
Balancing the Virulence and Antimicrobial Resistance in VISA DAP-R CA-MRSA Superbug

 and
and
Abstract
:1. Introduction
2. Results
2.1. Biological Fitness Costs of AMR and Virulence
2.2. Biofilm Production
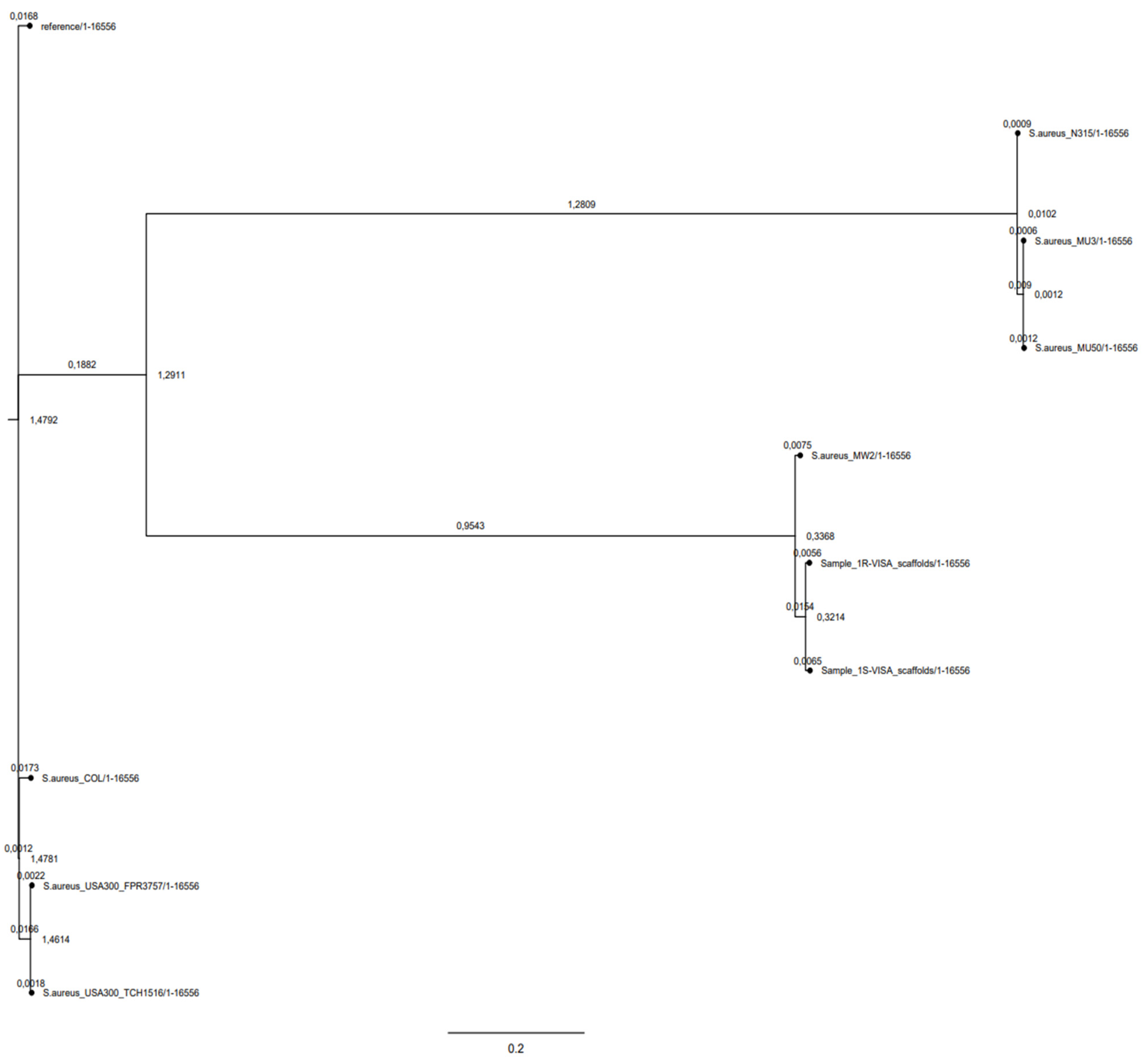
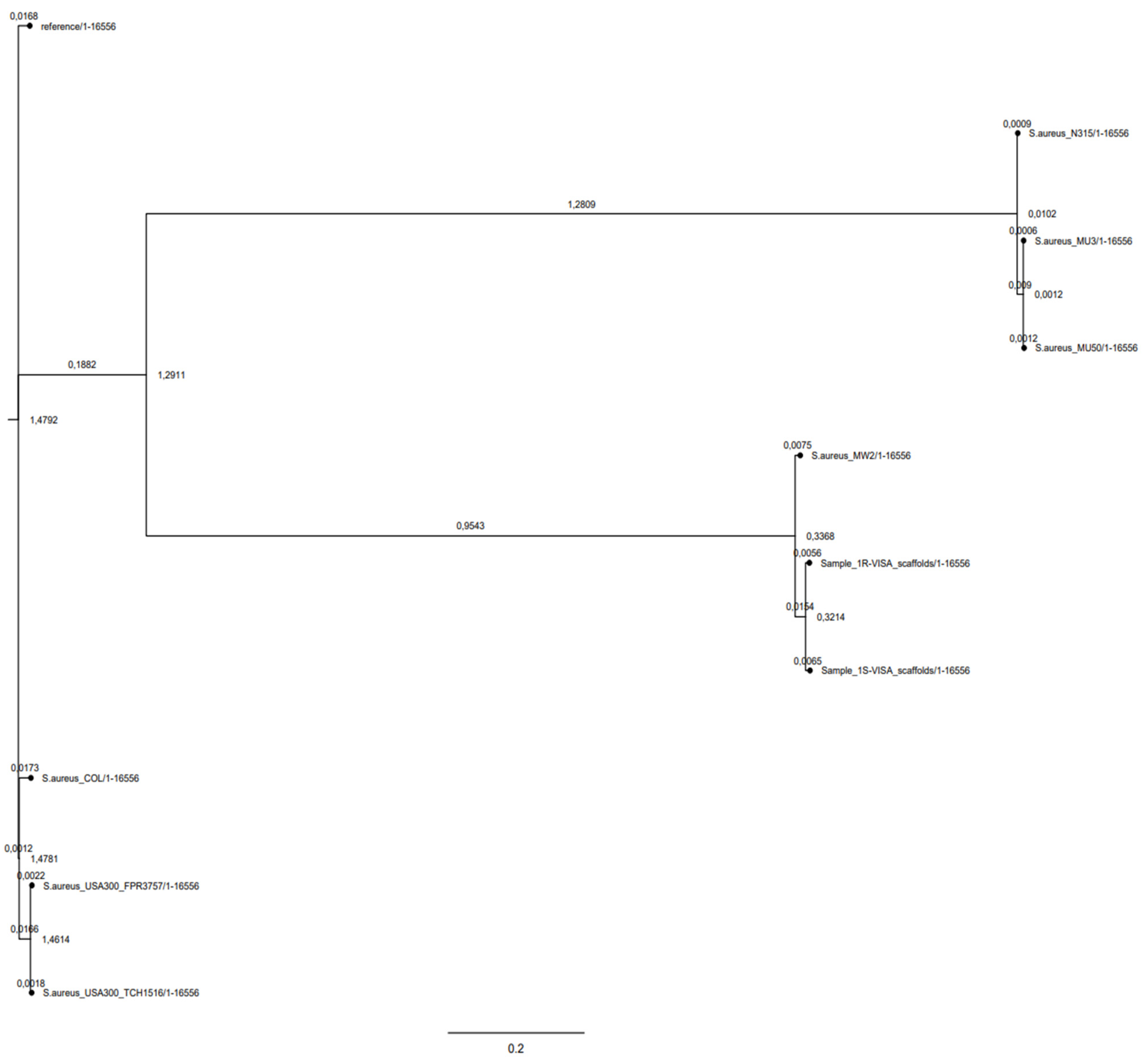
2.3. Phylogeny
2.4. Genomic Characterization
2.5. Genomic SNPs
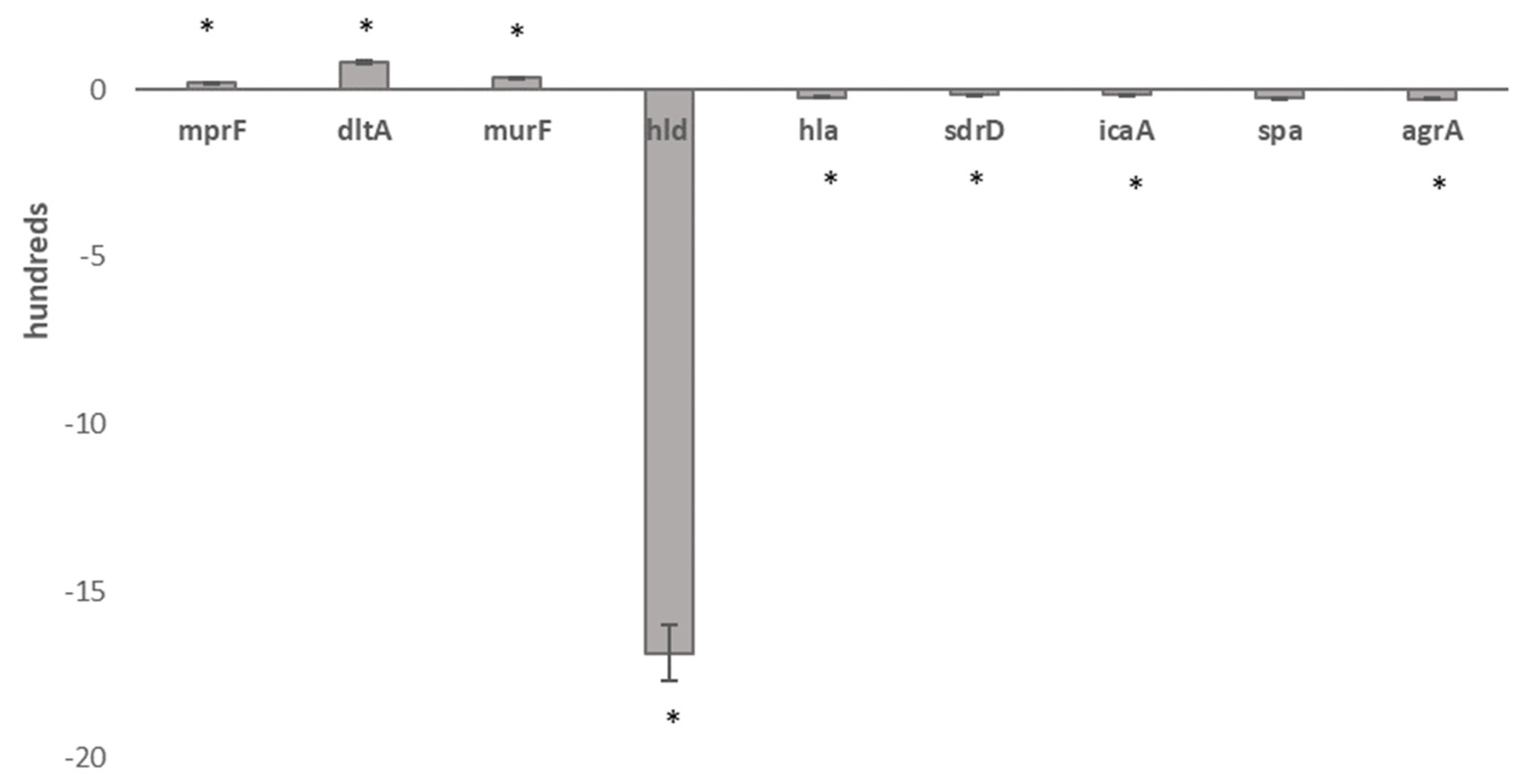
2.6. Comparative Transcriptomics
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Bacterial Strains
5.2. Biological Costs Determined by the Growth Kinetics Test
5.3. Biofilm Production
5.4. Whole-Genome Sequencing
5.5. De-Novo Genome Assembly
5.6. Gene Annotation
5.7. Single Nucleotide Variants (SNVs)
5.8. Whole-Genome Single Nucleotide Polymorphisms (wgSNPs) Effect Prediction
5.9. Phylogeny and Genomic Epidemiology
5.10. RNA-Seq
5.10.1. RNA-Seq Bacterial Cultures
5.10.2. RNA-Seq Libraries
5.10.3. RNA-Extraction
5.10.4. Tru-Seq Library Preparation
5.10.5. Short-Insert Library Preparation
5.10.6. Tru-Seq Library Raw Read Post-Processing
5.10.7. Short-Insert Library Raw Read Post-Processing
5.10.8. Tru-Seq and Short-Insert Read Analysis
5.11. DAVID Enrichment Analysis
5.12. Real-Time qPCR validation
5.13. DNA-Sequencing and RNA-Sequencing Data Accession Number
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. WHO Publishes List of Bacteria for which New Antibiotics Are Urgently Needed. 2017. Available online: https://www.who.int/news/item/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed on 1 January 2020).
- Patel, R.M.D. Biofilms and Antimicrobial Resistance. Clin. Orthop. Relat. Res. 2005, 437, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Seral, C.; Van Bambeke, F.; Tulkens, P.M. Quantitative analysis of gentamicin, azithromycin, telithromycin, ciprofloxacin, moxifloxacin, and oritavancin (LY333328) activities against intracellular Staphylococcus aureus in mouse J774 macrophages. Antimicrob. Agents Chemother. 2003, 47, 2283–2292. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, T.M.; Levy, S.B. Differential expression of over 60 chromosomal genes in Escherichia coli by constitutive expression of MarA. J. Bacteriol. 2000, 182, 3467–3474. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.K.; Fung, C.P.; Lin, J.C.; Chen, J.H.; Chang, F.Y.; Chen, T.L.; Siu, L.K. Klebsiella pneumoniae outer membrane porins OmpK35 and OmpK36 play roles in both antimicrobial resistance and virulence. Antimicrob. Agents Chemother. 2011, 55, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Moya, B.; Juan, C.; Albertí, S.; Pérez, J.L.; Oliver, A. Benefit of having multiple ampD genes for acquiring beta-lactam resistance without losing fitness and virulence in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2008, 52, 3694–3700. [Google Scholar] [CrossRef] [PubMed]
- Yeung, A.T.; Bains, M.; Hancock, R.E. The sensor kinase CbrA is a global regulator that modulates metabolism, virulence, and antibiotic resistance in Pseudomonas aeruginosa. J. Bacteriol. 2011, 193, 918–931. [Google Scholar] [CrossRef]
- David, M.Z.; Daum, R.S. Community-associated methicillin-resistant Staphylococcus aureus: Epidemiology and clinical consequences of an emerging epidemic. Clin. Microbiol. Rev. 2010, 23, 616–687. [Google Scholar] [CrossRef]
- Cuny, C.; Lothar, H.W.; Wolfgang, W. Livestock-Associated MRSA: The Impact on Humans. Antibiotics 2015, 4, 521–543. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Ando, R.; Matsumoto, T.; Ishii, Y.; Tateda, K. Association between cell growth and vancomycin resistance in clinical community-associated methicillin-resistant Staphylococcus aureus. Infect. Drug Resist. 2019, 12, 2379–2390. [Google Scholar] [CrossRef]
- Almeida, S.T.; Paulo, A.C.; Babo, J.; Borralho, J.; Figueiredo, C.; Gonçalves, B.; Lança, J.; Louro, M.; Morais, H.; Queiroz, J.; et al. Absence of methicillin-resistant Staphylococcus aureus colonization among immunocompetent healthy adults: Insights from a longitudinal study. PLoS ONE 2021, 16, e0253739. [Google Scholar]
- Burrus, V.; Waldor, M.K. Shaping bacterial genomes with integrative and conjugative elements. Res. Microbiol. 2004, 155, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Handel, A.; Regoes, R.R.; Antia, R. The role of compensatory mutations in the emergence of drug resistance. PLoS Comput. Biol. 2006, 2, e137. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.A.; Sharma-Kuinkel, B.K.; Maskarinec, S.A.; Eichenberger, E.M.; Shah, P.P.; Carugati, M.; Holland, T.L.; Fowler, V.G., Jr. Methicillin-resistant Staphylococcus aureus: An overview of basic and clinical research. Nat. Rev. Microbiol. 2019, 17, 203–218. [Google Scholar] [CrossRef] [PubMed]
- McGuinness, W.A.; Malachowa, N.; DeLeo, F.R. Vancomycin Resistance in Staphylococcus aureus. Yale J. Biol. Med. 2017, 90, 269–281. [Google Scholar]
- Singh, A.; Singh, S.; Singh, J.; Rahman, M.; Pathak, A.; Prasad, K.N. Survivability and fitness cost of heterogeneous vancomycin-intermediate Staphylococcus aureus. Indian J. Med. Microbiol. 2017, 35, 415–416. [Google Scholar] [CrossRef]
- Capone, A.; Cafiso, V.; Campanile, F.; Parisi, G.; Mariani, B.; Petrosillo, N.; Stefani, S. In Vivo development of daptomycin resistance in vancomycin-susceptible methicillin-resistant Staphylococcus aureus severe infections previously treated with glycopeptides. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 625–631. [Google Scholar] [CrossRef]
- Gillen, A.L.; Conrad, J.; Cargill, M. The Genesis and Emergence of Community-Associated Methicillin-Resistant Staphylococcus aureus (CA-MRSA): An Example of Evolution in Action? Answ. Res. J. 2015, 8, 391–401. [Google Scholar]
- Lindsay, J.A.; Holden, M.T. Staphylococcus aureus: Superbug, super genome? Trends Microbiol. 2004, 12, 378–385. [Google Scholar] [CrossRef]
- Malachowa, N.; DeLeo, F.R. Mobile genetic elements of Staphylococcus aureus. Cell. Mol. Life Sci. 2010, 67, 3057–3071. [Google Scholar] [CrossRef]
- Jamrozy, D.; Coll, F.; Mather, A.E.; Harris, S.R.; Harrison, E.M.; MacGowan, A.; Karas, A.; Elston, T.; Török, M.E.; Parkhill, J.; et al. Evolution of mobile genetic element composition in an epidemic methicillin-resistant Staphylococcus aureus: Temporal changes correlated with frequent loss and gain events. BMC Genom. 2017, 18, 684. [Google Scholar] [CrossRef]
- Maki, H.; McCallum, N.; Bischoff, M.; Wada, A.; Berger-Bächi, B. tcaA inactivation increases glycopeptide resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 2004, 48, 1953–1959. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.I.; Kim, J.W.; Kang, G.S.; Kim, H.S.; Yoo, J.S.; Lee, Y.S. Prevalence of amino acid changes in the yvqF, vraSR, graSR, and tcaRAB genes from vancomycin intermediate resistant Staphylococcus aureus. J. Microbiol. 2013, 51, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Cafiso, V.; Bertuccio, T.; Purrello, S.; Campanile, F.; Mammina, C.; Sartor, A.; Raglio, A.; Stefani, S. dltA overexpression: A strain-independent keystone of daptomycin resistance in methicillin-resistant Staphylococcus aureus. Int. J. Antimicrob. Agents 2014, 43, 26–31. [Google Scholar] [CrossRef]
- Cafiso, V.; Bertuccio, T.; Spina, D.; Purrello, S.; Campanile, F.; Di Pietro, C.; Purrello, M.; Stefani, S. Modulating activity of vancomycin and daptomycin on the expression of autolysis cell-wall turnover and membrane charge genes in hVISA and VISA strains. PLoS ONE 2012, 7, e29573. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.N.; Bayer, A.S.; Weidenmaier, C.; Grau, T.; Wanner, S.; Stefani, S.; Cafiso, V.; Bertuccio, T.; Yeaman, M.R.; Nast, C.C.; et al. Phenotypic and genotypic characterization of daptomycin-resistant methicillin-resistant Staphylococcus aureus strains: Relative roles of mprF and dlt operons. PLoS ONE 2014, 9, e107426. [Google Scholar] [CrossRef]
- Cafiso, V.; Stracquadanio, S.; Lo Verde, F.; De Guidi, I.; Zega, A.; Pigola, G.; Stefani, S. Genomic and Long-Term Transcriptomic Imprints Related to the Daptomycin Mechanism of Action Occurring in Daptomycin-and Methicillin-Resistant Staphylococcus aureus Under Daptomycin Exposure. Front. Microbiol. 2020, 11, 1893. [Google Scholar] [CrossRef]
- Li, M.; Cheung, G.Y.; Hu, J.; Wang, D.; Joo, H.S.; DeLeo, F.R.; Otto, M. Comparative analysis of virulence and toxin expression of global community-associated methicillin-resistant Staphylococcus aureus strains. J. Infect. Dis. 2010, 202, 1866–1876. [Google Scholar] [CrossRef]
- Traber, K.E.; Lee, E.; Benson, S.; Corrigan, R.; Cantera, M.; Shopsin, B.; Novick, R.P. agr function in clinical Staphylococcus aureus isolates. Microbiology 2008, 154 Pt 8, 2265. [Google Scholar] [CrossRef]
- Beceiro, A.; Tomás, M.; Bou, G. Antimicrobial resistance and virulence: A successful or deleterious association in the bacterial world? Clin. Microbiol. Rev. 2013, 26, 185–230. [Google Scholar] [CrossRef]
- EUCAST. Available online: https://www.eucast.org/eucast_news/news_singleview/?tx_ttnews%5Btt_news%5D=459cHash=160a5b91371e598957e10178fb3aa143 (accessed on 1 July 2022).
- Ramadhan, A.A.; Hegedus, E. Survivability of vancomycin resistant enterococci and fitness cost of vancomycin resistance acquisition. J. Clin. Pathol. 2005, 58, 744–746. [Google Scholar] [CrossRef]
- Cafiso, V.; Bertuccio, T.; Santagati, M.; Demelio, V.; Spina, D.; Nicoletti, G.; Stefani, S. agr-Genotyping and transcriptional analysis of biofilm-producing Staphylococcus aureus. FEMS Immunol. Med. Microbiol. 2007, 51, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Cafiso, V.; Lo Verde, F.; Zega, A.; Pigola, G.; Rostagno, R.; Borrè, S.; Stefani, S. Genomic Characterization of a New Biofilm-Forming and Adhesive ST398 Human-Adapted MSSA Lineage Causing Septic Knee Arthritis Following Surgical Reconstruction. Microorganisms 2021, 9, 305. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, l.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef] [PubMed]
- Bartels, M.D.; Petersen, A.; Worning, P.; Nielsen, J.B.; Larner-Svensson, H.; Johansen, H.K.; Andersen, L.P.; Jarløv, J.O.; Boye, K.; Larsen, A.R.; et al. Comparing whole-genome sequencing with Sanger sequencing for spa typing of methicillin-resistant Staphylococcus aureus. J. Clin. Microbiol. 2014, 52, 4305–4308. [Google Scholar] [CrossRef]
- Joensen, K.G.; Scheutz, F.; Lund, O.; Hasman, H.; Kaas, R.S.; Nielsen, E.M.; Aarestrup, F.M. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 2014, 52, 1501–1510. [Google Scholar] [CrossRef]
- Carattoli, A.; Hasman, H. PlasmidFinder and in silico pMLST: Identification and typing of plasmid replicons in whole-genome sequencing (WGS). In Horizontal Gene Transfer; Humana: New York, NY, USA, 2020; pp. 285–294. [Google Scholar]
- Johansson, M.; Bortolaia, V.; Tansirichaiya, S.; Aarestrup, F.M.; Roberts, A.P.; Petersen, T.N. Detection of mobile genetic elements associated with antibiotic resistance in Salmonella enterica using a newly developed web tool: MobileElementFinder. J. Antimicrob. Chemother. 2021, 76, 101–109. [Google Scholar] [CrossRef]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A fast phage search tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef]
- Van Der Mee-Marquet, N.; Corvaglia, A.R.; Valentin, A.S.; Hernandez, D.; Bertrand, X.; Girard, M.; Kluytmansf, J.; Donnio, P.Y.; Francois, P. Analysis of prophages harboured by the human-adapted subpopulation of Staphylococcus aureus CC398. Infect. Genet. Evol. 2013, 18, 299–308. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Strain | Sccmec Cassette | Agr-Group | DAP and Glycopeptide Resistance | PVL | Generation Time Average (min) | Lag Growth Phase Duration Average (h) |
|---|---|---|---|---|---|---|
| 1S CA-MRSA | IVa | III | VSSA, DAP-S | + | 30 | 1.0 |
| 1R CA-MRSA | Iva | III | VISA, DAP-R | + | 60 | 2.00 |
| MW2 CA-MRSA | Iva | III | VSSA, DAP-S | + | 30 | 2.30 |
| S. aureus ATCC 29213 | II | II | VSSA, DAP-S | - | 30 | 2.25 |
| Strains | MIC | Biofilm | δ-hemolysis | ||||
|---|---|---|---|---|---|---|---|
| CFX | DAP | VAN | TEC | RIF | |||
| 1-S | 32 | 0.5 | 1 | 1 | <0.06 | No Producer | Negative |
| 1-R | 48 | 2 | 8 | 32 | >256 | No Producer | Negative |
| Strain | MLST | PFGE | Agr-Type | Spa-Type | SCCmec | Plasmids | Phages | Virulome | Resistome | Resistance Gene SNPs | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | NUCL. Change | AA Change | AMR | |||||||||||
| 1-S | ST-1 [17] | USA 400 [17] | III [17] | t127 | IVa | rep5a (pN315) rep7a (repC-Cassette) rep7c (MSSA476) rep10 (pDLK1) rep16 (pSAS) | Phage Staph phiJB-NC028669 Phage Staph-ST398_4NC023499 | ebpS, ebh, vwb, eap, atl, fib, spa, clfA, clfB vwb, fnbA, fnbB, eap, sdrC, sdrD, sdrE sdrH, icaRABC, cna | Adherence | Aminoglycosides-R: ant(6)-Ia, aph(3′)-III β-lactams-R: blaZ, mecA Macrolides-R: ermC Tetracyclines-R: tetK | gyrA | 251C > T | S84L | FQs |
| grlA | 239 C > T | S80F | FQs | |||||||||||
| hla, hlgACB, hld [17], hlb, eta, lukE, lukSF-PV, set2, set3, set16, set17, set18, set19 set20, set21, set22, set23 set25, set26, SEnt-ike, SEntHSEntA, sal, tst | Toxins | |||||||||||||
| sspABC, geh, nuc hysA, lip, splABCF sspB2, sak, scn | Exoenzyme genes | |||||||||||||
| cap8J, cap8I, cap8H, cap8G, capF, cap8E, capD, cap8C, cap8B, cap5A, isb, capD, cap8P, capC, capB,capA, cap8O, cap8M, cap8M, cap8L, cap8K, cap5M | Host Immune Evasion | |||||||||||||
| essB, esaB, essA esaA, esxA, esxB, esaC, essC | Secretion System | |||||||||||||
| 1-R | ST-1 [17] | USA 400 [17] | III [17] | t127 | IVa | rep5a (pN315) rep7a (repC-Cassette) rep7c (MSSA476) rep10 (pDLK1) rep16 (pSAS) | Phage Staph phiJB-NC028669 Phage Staph-ST398_4NC023499 | ebpS, ebh, vwb, eap, atl, fib, spa, clfA, clfB vwb, fnbA, fnbB, eap, sdrC, sdrD, sdrE sdrH, icaRABC, cna | Adherence | Aminoglycosides-R: ant(6)-Ia, aph(3′)-III β-lactams-R: blaZ, mecA Macrolides-R: ermC Tetracyclines-R: tetK | gyrA | 251C > T | S84L | FQs |
| grlA | 239 C > T | S80F | FQs | |||||||||||
| rpoB | 1441C > T | H481Y | RIF [17] | |||||||||||
| mprF | 1033A > G | T345A | DAP [17] | |||||||||||
| hla, hlgACB, hld [17], hlb, eta, lukE, lukSF-PV, set2, set3, set16, set17, set18, set19 set20, set21, set22, set23 set25, set26, SEnt-ike, SEntHSEntA, sal, tst | Toxins | |||||||||||||
| sspABC, geh, nuc hysA, lip, splABCF sspB2, sak, scn | Exoenzyme genes | |||||||||||||
| cap8J, cap8I, cap8H, cap8G, capF, cap8E, capDcap8C, cap8B, cap5A, isb, capDcap8P, capC, capB, capA, cap8O, cap8M, cap8M, cap8Lcap8K, cap5M | Host Immune Evasion | |||||||||||||
| essB, esaB, essA esaA, esxA, esxB, esaC, essC | Secretion System | |||||||||||||
| Gene | Product | nsSNPs |
|---|---|---|
| HI-nsSNPs | ||
| MW1125 | YfhO family protein | Gly75 * |
| MW1482 | proline dipeptidase | Thr265Ala |
| MW2347 | 8-amino-7-oxononanoate synthase (bioF) | Glu272 * |
| MW1963 | Accessory gene regulator protein A (agrA) | Arg170 * |
| Gene | Product | nsSNPs |
|---|---|---|
| MI- nsSNPs | ||
| MW0014 | Cyclic-di-AMP phosphodiesterase | Ile186Met |
| MW0165 | N-acetylmuramic acid 6-phosphate etherase | Ile156Asn |
| MW0447 | Ribonuclease M5 | Asp98Glu |
| MW1054 | Exfoliative toxin A | Leu47Ile |
| MW1080 | Pseudouridine synthase | Glu294Val |
| MW1664 | Peroxiredoxin osmotic stress-related protein | Asn87Lys |
| MW1826 | DUF2154 domain-containing protein VraT | Ala59Glu |
| MW2107 | D-ornithine--citrate ligase SfnaD | Leu195Trp |
| MW2286 | Malate:quinone oxidoreductase 1 | Val280Glu |
| MW2393 | D-histidine (S)-2-aminobutanoyltransferase CntL | Glu31Leu |
| MW2533 | HTH-type transcriptional regulator | Gly41Asp |
| MW0131 | Capsular polysaccharide synthesis enzyme H | Tyr130Hys |
| MW0134 | Capsular polysaccharide synthesis enzyme K | Val120Glu |
| MW2070 | Cobalt-zinc-cadmium resistance protein czcD | Asp52Glu |
| MW1283 | Dihydrodipicolinate synthase DapA | Ala101Thr |
| MW1247 | Phosphatidylglycerol lysyl-transferase MprF | Thr345Ala Leu538Phe |
| MW2304 | Proton/sodium-glutamate symport protein | Val232Glu |
| MW1324 | Extracellular matrix-binding protein Ebh | Val1768Asp |
| MW2287 | L-lactate permease LctP | Ile178Phe |
| MW1307 | UDP-NAG--NAM-(pentapeptide) pyrophosphoryl-undecaprenol N-acetylglucosamine transferase MurG | Ile121ASn |
| MW0497 | DNA-directed RNA polymerase subunit beta RpoB | His481Tyr |
| MW0517 | Serine-aspartate repeat-containing protein SdrD | Thr1313Ser |
| Kegg Pathway | Gene Number in Cluster | Products | p-Value |
|---|---|---|---|
| Annotation Cluster 1 Enrichment Score: 1.06 | |||
| Glycolysis/Gluconeogenesis Microbial metabolism in diverse environments | 12 17 | 6-phosphofructokinase (pfkA) MW1642 Acetate--CoA ligase (acsA) MW1676 Bifunctional acetaldehyde-CoA/alcohol dehydrogenase (adhE) MW0123 Glucose-6-phosphate isomerase (pgi) MW0844 Glucokinase (glcK) MW1499 Glyceraldehyde-3-phosphate dehydrogenase (gap) MW0734 Phosphoglycerate kinase (pgk) MW0735 Pyruvate kinase (pykA) MW1641 Triosephosphate isomerase (tpiA) MW0736 PTS system transporter subunit IIA (crr) MW1312 Fructose-1,6-bisphosphate aldolase (fdaB) MW2525 Phosphoglucomutase (pgcA) MW2411–– Glutamate-1-semialdehyde aminotransferase (gsaB) MW1804 Carbamate kinase (arcC) MW2553 Delta-aminolevulinic acid dehydratase (hemB) MW1612 Dipeptidase PepV (pepV) MW1694 Formate--tetrahydrofolate ligase (fhs) MW1675 Fructose-1,6-bisphosphate aldolase (fdaB) MW2525 Fumarate hydratase (fumC) MW1792 3-hexulose-6-phosphate synthase (hxlA) MW0525 Citrate synthase (citZ) MW 1639 Phosphoglucomutase (pgcA) MW2411 Glyoxalase MW2442 Malate:quinone oxidoreductase (mqo1) MW2286 Nitrate reductase subunit beta (narH) MW2318 Putative translaldolase (tal) Mw1721 Respiratory nitrate reductase subunit gamma (narI) MW2316 Sulfite reductase (NADPH) flavoprotein subunit alpha (cysJ) MW2540 Uroporphyrinogen III synthase (hemD) MW1613 | 3.2 × 10−2 2.3 × 10−1 |
| Annotation Cluster 2 Enrichment Score: 0.88 | |||
| Mismatch repair Homologous recombination DNA replication | 7 | ATP-dependent DNA helicase PcrA MW1846 ATP-dependent DNA helicase RecG (recG) MW1110 DNA polymerase III PolC (polC) MW1147 DNA polymerase III subunit delta (holA) MW1538 DNA repair protein RecO (MW1518) DNA polymerase III subunit epsilon MW1835 DNA primase (SAOUHSC_01663) NAD-dependent DNA ligase (lig) MW1845 Single-stranded-DNA-specific exonuclease RecJ MW1586 Recombination and DNA strand exchange inhibitor protein (mutS2) MW1027 | 4.3 × 10−2 2.1 × 10−1 2.5 × 10−1 |
| Gene ID | Description | RPKM 1-R | Expression 1-R | RPKM 1-S | Expression 1-S | q-Value |
|---|---|---|---|---|---|---|
| AMR-RELATED TRAITS | ||||||
| Overexpression | ||||||
| MW2277 | Teicoplanin resistance-associated protein A (TcaA) | 33 | 54 | 0 | 0 | 0 |
| MW0005 | DNA gyrase subunit B (GyrB) | 42 | 68 | 0 | 0 | 0 |
| MW2005 | UDP-N-acetylmuramoylalanyl-D-glutamyl-2, 6-diaminopimelate- D-alanyl-D-alanyl ligase (MurF) | 36 | 59 | 0 | 0 | 0 |
| MW0814 | D-alanine--poly(phosphoribitol) ligase subunit 1 (DltA) | 83 | 135 | 0 | 0 | 0 |
| MW1247 | Oxacillin resistance-related FmtC protein (MprF) | 23 | 38 | 0 | 0 | 0 |
| Underexpression | ||||||
| MW0657 | Quinolone resistance protein (NorA) | 0 | 0 | 14 | 21 | 0 |
| MW1325 | Quinolone resistance protein (NorB) | 0 | 0 | 18 | 26 | 0 |
| MW0111 | Tetracycline resistance protein (Tet38) | 0 | 0 | 93 | 0 | 0 |
| VIRULENCE-RELATED TRAITS | ||||||
| Adhesion | ||||||
| Underexpression | ||||||
| MW0516 | Ser-Asp-rich fibrinogen-binding bone sialoprotein-binding protein SdrC | 0 | 0 | 20 | 30 | 0 |
| MW0517 | Ser-Asp-rich fibrinogen-binding bone sialoprotein-binding protein SdrD | 0 | 0 | 12 | 18 | 0 |
| MW1880 | Truncated cell surface protein map-w (EapP/Map) | 0 | 0 | 11 | 16 | 0 |
| Effector delivery system | ||||||
| Underexpression | ||||||
| MW0259 | Type VII secretion protein EsaA | 0 | 0 | 17 | 26 | 0 |
| Exotoxins | ||||||
| Underexpression | ||||||
| MW1959 | Delta-hemolysin (hld) | 0 | 0 | 1685 | 2516 | 0 |
| MW1044 | Alpha-hemolysin (hla) | 0 | 0 | 21 | 31 | 0 |
| MW2344 | Gamma-hemolysin component B (hlgB) | 0 | 0 | 21 | 31 | 0 |
| MW2343 | Gamma-hemolysin component C (hlgC) | 0 | 0 | 38 | 57 | 0 |
| MW0051 | Enterotoxin H (seh) | 0 | 0 | 28 | 42 | 0 |
| MW1552 | Enterotoxin homologous Protein | 0 | 0 | 36 | 37500 | 0 |
| Membrane-acting toxin and Superantigen | ||||||
| Underexpression | ||||||
| MW0084 | Immunoglobulin G binding protein A precursor (SpA) | 0 | 0 | 23 | 34 | 6.92 × 10−94 |
| Exoenzyme s | ||||||
| Underexpression | ||||||
| MW2590 | Triacylglycerol lipase precursor (Lip) | 0 | 0 | 7 | 11 | 2.97 × 10−8 |
| MW1754 | Serine protease SplB | 0 | 0 | 28 | 42 | 0 |
| Immune modulatory proteins | ||||||
| Overexpression | ||||||
| MW0137 | Capsular polysaccharide synthesis enzyme Cap8N | 1 | 27 | 0 | 0 | 0 |
| MW2341 | IgG-binding protein SBI (sbi) | 24 | 40 | 0 | 0 | 8.22 × 10−3 |
| Underexpression | ||||||
| MW0126 | Capsular polysaccharide synthesis enzyme Cap8C | 0 | 0 | 0 | 20 | 1.01 × 10−3 |
| MW0130 | Capsular polysaccharide synthesis enzyme Cap8G | 0 | 0 | 0 | 20 | 1.35 × 10−3 |
| MW0139 | Capsular polysaccharide synthesis enzyme Cap8P | 0 | 0 | 14 | 21 | 0 |
| MW2559 | Immunodominant antigen B (IsaB) | 0 | 0 | 82 | 122 | 0 |
| Biofilm | ||||||
| Underexpression | ||||||
| MW2586 | Poly-beta-1,6-N-acetyl-D-glucosamine synthase icaA | 0 | 0 | 13 | 20 | 0 |
| AMR AND VIRULENCE TRANSCRIPTIONAL REGULATORS | ||||||
| Overexpression | ||||||
| MW1824 | Two-component response regulator VraR | 232 | 378 | 0 | 0 | 0 |
| MW1825 | Two-component sensor histidine kinase VraS | 270 | 438 | 0 | 0 | 0 |
| MW1988 | RNA polymerase sigma factor SigB | 2 | 67 | 0 | 4 | 5.39 × 10−3 |
| MW0668 | Response regulator SaeR | 89 | 145 | 0 | 0 | 0 |
| MW1705 | Repressor toxin Rot | 72 | 117 | 0 | 0 | 1.87 × 10−3 |
| Underexpression | ||||||
| MW0018 | Response regulatory protein WalR | 0 | 0 | 27 | 0 | 0 |
| MW0019 | Sensor protein kinase WalK | 0 | 0 | 23 | 0 | 0 |
| MW1446 | Respiratory response protein SrrA | 0 | 0 | 139 | 208 | 0 |
| MW1960 | Accessory gene regulator B (AgrB) | 0 | 7 | 30 | 789 | 2.9 × 10−53 |
| MW1962 | Accessory gene regulator C (AgrC) | 0 | 0 | 8 | 212 | 0 |
| MW1963 | Accessory gene regulator A (AgrA) | 0 | 11 | 27 | 717 | 2.5 × 10−57 |
| MW0085 | HTH-type transcriptional regulator SarS (SarS) | 0 | 0 | 26 | 39 | 0 |
| MW0621 | Response regulator protein GraR | 0 | 0 | 32 | 48 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salemi, R.; Zega, A.; Aguglia, E.; Lo Verde, F.; Pigola, G.; Stefani, S.; Cafiso, V. Balancing the Virulence and Antimicrobial Resistance in VISA DAP-R CA-MRSA Superbug. Antibiotics 2022, 11, 1159. https://doi.org/10.3390/antibiotics11091159
Salemi R, Zega A, Aguglia E, Lo Verde F, Pigola G, Stefani S, Cafiso V. Balancing the Virulence and Antimicrobial Resistance in VISA DAP-R CA-MRSA Superbug. Antibiotics. 2022; 11(9):1159. https://doi.org/10.3390/antibiotics11091159
Chicago/Turabian StyleSalemi, Rossella, Alessandra Zega, Elvira Aguglia, Flavia Lo Verde, Giuseppe Pigola, Stefania Stefani, and Viviana Cafiso. 2022. "Balancing the Virulence and Antimicrobial Resistance in VISA DAP-R CA-MRSA Superbug" Antibiotics 11, no. 9: 1159. https://doi.org/10.3390/antibiotics11091159
APA StyleSalemi, R., Zega, A., Aguglia, E., Lo Verde, F., Pigola, G., Stefani, S., & Cafiso, V. (2022). Balancing the Virulence and Antimicrobial Resistance in VISA DAP-R CA-MRSA Superbug. Antibiotics, 11(9), 1159. https://doi.org/10.3390/antibiotics11091159