74 (d, JH5-H6 = 8.1Hz, 1H, H5), 5.03 (s, 1H, H1″), 4.30 (td, JH5′-H6′ = 6.3 Hz, JH5′-H4′ = 2.2 Hz, 1H, H5′), 4.14–4.11 (m, 2 H, H2′, H3′), 4.07–4.04 (m, 1H, H4′), 3.98 (dd, JH3″-H2″ = 6.6 Hz, JH3″-H4″ = 4.8 Hz, 1H, H3″), 3.93 (dd, JH2″-H3″ = 4.6 Hz, JH2″-H1″ = 1.2 Hz, 1H, H2″), 3.88 (td, JH4″-H5″ = 7.1 Hz, JH4″-H3″ = 3.5 Hz, 1H, H4″), 3.24 (dd, JH6′a-H5′ = 8.7 Hz, JH6′a-H6′b = 6.3 Hz, 1H, H6′a), 3.19 (dd, JH6′b-H5′ = 8.7 Hz, JH6′b-H5′ = 6.3 Hz, 1H, H6′b), 3.06–3.00 (m, 1H, H1*), 2.91 (dd, JH5″a-H4″ = 13.3 Hz, JH5″a-H5″b = 3.7 Hz, 1H, H5″a), 2.79 (dd, JH5″b-H4″ = 13.3 Hz, JH5″b-H5″a = 7.5 Hz, 1H, H5″b), 1.79–1.69 (m, 4 H, H2*,H2**), 1.35–1.17 ( m, 34 H, H3**-H10**, H3*-H10*), 0.91–0.88 ( 6H, H11*, H11**); 13C NMR (125 MHz, MeOD) δ 184.5 (C8′), 168.6 (C7′), 166.2 (C4), 152.2 (C2), 142.0 (C6), 111.1 (C1″), 102.4 (C5), 91.0 (C1′), 86.3 (C4′), 84.5 (C4″), 78.5 (C5′), 76.8 (C2”), 75.8 (C2′), 73.3 (C3″), 71.3 (C3′), 45.3 (C5”), 39.6 (C1*), 34.6 (C2*), 34.5 (C2**), 33.0 (C9*, C9**), 30.8, 30.7, 30.4, 30.4 (C4*-C8*, C4**-C8**), 28.2 (C3*, C3**), 23.7 (C10*, C10**), 14.4 (C11*, C11**); HRMS ESI+ calcd. for C38H66N5O10+ (M + H)+ 752,4804 found 752.4804.
3.1.23. Oxadiazole 12j
To a solution of oxadiazole 12h (2.5 mg, 3.7 µmol, 1 equiv.) in methanol (0.4 mL) at 0 °C was added a solution of NH4HCO3 in water (0.1 M, 0.5 mL) and trimethylamine (14 µL, 99 µmol, 27 equiv.). After 48 h at 0 °C, the reaction mixture was diluted in 5 mL water, and the solution was freeze-dried. The residue was purified on a reverse phase column (Sep-Pak Cartridges C18) using a mixture of an aqueous solution NH4HCO3 (0.1 M) and acetonitrile (1/0 to 1/1, v/v). The fractions containing the product were freeze-dried to afford product 12j as a white powder (0.4 mg) and as a 1/1 mixture of stereoisomers in 17% yield: Rf 0.07 (DCM/MeOH/NH4OH 14% 80/18/2); 1H NMR (500 MHz, MeOD) δ 7.87 (d, JH5-H6 = 8.1 Hz, 0.5H, H6), 7.85 (d, JH5-H6 = 8.1 Hz, 0.5H, H6), 5.80 (d, JH1′-H2′ = 3.4 Hz, 0.5H, H1′), 5.79 (d, JH1′-H2′ = 3.4 Hz, 0.5H, H1′), 5.74 (d, JH5-H6 = 8.1 Hz, 0.5H, H5), 5.74 (d, JH5-H6 = 8.1 Hz, 0.5H, H5), 5.12 (s, 0.5H, H1″), 5.09 (s, 0.5H, H1″), 4.37–4.31 (m, 1H, H5′), 4.18–3.85 (m, 6H, H1*, H2′, H4″, H4′, H3″, H2″), 3.14–2.72 (m, 4H, H6′a, H5″a, H6′b, H5″b), 2.18–1.98 (m, 2H, H2*), 1.40–1.22 (m, 14H, H3*-H9*), 0.90 (t, JH9*-H10* = 6.9 Hz, 3H, H10*); 13C NMR (125 MHz, MeOD) δ 181.4 (C2**), 170.3 (C8′), 169.0 (C7′), 166.3 (C4), 152.3, 152.2(C2), 142.4, 142.2 (C6), 110.4, 110.0 (C1″), 102.57, 102.51 (C5), 91.7 (C1′), 86.4 (C4′), 80.0 (C4″), 78.1 (C5′), 76.5 (C2″), 75.55, 75.49 (C1*), 73.7, 73.6 (C3′), 71.4, 71.3 (C2′), 44.94, 44.87 (C5″), 33.1, 32.1, 31.9, 30.8, 30.7, 30.6, 30.5, 26.9, 23.7 (C2*-C9*, C6′), 14.5 (C10*); HRMS ESI+ calcd. for C28H44N5O12+ (M + H)+ 642.2942 found 642.2979.




 ,
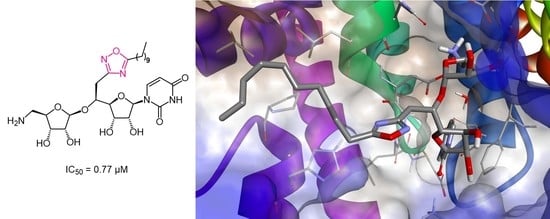
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}