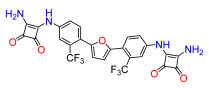
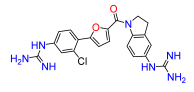
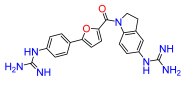
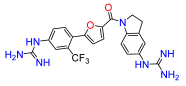
4. Materials and Methods
4.1. General Experimental Information for Synthesis and Compound Characterization
General reagent and solvents for the synthesis of compounds were purchased from commercial sources and used as supplied, unless otherwise stated.
Purification by flash column chromatography was performed on a Selekt (Biotage, Uppsala, Sweden) automated instrument with Sfär KP-amino D or Sfär silica D cartridges (Biotage, Uppsala, Sweden), mobile phase consist of pentane (solvent A) and ethyl acetate (solvent B). The final compounds were purified by reverse phase flash column chromatography performed on an Isolera (Biotage, Uppsala, Sweden) automated instrument with Sfär C18 D cartridges (Biotage, Uppsala, Sweden), mobile phase consist of water (solvent A) and acetonitrile (solvent B). The standard gradient consisted of x% solvent B for 1 columns volume, x% to y% B for 10 column volumes, and then y% B for 2 column volumes. x and y are defined in the characterization section of the compound the interest.
All NMR spectra (1H and 13C) were recorded on a Varian 400 MHz spectrometer (Varian, Palo Alto, CA, USA) at 25 °C. Samples were dissolved (0.5 mL) in deuterated chloroform, methanol or dimethylsulfoxide (CDCl3, CD3OD, DMSO-d6). The residual solvent peaks specific to that to the deuterated solvent was used as an internal reference; CDCl3: 7.26 ppm (1H NMR) and 77.20 ppm (13C NMR); CD3OD: 3.31 ppm (1H NMR) and 49.00 ppm (13C NMR); DMSO-d6: 2.50 ppm (1H NMR) and 39.52 ppm (13C NMR). Data are presented as follows: chemical shift in ppm, multiplicity (br = broad, s = singlet, d = doublet, dd = doublet of doublet, t = triplet, q = quartet, m = multiplet), coupling constants in Hz and integration. High resolution mass spectra (HRMS) were recorded on an Agilent 1290 infinity LC system tandem to an Agilent 6520 Accurate Mass Q-TOF spectrometer (Agilent, Santa Clara, CA, USA).
4.2. General Procedure A
In a sealed 20 mL microwave vial, 2,5-Bis-(trimethylstannyl)furan (0.5 mmol), aryl bromide (1.0 mmol) and tetrakis(triphenyphosphine)-palladium(0) (0.025 mmol) in anhydrous dimethylformamide (10 mL) was evacuated and backfilled with N2 (×3) and heated for 14 h at 100 °C. Upon cooling, the mixture was filtered through Celite, the Celite rinsed with chloroform, and the residue was reduced under vacuum. Then 100 mL of chloroform, and 50 mL of 10% aqueous potassium fluoride was added and the mixture was stirred at room temperature for 0.5 h. The organic layer was separated and dried over sodium sulfate, filtered, and concentrated under reduced pressure. The crude product was purified by flash chromatography (25 g Sfär silica D cartridge, 15–75% B, Rf = 12 column volumes) to give the desired compounds.
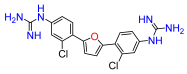
2,5-Bis(4-nitro-2-chlorophenyl)furan (1a). Following general procedure A, 1-bromo-2-chloro-4-nitrobenzene was used as aryl bromide. Afforded the title compound as an orange solid, yield 45%.
1H NMR (400 MHz, DMSO-
d6,
δ, ppm): 8.45 (d,
J = 2.2 Hz, 2H), 8.38 (d,
J = 8.8 Hz, 2H), 8.32 (dd,
J = 8.8; 2.2 Hz, 2H), 7.72 (s, 2H). Data is consistent with that previously reported [
8].
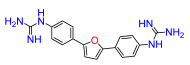
2,5-Bis(4-nitrophenyl)furan (3a). Following general procedure A, 1-bromo-4-nitrobenzene was used as aryl bromide. Afforded the title compound as orange solid, yield 33%.
1H NMR (400 MHz, DMSO-
d6,
δ, ppm): 8.35 (d,
J = 9.0 Hz, 4H), 8.18 (d,
J = 9.0 Hz, 4H), 7.44 (s, 2H). Data is consistent with that previously reported [
8].
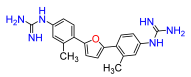
2,5-Bis(4-nitro-2-methylphenyl)furan (4a). Following general procedure A, 1-bromo-2-methyl-4-nitrobenzene was used as aryl bromide. Afforded the title compound as tan solid, yield 62.2%.
1H NMR (400 MHz, DMSO-
d6,
δ, ppm): 8.27 (d,
J = 2.0 Hz, 2H), 8.19 (dd,
J = 8.9; 2.0 Hz, 2H), 8.15 (d,
J = 8.9 Hz, 2H), 7.36 (s, 2H), 2.71 (s, 6H). Data is consistent with that previously reported [
8].
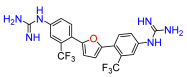
2,5-Bis(4-nitro-2-trifluoromethylphenyl)furan (5a). Following general procedure A, 2-bromo-5-nitrobenzotrifluoride was used as aryl bromide. Afforded the title compound as golden solid, yield 15%.
1H NMR (400 MHz, DMSO-
d6,
δ, ppm): 8.63 (d,
J = 8.7 Hz, 2H), 8.59 (s, 2H), 8.25 (d,
J = 8.7 Hz, 2H), 7.40 (s, 2H). Data is consistent with that previously reported [
8].
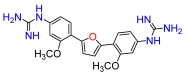
2,5-Bis(2-methoxy-4-nitro-phenyl)furan (6a). Following general procedure A, 1-bromo-2-methoxy-4-nitrobenzene was used as aryl bromide. Afforded the title compound as orange solid, yield 80%.
1H NMR (400 MHz, DMSO-
d6,
δ, ppm): 8.27 (d,
J = 8.7 Hz, 2H), 7.97 (dd,
J = 8.5; 2.0 Hz, 2H), 7.22 (d,
J = 2.0 Hz, 2H), 7.41 (s, 2H), 4.11 (s, 6H). Data is consistent with that previously reported [
8].
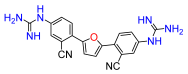
2,5-Bis(2-cyano-4-nitro-phenyl)furan (7a). Following general procedure A, 2-bromo-4-nitrobenzonitrile was used as aryl bromide. Afforded the title compound as pale yellow solid, yield 46%. 1H NMR (400 MHz, DMSO-d6, δ, ppm): 8.89 (d, J = 2.1 Hz, 2H), 8.63 (dd, J = 8.6; 2.1 Hz, 2H), 8.43 (d, J = 8.6 Hz, 2H), 7.86 (s, 2H).
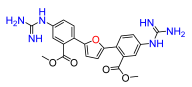
2,5-Bis(4-nitro-2-(methyl carboxy)-phenyl)furan (8a). Following general procedure A, methyl 2-bromo-5-nitrobenzoate was used as aryl bromide. Afforded the title compound as orange solid, yield 68%. 1H NMR (400 MHz, DMSO-d6, δ, ppm): 8.48–8.45 (m, 4H), 8.06 (m, 2H), 7.31 (s, 2H), 3.83 (s, 6H).
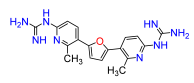
2,5-Bis(2-methyl-6-nitropyridin-3-yl)furan (9a). Following general procedure A, 3-bromo-2-methyl-6-nitropyridine was used as aryl bromide. Afforded the title compound as tan solid, yield 63%. 1H NMR (400 MHz, DMSO-d6, δ, ppm): 8.36 (d, J = 6.6 Hz, 2H), 8.33 (d, J = 6.6 Hz, 2H), 6.98 (s, 2H), 2.95 (s, 6H).
4.3. General Procedure B
To a solution of 2,5-Bis(4-nitrophenyl)furan derivatives (1a, 3a–9a) (0.3 mmol) in THF (3 mL) and EtOH (3 mL), ammonium chloride (3 mL, 0.3 M) and iron (1.75 mmol) were added After stirring at 60 °C for 4 h, the reaction was allowed to cool to room temperature and the heterogeneous mixture filtered through Celite and the Celite was rinsed with ethyl acetate. The solution was concentrated to half-volume, then diluted with ethyl acetate (20 mL) and washed with sodium hydroxide solution (1 M, 20 mL). The organic layer was separated, the aqueous phase was extracted with ethyl acetate (2×), the combined organic phases dried over sodium sulfate, filtered and the solvent evaporated. The crude product was purified by flash chromatography (11 g Sfär KP-amino D cartridge, 15–90% B, Rf = 10 column volumes) to give the desired compounds.
2,5-Bis(4-amino-2-chlorophenyl)furan (1b). Compound
1a was reacted according to General Procedure B, yield 82%, red solid.
1H NMR (400 MHz, CDCl
3,
δ, ppm): 7.58 (d,
J = 8.5 Hz, 2H), 6.83 (s, 2H), 6.69 (d,
J = 2.2 Hz, 2H), 6.62 (dd,
J = 8.5; 2.2 Hz, 2H), 5.64 (brs, 4H). Data is consistent with that previously reported [
8].
2,5-Bis(4-amino-phenyl)furan (3b). Compound
3a was reacted according to General Procedure B, yield 96%, red solid.
1H NMR (400 MHz, CDCl
3,
δ, ppm): 7.53 (d,
J = 8.7 Hz, 4H), 6.70 (d,
J = 8.7 Hz, 4H), 6.49 (s, 2H), 3.72 (br s, 4H). Data is consistent with that previously reported [
8].
2,5-Bis(4-amino-2-methylphenyl)furan (4b). Compound
4a was reacted according to General Procedure B, yield 76%, red solid.
1H NMR (400 MHz, DMSO,
δ, ppm): 7.41 (d,
J = 8.0 Hz, 2H), 6.51 (m, 4H), 6.47 (s, 2H), 5.20 (s, 4H), 2.38 (s, 6H). Data is consistent with that previously reported [
8].
2,5-Bis(4-amino-2-trifluoromethylphenyl)furan (5b). Compound
5a was reacted according to General Procedure B, yield 96%, red solid.
1H NMR (400 MHz, CDCl
3,
δ, ppm): 7.59 (d,
J = 8.4 Hz, 2H), 7.00 (d,
J = 2.4 Hz, 2H), 6.81 (dd,
J = 8.4; 2.4 Hz, 2H), 6.62 (s, 2H), 3.90 (br s, 4H). Data is consistent with that previously reported [
8].
2,5-Bis(4-amino-2-methoxyphenyl)furan (6b). Compound
6a was reacted according to General Procedure B, yield 90%, dark red solid.
1H NMR (400 MHz, CDCl
3,
δ, ppm): 7.73 (d,
J = 8.3, Hz, 2H), 6.77 (s, 2H), 6.36 (dd,
J = 8.3; 1.9 Hz, 2H), 6.28 (d,
J = 1.9 Hz, 2H), 3.88 (s, 6H), 3.73 (br s, 4H). Data is consistent with that previously reported [
8].
2,5-Bis(4-amino-2-cyanophenyl)furan (7b). Compound 7a was reacted according to General Procedure B, yield 75%, yellow solid. 1H NMR (400 MHz, DMSO-d6, δ, ppm): 7.82 (d, J = 9.2 Hz, 2H), 7.07 (s, 2H), 7.05–7.02 (m, 4H), 5.39 (br s, 4H).
2,5-Bis(4-amino-2-(methyl carboxy)-phenyl)furan (8b). Compound 8a was reacted according to General Procedure B, yield 95%, orange solid. 1H NMR (400 MHz, CDCl3, δ, ppm): 7.32 (d, J = 9.0 Hz, 2 H), 6.73–6.70 (m, 4H), 6.39 (s, 2H), 5.58 (br s, 4H), 3.69 (s, 6H).
2,5-Bis(6-amino-2-methylpyridin-3-yl)furan (9b). Compound 9a was reacted according to General Procedure B, yield 88%, orange solid. 1H NMR (400 MHz, CDCl3, δ, ppm): 7.68 (d, J = 8.5 Hz, 2 H), 6.54 (s, 2H), 6.37 (d, J = 8.5 Hz, 2 H), 5.07 (br s, 4H), 2.46 (s, 6H).
2,5-Bis-(4-cyanophenyl)furan (10a) A sealed 20 mL microwave vial containing 4-bromobenzonitrile (1.5 mmol), furan (4.5 mmol), potassium acetate (3.0 mmol) and palladium(II) acetate (0.015 mmol) in dimethylacetamide (5 mL) was evacuated and backfilled with N
2 (×3), and heated for 20 h at 150 °C. Upon cooling to room temperature, the residue filtered through Celite and the Celite rinsed with ethyl acetate. The solution was washed with water and brine, dried over sodium sulfate, filtered, and concentrated under reduced pressure. The crude product was purified by flash chromatography (25 g Sfär silica D cartridge, 15–75% B, R
f = 12 column volumes) to give the compound as a dark yellow solid, yield 29%.
1H NMR (400 MHz, DMSO-
d6,
δ, ppm): 8.03 (d,
J = 8.5 Hz, 4H), 7.90 (d,
J = 8.5 Hz, 4H), 7.42 (s, 2H). Data is consistent with that previously reported [
36].
2,5-Bis(4-aminomethylphenyl)furan (10b) To a solution of 2,5-Bis(4-cyanophenyl)furan (
10a) (0.5 mmol) in THF (10 mL) was added to Lithium aluminum hydride (4.7 mmol) in tetrahydrofuran (15 mL) at 0 °C, and stirred at room temperature overnight. Sodium hydroxide (5 mL, 10% solution) was added and stirred at 0 °C after 30 min, water (10 mL) was added to give a granular precipitate. The mixture was filtered, and the precipitate washed copiously with ether and dried in vacuo to give the product as a pale-yellow solid, yield 60%, that were used without further purification.
1H NMR (400 MHz, DMSO-
d6,
δ, ppm): 7.74 (d,
J = 8.1 Hz, 4H), 7.40 (d,
J = 8.1 Hz, 4H), 7.00 (s, 2H), 3.73 (s, 4H). Data is consistent with that previously reported [
37].
4.4. General Procedure C
A solution of 2,5-Bis(4-aminoaryl)furan derivatives (1b, 3b–9b) or 2,5-Bis(4-aminomethylphenyl)furan (10b) (0.10 mmol) mercury (II) chloride (0.21 mmol), 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea (0.19 mmol) and triethylamine (0.48 mmol).in either dichloromethane or dimethylformamide (5 mL) was stirred at 0 °C for 1 h and then at room temperature for 18 h. Then, the reaction mixture was diluted with ethyl acetate filtered through Celite and the Celite rinsed with ethyl acetate. The organic phase was washed with water (2×), and brine (2×), dried over sodium sulfate, filtered, and concentrated under reduced pressure. The crude product was purified by flash chromatography (10 g Sfär D cartridge, 15–80% B, Rf = 10 column volumes) to give the desired compounds.
2,5-Bis(2-chloro-4-N,N′-di-(tert-butoxycarbonyl)guanidinophenyl)furan (1c). Compound
1b was reacted according to General Procedure C
. Afforded the title compound as pale-yellow solid, yield 73%.
1H NMR (400 MHz, CDCl
3,
δ, ppm): 11.61 (brs, 2H), 10.45 (brs, 2H), 7.90 (d,
J = 8.7, Hz, 2H), 7.81 (d,
J = 2.1 Hz, 2H), 7.65 (dd,
J = 8.7; 2.1, 2H), 7.19 (s, 2H), 1.54 (s, 18H), 1.53 (s, 18H). Data is consistent with that previously reported [
8].
2,5-Bis(4-N,N′-di-(tert-butoxycarbonyl)guanidinophenyl)furan (3c). Compound
3b was reacted according to General Procedure C
. Afforded the title compound as pale yellow solid yield 63%.
1H NMR (400 MHz, CDCl
3,
δ, ppm): 11.65 (br s, 2H), 10.32 (br s, 2H), 7.60 (br d,
J = 8.6 Hz, 4H), 7.33–7.29 (m, 4H), 7.10 (m, 2H), 1.53 (s, 18H), 1.50 (s, 18H). Data is consistent with that previously reported [
8].
2,5-Bis(2-methyl-4-N,N′-di-(tert-butoxycarbonyl)
guanidinophenyl)furan (4c). Compound
4b was reacted according to General Procedure C
. Afforded the title compound as pale-yellow solid, yield 56%.
1H NMR (400 MHz, CDCl
3,
δ, ppm): 11.63 (br s, 2H), 10.35 (br s, 2H), 7.74 (d,
J = 8.6 Hz, 2H), 7.64 (m, 2H), 7.42 (br s, 2H), 6.61 (br s, 2H), 2.53 (s, 6H), 1.54 (s, 18H), 1.52 (s, 18H). Data is consistent with that previously reported [
8].
2,5-Bis(2-trifluoromethyl-4-N,N′-di-(tert-butoxycarbonyl)guanidinophenyl)furan (5c). Compound
5b was reacted according to General Procedure C. Afforded the title compound as yellow/orange solid, yield 81%.
1H NMR (400 MHz, CDCl
3,
δ, ppm): 11.61 (br s, 2H), 10.55 (br s, 2H), 8.03 (dd,
J = 8.6; 2.2, Hz, 2H), 7.95 (d,
J = 2.2 Hz, 2H), 7.84 (d,
J = 8.5, 2H), 6.79 (s, 2H), 1.55 (s, 18H), 1.52 (s, 18H). Data is consistent with that previously reported [
8].
2,5-Bis(2-methoxy-4-N,N′-di-(tert-butoxycarbonyl)guanidinophenyl)furan (6c). Compound
6b was reacted according to General Procedure C Afforded the title compound as red solid, yield 93%.
1H NMR (400 MHz, CDCl
3,
δ, ppm): 11.58 (br s, 2H), 10.38 (br s, 2H), 7.88 (d,
J = 8.5, Hz, 2H), 7.60 (br s, 2H), 7.16 (br d,
J = 8.5, 2H), 6.97 (s, 2H), 3.96 (s, 6H), 1.54 (s, 18H), 1.51 (s, 18H). Data is consistent with that previously reported [
8].
2,5-Bis(2-cyano-4-N,N′-di-(tert-butoxycarbonyl)guanidinophenyl)furan (7c). Compound 7b was reacted according to General Procedure C. Afforded the title compound as pale yellow solid, yield 94%. 1H NMR (400 MHz, CDCl3, δ, ppm): 11.61 (br s, 2H), 10.57 (br s, 2H), 8.14 (br s, 2H), 8.06 (d, J = 8.5, 2H), 6.68 (s, 2H), 2.70 (s, 6H), 1.55 (s, 18H), 1.54 (s, 18H).
2,5-Bis(2-(methylcarboxy)-4-N,N′-di-(tert-butoxycarbonyl)guanidinophenyl)furan (8c). Compound 8b was reacted according to General Procedure C. Afforded the title compound as orange solid, yield 95%. 1H NMR (400 MHz, CDCl3, δ, ppm): 11.60 (br s, 2H), 10.50 (br s, 2H), 8.00 (dd, J = 8.6; 2.3, 2H), 7.76 (d, J = 2.3, 2H), 7.64 (br d, J = 8.6, 2H), 3.78 (s, 2H), 6.63 (s, 2H), 1.53 (s, 18H), 1.51 (s, 18H).
2,5-Bis(2-(methyl)-6-N,N′-di-(tert-butoxycarbonyl)guanidinopiridin-3-yl)furan (9c). Compound 9b was reacted according to General Procedure C. Afforded the title compound as tan solid, yield 31%. 1H NMR (400 MHz, CDCl3, δ, ppm): 11.54 (br s, 2H), 10.83 (br s, 2H), 8.31 (br s, 2H), 7.87 (d, J = 8.8, 2H), 7.84 (br d, J = 8.8, 2H), 7.30 (s, 2H), 1.54 (s, 18H), 1.52 (s, 18H).
2,5-Bis(4-N,N′-di-(tert-butoxycarbonyl)guanidinomethylphenyl)furan (10c). Compound 10b was reacted according to General Procedure C. Afforded the title compound as yellow oil, yield 76%. 1H NMR (400 MHz, CDCl3, δ, ppm): 11.55 (br s, 2H), 8.61 (t, J = 4.9 Hz, 2H), 7.70 (d, J = 8.1 Hz, 4H), 7.34 (d, J = 8.1 Hz, 4H), 6.71 (s, 2H), 4.64 (d, J = 4.9 Hz, 4H), 1.51 (s, 18H), 1.47 (s, 18H).
4.5. General Procedure D
To a solution of 2,5-Bis(4-N,N’-di-(tert-butoxycarbonyl)guanidinophenyl)furan derivatives (1c, 3c–9c) or 2,5-Bis(4-N,N′-di-(tert-butoxycarbonyl)guanidinomethylphenyl)furan (10c) (0.05 mmol) in dichloromethane (1 mL) was added HCl (4 M in dioxane, 2 mL). The solution was stirred at room temperature for 16 h. The mixture was concentrated under reduced pressure and the residue was purified by reverse phase flash chromatography (6 g Sfär C18 D cartridge, 0–45% B, Rf = 10 column volumes) to give the desired compounds.
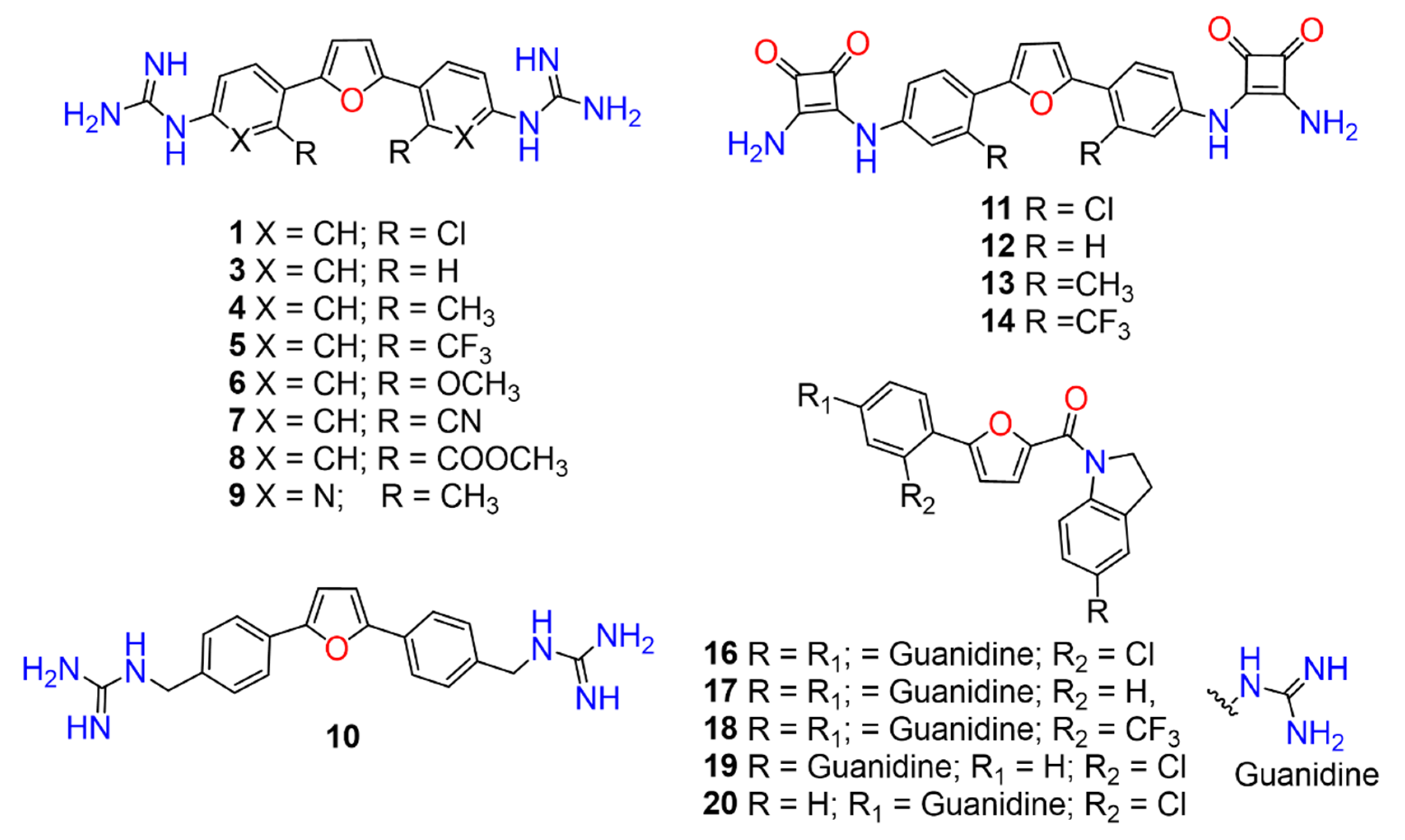
2,5-Bis(2-chloro-4-guanidinophenyl)furan (1). Compound
2c was reacted according to General Procedure D, yield 89%, tan solid.
1H NMR (400 MHz, CD
3OD,
δ, ppm): 8.11 (d,
J = 8.6 Hz, 6H), 7.51 (dt,
J = 2.2 Hz, 2H), 7.38 (br dd,
J = 8.6; 2.2 Hz, 2H), 7.35 (br s, 2H).
13C NMR (100 MHz, CD
3OD,
δ, ppm): 157.90, 150.71, 136.50, 132.09, 130.24, 128.61, 128.15, 124.84, 114.52. Data is consistent with that previously reported [
8].
2,5-Bis(guanidinophenyl)furan (3). Compound
3c was reacted according to General Procedure D, yield 85%, light brown solid.
1H NMR (400 MHz, CD
3OD,
δ, ppm): 7.96 (d,
J = 8.8 Hz, 4H), 7.42 (d,
J = 8.8 Hz, 4H), 7.03 (s, 2H).
13C NMR (100
1H NMR (400 MHz, DMSO-
d6,
δ, ppm): 158.06, 15410.13, (s, 2H), 7.56 (brs, 6H), 7.44 (t,
J = 7. Hz, 4H), 7.29 (t,
J = 7.5 Hz, 2H), 7.23 (d,
J = 7.6 Hz, 4H).
13C NMR (100 MHz, DMSO-
d6,
δ, ppm): 156.04, 135.22, 131.1726, 129.66, 126.86, 126.19, 109.53.32, 124.25. Data is consistent with that previously reported [
8].
2,5-Bis(4-guanidino-2-methylphenyl)furan (4). Compound
4c was reacted according to General Procedure D, yield 90.2%, light brown solid.
1H NMR (400 MHz, CD
3OD,
δ, ppm): 7.95 (d,
J = 8.1 Hz, 2H), 7.29 (m, 4H), 6.93 (s, 2H), 2.67 (s, 6H).
13C NMR (100 MHz, CD
3OD,
δ, ppm): 158.01, 153.45, 137.74, 135.15, 130.41, 129.34, 128.89, 123.91, 112.50, 22.25. Data is consistent with that previously reported [
8].
2,5-Bis(4-guanidino-2-trifluoromethylphenyl)furan (5). Compound
5c was reacted according to General Procedure D, yield 60%, orange solid.
1H NMR (400 MHz, DMSO-
d6,
δ, ppm): 10.53 (s, 2H), 7.93 (d,
J = 8.6 Hz, 2H), 7.87 (br s, 6H), 7.70 (br s, 2H), 7.65 (d,
J = 8.6 Hz, 2H), 7.00 (s, 2H).
13C NMR (100 MHz, DMSO-
d6,
δ, ppm): 156.34, 150.63, 136.68, 131.84, 128.02, 126.51, 126.20, 125.70, 122.52, 113.03. Data is consistent with that previously reported [
8].
2,5-Bis(4-guanidino-2-methoxyphenyl)furan (6). Compound
6c was reacted according to General Procedure D, yield 90%, red solid.
1H NMR (400 MHz, CD
3OD,
δ, ppm): 8.02 (d,
J = 7.8 Hz, 2H), 7.07 (s, 2H), 7.02 (d,
J = 2.0 Hz, 2H), 6.99 (dd,
J = 8.3; 2.0 Hz, 2H), 4.00 (s, 6H).
13C NMR (100 MHz, CD
3OD,
δ, ppm): 157.92, 157.80, 149.50, 135.71, 127.56, 120.06, 118.06, 114.01, 109.49, 56.24. Data is consistent with that previously reported [
8].
2,5-Bis(2-cyano-4-guanidinophenyl)furan (7). Compound 7c was reacted according to General Procedure D, yield 86%, orange solid. 1H NMR (400 MHz, CD3OD, δ, ppm): 8.23 (d, J = 8.7 Hz, 2H), 7.79 (d, J = 2.1 Hz, 2H), 7.68 (dd, J = 8.7; 2.1 Hz, 2H), 7.49 (s, 2H). 13C NMR (100 MHz, CD3OD, δ, ppm): 157.88, 151.77, 136.35, 131.65, 131.37, 130.90, 129.34, 119.04, 113.89, 109.16. HRMS (ESI), found 385.1525 C20H16N8O2, [M + H]+, requires 385.1525.
2,5-Bis(4-guanidino-2-(methyl carboxy)phenyl)furan (8). Compound 8c was reacted according to General Procedure D, yield 90%, orange solid. 1H NMR (400 MHz, CD3OD, δ, ppm): 7.80 (d, J = 8.4 Hz, 2H), 7.55 (d, J = 2.0 Hz, 2H), 7.51 (dd, J = 8.4; 2.0 Hz, 2H), 6.83 (s, 2H), 3.79 (s, 6H). 13C NMR (100 MHz, CD3OD, δ, ppm): 169.93, 157.86, 153.12, 135.96, 132.43, 130.39, 128.82, 128.45, 126.54, 112.08, 53.32. HRMS (ESI), found 451.1731 C22H22N6O5, [M + H]+, requires 451.1730.
2,5-Bis(6-guanidino-2-methylpyridin-3-yl)furan (9). Compound 9c was reacted according to General Procedure D, yield 78%, tan solid. 1H NMR (400 MHz, CD3OD, δ, ppm): 8.29 (d, J = 8.5 Hz, 2H), 7.08 (d, J = 8.5 Hz, 2H), 7.00 (s, 2H), 2.85 (s, 6H). 13C NMR (100 MHz, CD3OD, δ, ppm): 157.07, 153.62, 151.95, 151.26, 138.71, 123.01, 112.94, 112.01, 24.79. HRMS (ESI), found 365.1837 C18H20N8O, [M + H]+, requires 365.1838.
2,5-Bis(4-guanidinomethylphenyl)furan (10). Compound 10 was reacted according to General Procedure D, yield 79%, pale yellow solid. 1H NMR (400 MHz, CD3OD, δ, ppm): 7.81 (d, J = 7.8 Hz, 4H), 7.41 (d, J = 7.8 Hz, 4H), 6.91 (s, 2H), 4.44 (s, 4H). 13C NMR (100 MHz, CD3OD, δ, ppm): 158.73, 154.34, 136.80, 131.74, 129.01, 128.85, 125.17, 125.00, 108.91, 45.75. HRMS (ESI), found 363.1930 C20H22N6O, [M + H]+, requires 363.1933.
4.6. General Procedure E
To a solution of 2,5-Bis(4-aminophenyl)furan derivatives (1b, 3b–5b) (0.2 mmol) in 2.5 mL of ethanol was added 3,4-diethoxy-3-cyclobutene-1,2-dione (0.40 mmol) and zinc trifluoromethanesulfonate (0.08 mmol) in ethanol (2.5 mL). The solution was stirred at room temperature for 3 h. Then, the reaction mixture was evaporated under reduced pressure and the residue was subsequently dissolved in ethyl acetate and washed several times with aqueous ammonium chloride (1 M), and then with water. The organic layer was separated, dried over sodium sulfate, filtered, and triturated several times with pentane and diethyl ether. The resultant solid was dried under vacuum to give the desired compounds.
2,5-Bis(2-chloro-4-((3,4-dioxo-2-(ethoxy)cyclobut-1-en-1-yl)amino)phenyl)furan (11a). Compound 1b was reacted according to General Procedure E, yield 52%, yellow solid. 1H NMR (400 MHz, CDCl3, δ, ppm): 8.24 (br s, 2H), 7.38 (m, 2H), 7.29 (br d, J = 8.0 Hz, 2H), 7.24 (d, J = 8.0 Hz, 2H), 7.14 (m, 2H), 4.92 (q, J = 6.8 Hz, 4H), 1.53 (t, J = 6.8 Hz, 6H).
2,5-Bis(4-((3,4-dioxo-2-(ethoxy)cyclobut-1-en-1-yl)amino)phenyl)furan (12a). Compound 3b was reacted according to General Procedure E, yield 36%, red solid. 1H NMR (400 MHz, DMSO-d6, δ, ppm): 10.87 (br s, 2H), 7.80 (d, J = 7.8 Hz, 4H), 7.46 (d, J = 7.8 Hz, 4H), 7.01 (s, 2H), 4.78 (q, J = 6.9 Hz, 4H), 1.45 (t, J = 6.9 Hz, 6H).
2,5-Bis(2-methyl-4-((3,4-dioxo-2-(ethoxy)cyclobut-1-en-1-yl)amino)phenyl)furan (13a). Compound 4b was reacted according to General Procedure E), yield 57%, yellow solid. 1H NMR (400 MHz, CDCl3, δ, ppm): 9.01 (br s, 2H), 7.22–7.16 (m, 6H), 6.95 (d, J = 7.1 Hz, 2H), 4.85 (q, J = 7.0 Hz, 4H), 2.33 (s, 6H), 1.47 (t, J = 7.0 Hz, 6H).
2,5-Bis(4-((3,4-dioxo-2-(ethoxy)cyclobut-1-en-1-yl)amino)phenyl)furan (14a). Compound 5b was reacted according to General Procedure E, yield 75%, orange solid. 1H NMR (400 MHz, DMSO-d6, δ, ppm): 11.11 (br s, 2H), 7.96 (br s, 2H), 7.88 (d, J = 8.5 Hz, 2H), 7.72 (m, 2H), 6.92 (s, 2H), 4.81 (q, J = 7.0 Hz, 4H), 1.43 (t, J = 7.0 Hz, 6H).
4.7. General Procedure F
To a solution of 2,5-Bis(4-((3,4-dioxo-2-(ethoxy)cyclobut-1-en-1-yl)amino)phenyl)furan derivatives (11a–14a) (0.1 mmol) in 1 mL of methanol was treated with ammonia (7 M in methanol, 200 µL, 1.45 mmol) and stirred at room temperature for 12 h. Then, the solvent was evaporated under reduced pressure and the residue was triturated several times with pentane and diethyl ether. The resultant solid was dried under vacuum to give the desired compounds.
2,5-Bis(2-chloro-4-((3,4-dioxo-2-(amino)cyclobut-1-en-1-yl)amino)phenyl)furan (11). Compound 11a was reacted according to General Procedure F, yield 64%, yellow solid. 1H NMR (400 MHz, DMSO-d6, δ, ppm): 9.78 (s, 2H), 7.61 (br s, 2H), 7.27 (t, J = 8.1 Hz, 2H), 7.20 (m, 2H), 6.97 (d, J = 7.8 Hz, 2H). 13C NMR (100 MHz, DMSO-d6, δ, ppm): 184.76, 181.18, 171.37, 163.99, 140.73, 133.86, 130.95, 121.98, 117.62, 116.36. HRMS (ESI), found 509.0399 C24H14Cl2N4O5, [M + H]+, requires 509.0420.
2,5-Bis(4-((3,4-dioxo-2-(amino)cyclobut-1-en-1-yl)amino)phenyl)furan (12). Compound 12a was reacted according to General Procedure F, yield 56%, red solid. 1H NMR (400 MHz, DMSO-d6, δ, ppm): 10.05 (s, 2H), 7.77 (d, J = 8.4 Hz, 4H), 7.52 (d, J = 8.4 Hz, 4H), 6.94 (s, 2H). 13C NMR (100 MHz, DMSO-d6, δ, ppm): 184.44, 181.18, 171.25, 164.12, 152.11, 138.45, 124.53, 118.24, 109.55, 107.14. HRMS (ESI), found 441.1190 C24H16N4O5, [M + H]+, requires 440.1119.
2,5-Bis(2-methyl-4-((3,4-dioxo-2-(amino)cyclobut-1-en-1-yl)amino)phenyl)furan (13). Compound 13a was reacted according to General Procedure F, yield 52%, yellow solid. 1H NMR (400 MHz, DMSO-d6, δ, ppm): 9.62 (s, 2H), 7.20–7.16 (m, 6H), 6.79 (d, 7.0 Hz, 2H), 2.24 (s, 6H). 13C NMR (100 MHz, DMSO-d6, δ, ppm): 184.45, 181.20, 171.03, 164.58, 139.07, 138.70, 129.21, 123.29, 118.49, 115.15, 21.21. HRMS (ESI), found 469.1490 C26H20N4O5, [M + H]+, requires 469.1512.
2,5-Bis(2-trifluoromethyl-4-((3,4-dioxo-2-(amino)cyclobut-1-en-1-yl)amino)phenyl)furan (14). Compound 14a was reacted according to General Procedure F, yield 77%, orange solid. 1H NMR (400 MHz, DMSO-d6, δ, ppm): 10.14 (s, 2H), 8.02 (d, J = 1.8 Hz, 2H), 7.85 (d, J = 8.6 Hz, 2H), 7.70 (dd, J = 8.6; 1.8 Hz, 2H), 6.87 (s, 2H). 13C NMR (100 MHz, DMSO-d6, δ, ppm): 185.00, 181.29, 171.58, 163.58, 150.09, 139.48, 131.31, 126.04, 125.41, 121.60, 121.20, 115.93, 115.59. HRMS (ESI), found 577.0919 C26H14F6N4O5, [M + H]+, requires 577.0947.
4.8. General Procedure G
To a solution of 2-furoyl chloride (2.8 mmol) in dichloromethane (15 mL) was added 5-nitroindoline or indoline (2.8 mmol) and triethylamine (1.6 mL), the mixture was stirred at room temperature for 3 h. The reaction mixture was monitored by GCMS, until the starting material was consumed. Then, the mixture was concentrated under reduced pressure and the crude product was purified by flash chromatography (25 g Sfär silica D cartridge, 15–50% B, Rf = 8 column volumes) to give the desired compounds.
Furan-2-yl(5-nitroindolin-1-yl)methanone (2). Synthesized from 5-nitroindoline, yield 50%, dark yellow solid. 1H NMR (400 MHz, DMSO-d6, δ, ppm): 8.22 (m, 1H), 8.17 (m, 2H), 8.02 (m, 1H), 7.38 (m, 1H), 6.76 (m, 1H), 4.55 (t, J = 8.3 Hz, 2H), 3.31 (m, 2H).
Furan-2-yl(indolin-1-yl)methanone (15). Synthesized from indoline, yield 59%, white solid. 1H NMR (400 MHz, DMSO-d6, δ, ppm): 8.04 (br d, J = 7.4 Hz, 1H), 7.92 (m, 1H), 7.26 (br d, J = 7.4 Hz, 1H), 7.23 (br d, J = 3.6 Hz, 1H), 7.17 (br t, J = 8.0 Hz, 1H), 7.03 (br t, J = 7.4 Hz, 1H), 6.69 (m, 1H), 4.38 (t, J = 8.2 Hz, 2H), 3.17 (t, J = 8.2 Hz, 2H).
4.9. General Procedure H
In a sealed 20 mL microwave vial, the aryl bromide (1.1 mmol), furan-2-yl(indolin-1-yl)methanone (2 or 15) (0.9 mmol), potassium acetate (2.75 mmol), and palladium(II) acetate (0.02 mmol) were dissolved in dimethylacetamide (5 mL) and the resulting reaction mixture was evacuated and backfilled with nitrogen several times. The reaction was stirred at 150 °C for 20 h. Upon cooling to room temperature, the residue filtered through Celite and the Celite rinsed with ethyl acetate. The organic phase was washed with water and brine, dried over sodium sulfate, concentrated under reduced pressure. The crude product was purified by flash chromatography (25 g Sfär silica D cartridge, 25–100% B, Rf = 12 column volumes) to give the desired compounds.
(5-(2-chloro-4-nitrophenyl)furan-2-yl)(5-nitroindolin-1-yl)methanone (16a). Compound 2 was reacted according to General Procedure H, 1-bromo-2-chloro-4-nitrobenzene was used as aryl bromide, yield 50%, red solid. 1H NMR (400 MHz, DMSO-d6, δ, ppm): 8.45 (m, 1H), 8.32 (m, 1H), 8.27 (m, 1H), 8.22 (m, 1H), 8.18 (m, 3H), 7.64 (dd, J = 3.8; 1.1 Hz, 1H), 7.59 (dd, J = 3.8; 1.1 Hz, 1H), 4.70 (t, J = 8.4 Hz, 2H), 3.33 (m, 2H).
(5-nitroindolin-1-yl)(5-(4-nitrophenyl)furan-2-yl)methanone (17a). Compound 2 was reacted according to General Procedure H, 1-bromo-4-nitrobenzene was used as the aryl bromide. Afforded the title compound as a yellow solid yield 43%. 1H NMR (400 MHz, DMSO-d6, δ, ppm): 8.34 (dd, J = 9.0; 1.0 Hz, 2H), 8.32 (m, 1H), 8.18 (m, 2H), 8.12 (dd, J = 9.0; 1.0 Hz, 2H), 7.56 (d, J = 1.0 Hz, 2H), 4.71 (t, J = 8.4 Hz, 2H), 3.35 (m, 2H).
(5-(2-trifluoromethyl-4-nitrophenyl)furan-2-yl)(5-nitroindolin-1-yl)methanone (18a). Compound 2 was reacted according to General Procedure H, 2-bromo-5-nitrobenzotrifluoride was used as the aryl bromide. Afforded the title compound as a brown solid, yield 31%. 1H NMR (400 MHz, DMSO-d6, δ, ppm): 8.59 (m, 2H), 8.24 (d, J = 6.28 Hz, 2H), 8.16 (m, 2H), 7.56 (d, J = 3.6 Hz, 1H), 7.30 (d, J = 3.6 Hz, 1H), 4.71 (t, J = 8.7 Hz, 2H), 3.30 (m, 2H).
(5-(2-chloro-phenyl)furan-2-yl)(5-nitroindolin-1-yl)methanone (19a). Compound 2 was reacted according to General Procedure H, 1-bromo-2-chlorobenzene was used as the aryl bromide. Afforded the title compound as a yellow solid, yield 38.3%. 1H NMR (400 MHz, CDCl3, δ, ppm): 8.35 (br d, J = 8.8 Hz, 1H), 8.16 (br d, J = 7.2 Hz, 1H), 8.10 (m, 1H), 7.82 (br d, J = 7.2 Hz, 1H), 7.50 (d, J = 7.6Hz, 1H), 7.43 (d, J = 3.5 Hz, 1H), 7.38 (t, J = 7.6 Hz, 1H), 7.32 (t, J = 7.6 Hz, 1H), 7.23 (d, J = 3.5 Hz, 1H), 4.72 (t, J = 8.4 Hz, 2H), 3.37 (t, J = 8.4 Hz, 1H).
(5-(2-chloro-4-nitrophenyl)furan-2-yl)(5-indolin-1-yl)methanone (20a). Compound 15 was reacted according to General Procedure H, 1-bromo-2-chloro-4-nitrobenzene was used as the aryl bromide. Afforded the title compound as a yellow solid, yield 36.5%. 1H NMR (400 MHz, DMSO-d6, δ, ppm): 8.38 (d, J = 2.3 Hz, 1H), 8.22 (m, 1H), 8.07 (br d, J = 8.8 Hz, 1H), 7.50 (d, J = 3.8 Hz, 1H), 7.35 (m, 1H), 7.26 (m, 3H), 7.10 (br t, J = 7.5 Hz, 1H), 4.55 (t, J = 8.1 Hz, 2H), 3.31 (t, J = 8.1 Hz, 2H).
4.10. General Procedure I
To a solution of (5-nitroindolin-1-yl)(5-(4-nitrophenyl)furan-2-yl)methanone derivatives (16a–18a) or (5-nitroindolin-1-yl)(5-(phenyl)furan-2-yl)methanone (19a) or (indolin-1-yl)(5-(4-nitrophenyl)furan-2-yl)methanone (20a) (0.3 mmol) in tetrahydrofuran (3 mL) was added ethanol (3 mL) following was added 3 mL of ammonium chloride (0.3 M) and iron (1.75 mmol). After stirring at 60 °C for 4 h, the reaction was allowed to cool to room temperature and the heterogeneous mixture filtered through Celite and the Celite was rinsed with ethyl acetate. The solution was concentrated to half-volume, then diluted with ethyl acetate (20 mL) and washed with sodium hydroxide solution (1 M, 20 mL). The organic layer was separated, the aqueous phase was extracted with ethyl acetate (2×), the combined organic phases dried over sodium sulfate, filtered and the solvent evaporated. The crude products were used directly in the next step without further purification.
(5-(4-amino-2-chlorophenyl)furan-2-yl)(5-aminoindolin-1-yl)methanone (16b). Compound 16a was reacted according to General Procedure I, yield 75%, yellow solid. 1H NMR (400 MHz, DMSO-d6, δ, ppm): 7.81 (br s, 1H), 7.53 (d, J = 8.7 Hz, 1H), 7.20 (dd, J = 3.6; 1.0 Hz, 1H), 6.90 (dd, J = 3.6; 1.0 Hz, 1H), 6.69 (dd, J = 2.2; 1.0 Hz, 1H), 6.62 (m, 1H), 6.49 (br s, 1H), 6.36 (m, 1H), 5.81 (br s, 2H), 4.94 (br s, 2H), 4.40 (t, J = 8.2 Hz, 2H), 3.08 (t, J = 8.2 Hz, 2H).
(5-aminoindolin-1-yl)(5-(4-aminophenyl)furan-2-yl)methanone (17b). Compound 17a was reacted according to General Procedure I, yield 84%, fluffy yellow solid. 1H NMR (400 MHz, DMSO-d6, δ, ppm): 7.83 (br d, J = 7.5 Hz, 1H), 7.48 (d, J = 8.4 Hz, 2H), 7.18 (dd, J = 3.6; 0.5 Hz, 1H), 6.76 (dd, J = 3.6; 0.5 Hz, 1H), 6.63 (d, J = 8.4 Hz, 2H), 6.51 (d, J = 1.8 Hz, 1H), 6.38 (dd, J = 8.4; 1.8 Hz, 1H), 5.51 (br s, 2H), 4.94 (br s, 2H), 4.43 (t, J = 8.2 Hz, 2H), 3.10 (t, J = 8.2 Hz, 2H).
(5-(4-amino-2-trifluoromethylphenyl)furan-2-yl)(5-aminoindolin-1-yl)methanone (18b). Compound 18a was reacted according to General Procedure I, yield 78.7%, tan solid. 1H NMR (400 MHz, DMSO-d6, δ, ppm): 8.09 (br s, 1H), 7.49 (d, J = 8.4 Hz, 1H), 7.25 (d, J = 4.1 Hz, 1H), 7.02 (d, J = 2.4 Hz, 1H), 6.84 (dd, J = 8.4; 2.4 Hz, 1H), 6.61 (d, J = 4.1 Hz, 1H), 6.56 (m, 2H), 4.69 (t, J = 8.2 Hz, 2H), 4.04 (br s, 2H), 3.13 (t, J = 8.2 Hz, 2H).
(5-(2-chlorophenyl)furan-2-yl)(5-aminoindolin-1-yl)methanone (19b). Compound 19a was reacted according to General Procedure I, yield 90%, yellow solid. 1H NMR (400 MHz, CDCl3, δ, ppm): 8.11 (m, 1H), 7.84 (dd, J = 7.7; 1.5 Hz, 1H), 7.35 (td, J = 7.7; 1.5 Hz, 1H), 7.29 (d, J = 3.7; Hz, 1H), 7.28 (dd, J = 9.5; 1.6 Hz, 1H), 7.20 (d, J = 3.7; Hz, 1H), 6.57 (m, 2H), 4.53 (t, J = 8.3 Hz, 2H), 3.18 (t, J = 8.3 Hz, 2H).
(5-(4-amino-2-chlorophenyl)furan-2-yl)(indolin-1-yl)methanone (20b). Compound 20a was reacted according to General Procedure I, yield 84%, yellow solid. 1H NMR (400 MHz, CDCl3, δ, ppm): 8.23 (m, 1H), 7.56 (dd, J = 8.5; 4.8 Hz, 1H), 7.20 (m, 3H), 7.03 (td, J = 7.4; 3.3 Hz, 1H), 6.96 (t, J = 3.9 Hz, 1H), 6.71 (dd, J = 4.8; 2.2 Hz, 2H), 6.59 (m, 1H), 4.47 (m, 2H), 3.21 (m, 2H).
4.11. General Procedure J
To a solution of (5-aminoindolin-1-yl)(5-(4-aminophenyl)furan-2-yl)methanone derivatives (16b–18b) or (5-aminoindolin-1-yl)(5-(phenyl)furan-2-yl)methanone (19b) or (indolin-1-yl)(5-(4-aminophenyl)furan-2-yl)methanone (20b) (0.10 mmol) in dimethylformamide (5 mL) at 0 °C was added mercury (II) chloride (0.21 mmol), 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea (0.19 mmol) and triethylamine (0.48 mmol). The resulting mixture was stirred at 0 °C for 1 h and 18 h at room temperature. The reaction mixture was diluted with ethyl acetate, filtered through Celite and the Celite rinsed with ethyl acetate. The organic phase was washed with water (2×), and brine (2×), dried over sodium sulfate, filtered, and concentrated under reduced pressure. The crude product was purified by flash chromatography (10 g Sfär silica D cartridge, 10–45% B, Rf = 10 column volumes) to give the desired compounds.
(5-(2-chloro-4-N,N′-di-(tert-butoxycarbonyl)guanidinophenyl)furan-2-yl)(5-N,N′-di-(tert-butoxycarbonyl)guanidinoindolin-1-yl)methanone (16c). Compound 16b was reacted according to General Procedure J, yield 21%, yellow solid. 1H NMR (400 MHz, CDCl3, δ, ppm): 11.63 (m, 2H), 10.51 (d, J = 9.8 Hz, 1H), 10.34 (d, J = 9.8 Hz, 1H), 8.19 (br s, 1H), 7.80–7.67 (m, 4H), 7.31 (m, 1H), 7.20 (m, 1H), 7.15 (m, 1H), 4.57 (q, J = 8.6 Hz, 2H), 3.27 (q, J = 8.6 Hz, 2H), 1.51 (br s, 36H).
(5-(4-N,N′-di-(tert-butoxycarbonyl)guanidinophenyl)furan-2-yl)(5-N,N′-di-(tert-butoxycarbonyl)guanidinoindolin-1-yl)methanone (17c). Compound 17b was reacted according to General Procedure J, yield 45%, yellow solid. 1H NMR (400 MHz, CDCl3, δ, ppm): 11.65 (m, 2H), 10.46 (s, 1H), 10.34 (s, 1H), 8.19 (br s, 1H), 7.74 (br s, 1H), 7.70 (d, J = 8.6 Hz, 2H), 7.66 (d, J = 8.6 Hz, 2H), 7.22 (m, 1H), 7.20 (br d, J = 8.6 Hz, 1H), 6.71 (m, 1H), 4.59 (t, J = 8.1 Hz, 2H), 3.28 (q, J = 8.1 Hz, 2H), 1.52 (br s, 36H).
(5-(2-trifluoromethyl-4-N,N′-di-(tert-butoxycarbonyl)guanidinophenyl)furan-2-yl)(5-N,N′-di-(tert-butoxycarbonyl)guanidinoindolin-1-yl)methanone (18c). Compound 18b was reacted according to General Procedure J, yield 49%, tan solid. 1H NMR (400 MHz, CDCl3, δ, ppm): 11.64 (br s, 1H), 11.61 (br s, 1H), 10.61 (s, 1H), 10.34 (s, 1H), 8.09 (dd, J = 8.6; 2.1 Hz, 1H), 7.94 (d, J = 2.1 Hz, 1H), 7.74 (br s, 1H), 7.73 (d, J = 8.6 Hz, 1H), 7.32 (d, J = 3.6 Hz, 1H), 7.21 (dd, J = 8.6; 2.0 Hz, 1H), 6.77 (d, J = 3.6 Hz, 1H), 4.54 (q, J = 8.2 Hz, 2H), 3.25 (q, J = 8.2 Hz, 2H), 1.53 (br s, 36H).
(5-(2-chlorophenyl)furan-2-yl)(5-N,N′-di-(tert-butoxycarbonyl)guanidinoindolin-1-yl)methanone (19c). Compound 19b was reacted according to General Procedure J, yield 27%, yellow solid. 1H NMR (400 MHz, CDCl3, δ, ppm): 11.65 (s, 1H), 10.34 (s, 1H), 8.19 (br s, 1H), 7.83 (dd, J = 7.9; 1.4 Hz, 1H), 7.73 (br s, 1H), 7.46 (dd, J = 7.9; 1.3 Hz, 1H), 7.35 (td, J = 7.9; 1.3 Hz, 1H), 7.31 (d, J = 3.7 Hz, 1H), 7.27 (dd, J = 7.9; 1.7 Hz, 1H), 7.20 (m, 2H), 4.55 (t, J = 8.3 Hz, 2H), 3.27 (t, J = 8.3 Hz, 2H), 1.53 (s, 9H), 1.50 (s, 9H).
(5-(2-chloro-4-N,N′-di-(tert-butoxycarbonyl)guanidinophenyl)furan-2-yl)(indolin-1-yl)methanone (20c). Compound 20b was reacted according to General Procedure I, yield 53%, yellow solid. 1H NMR (400 MHz, CDCl3, δ, ppm): 11.62 (s, 1H), 10.52 (s, 1H), 8.23 (br s, 1H), 7.81 (d, J = 4.8 Hz, 1H), 7.89 (d, J = 1.7 Hz, 1H), 7.71 (dd, J = 8.3; 2.1 Hz, 1H), 7.33 (d, J = 3.8 Hz, 1H), 7.23 (m, 1H), 7.16 (d, J = 3.8 Hz, 1H), 7.06 (m, 2H), 4.58 (t, J = 8.2 Hz, 2H), 3.28 (t, J = 8.2 Hz, 2H), 1.55 (s, 18H).
4.12. General Procedure K
To a solution of (5-(4-N,N′-di-(tert-butoxycarbonyl)guanidinophenyl)furan-2-yl)(5-N,N′-di-(tert-butoxycarbonyl)guanidinoindolin-1-yl)methanone derivatives (16c–18c) or (5-phenyl)furan-2-yl)(5-N,N′-di-(tert-butoxycarbonyl)guanidinoindolin-1-yl)methanone (19c) or (5-(4-N,N′-di-(tert-butoxycarbonyl)guanidinophenyl)furan-2-yl)(inoindolin-1-yl)methanone (20c) (0.05 mmol) in dichloromethane (1 mL) was added HCl (4 M in dioxane, 2 mL). The solution was stirred at room temperature for 16 h. The mixture was concentrated under reduced pressure and the residue was purified by reverse phase flash chromatography (6 g Sfär C18 D cartridge, 0–50% B, Rf = 10 column volumes) to give the desired compounds.
(5-(2-chloro-4-guanidinophenyl)furan-2-yl)(5-guanidinoindolin-1-yl)methanone (16). Compound 16c was reacted according to General Procedure K, yield 86%, yellow solid. 1H NMR (400 MHz, CD3OD, δ, ppm): 8.26 (br d, J = 8.5 Hz, 1H), 8.09 (d, J = 8.5 Hz, 1H), 7.51 (d, J = 2.0 Hz, 1H), 7.43 (d, J = 3.8 Hz, 1H), 7.39 (dd, J = 8.5; 2.0 Hz, 1H), 7.36 (d, J = 3.8 Hz, 1H), 7.24 (br s, 1H), 7.15 (dd, J = 8.5; 2.0 Hz, 1H), 4.64 (t, J = 8.3 Hz, 2H), 3.35 (t, J = 8.3 Hz, 2H). 13C NMR (100 MHz, CD3OD, δ, ppm): 158.93, 158.27, 157.79, 153.38, 148.10, 143.80, 137.63, 136.03, 132.88, 132.18, 131.07, 127.81, 127.78, 125.92, 124.66, 123.69, 120.54, 119.62, 114.99, 51.31, 29.50. HRMS (ESI), found 438.1441 C21H20ClN7O2, [M + H]+, requires 438.1445.
(5-(4-guanidinophenyl)furan-2-yl)(5-guanidinoindolin-1-yl)methanone (17). Compound 17c was reacted according to General Procedure K, yield 35%, pale yellow solid. 1H NMR (400 MHz, CD3OD, δ, ppm): 8.25 (d, J = 8.6 Hz, 1H), 7.96–7.93 (m, 2H), 7.39 (m, 3H), 7.24 (m, 1H), 7.14 (dd, J = 8.5; 2.0 Hz, 1H), 7.36 (d, J = 3.8 Hz, 1H), 7.24 (br s, 1H), 7.15 (dd, J = 8.5; 2.3 Hz, 1H), 7.08 (d, J = 3.8 Hz, 1H), 4.64 (t, J = 8.2 Hz, 2H), 3.35 (t, J = 8.2 Hz, 2H). 13C NMR (100 MHz, CD3OD, δ, ppm): 159.08, 158.27, 157.89, 157.03, 148.02, 143.87, 136.71, 135.98, 132.09, 129.76, 127.26, 126.56, 125.89, 123.67, 121.10, 119.58, 108.96, 51.24, 29.50. HRMS (ESI), found 404.1833 C21H21N7O2, [M + H]+, requires 404.1835.
(5-(2-trifluoromethy-4-guanidinophenyl)furan-2-yl)(5-guanidinoindolin-1-yl)methanone (18). Compound 18c was reacted according to General Procedure K, yield 80%, tan solid. 1H NMR (400 MHz, CD3OD, δ, ppm): 8.26 (br d, J = 8.6 Hz, 1H), 8.02 (d, J = 8.5 Hz, 1H), 7.79 (d, J = 2.1 Hz, 1H), 7.70 (dd, J = 2.1; 8.5 Hz, 1H), 7.44 (d, J = 3.6 Hz, 1H), 7.25 (d, J = 2.0 Hz, 1H), 7.16 (dd, J = 2.1; 8.6 Hz, 1H), 7.00 (d, J = 3.6 Hz, 1H), 4.61 (t, J = 8.3 Hz, 2H), 3.31 (m, 2H). 13C NMR (100 MHz, CD3OD, δ, ppm): 158.90, 158.29, 157.90, 153.66, 149.33, 143.78, 137.84, 136.05, 133.87, 132.19, 127.84, 129.31, 125.95, 124.13 (d, J = 5.38 Hz), 123.72, 123.39 120.49, 119.62, 113.62 (q, J = 6.3 Hz), 51.12, 29.43. HRMS (ESI), found 472.1708 C22H20F3N7O2, [M + H]+, requires 472.1709.
(5-(2-chlorophenyl)furan-2-yl)(5-guanidinoindolin-1-yl)methanone (19). Compound 19c was reacted according to General Procedure K, yield 81%, yellow solid. 1H NMR (400 MHz, CD3OD, δ, ppm): 8.24 (br d, J = 8.5 Hz, 1H), 7.96 (dd, J = 7.9; 1.5 Hz, 1H), 7.54 (dd, J = 7.9; 1.5; Hz, 1H), 7.45 (dt, J = 1.5; 7.6 Hz, 1H), 7.39 (d, J = 3.8 Hz, 1H), 7.37 (dt, J = 1.6; 7.6 Hz, 1H), 7.30 (d, J = 3.8 Hz, 1H), 7.23 (br d, J = 1.9, 1H), 7.13 (dd, J = 1.9; 8.5 1H), 4.63 (t, J = 8.3 Hz, 2H), 3.34 (t, J = 8.3 Hz, 2H). 13C NMR (100 MHz, CD3OD, δ, ppm): 158.88, 158.24, 154.19, 147.94, 143.81, 135.95, 132.05, 132.03, 130.99, 129.87, 129.17, 128.55, 125.83, 123.60, 120.61, 119.55, 113.68, 51.18, 29.44. HRMS (ESI), found 381.1118 C20H17ClN4O2, [M + H]+, requires 381.1118.
(5-(2-chloro-4-guanidinophenyl)furan-2-yl)(indolin-1-yl)methanone (20). Compound 20c was reacted according to General Procedure K, yield 58%, yellow solid. 1H NMR (400 MHz, CD3OD, δ, ppm): 10.27 (br s, 1H), 8.2009 (br d, J = 8.6 Hz, 1H), 7.93 (dd94 (d, J = 7.8; 1.6 Hz, 1H), 7.51 (dd74 (br s, 2H), 7.47 (d, J = 7.8; 1.3 Hz, 1H), 7.41 (td, J = 7.8; 1.32.1 Hz, 1H), 7.35 (m, 2H), 7.39 (d, J = 3.7 Hz, 1H), 7.34 (dd, J = 8.6; 2.1 Hz, 1H), 7.29 (br s, 1H), 7.26 (d, J = 3.87 Hz, 1H), 7.19 (m, 1H), 7.10 (dd, J = 8.6; 2.2 Hz04 (m, 1H), 4.52 (t, J = 8.2 Hz, 2H), 3.22 (t, J = 8.2 Hz, 2H). 13C NMR (100 MHz, DMSO-d6, δ, ppm): 165.72, 165.22, 160.34, 156.63, 152.50, 146.37, 141.79, 139.86, 139.04, 136.43, 134.99, 134.38, 133.58, 132.43, 128.28, 126.37, 120.54, 121.84, 58.50, 37.63. HRMS (ESI), found 381.1118 C20H17ClN4O2, [M + H]+, requires 381.1118.
4.13. Antibacterial Activity Assays
All compounds were dissolved at 50 mM in 100% DMSO and stored at −20 °C until analysis. The antibacterial activity of all compounds was determined against non-pathogenic Gram-positive bacteria
B. subtilis (NBRC/ATCC #111470), and four Gram-negative bacteria;
E. coli MG1655 (CGSC #6300),
P. putida (NBRC/ATCC #100650),
P. carotovorum (NBRC/ATCC #3380) and
P. caledonica (NBRC/ATCC #102488). The bacteria were cultured as previously described by Mueller and Hinton [
38] and Doyle [
39]. For in vitro determination of antibacterial activity, a culture of bacterial cells was grown to OD 600
nm = 0.5. The bacterial culture was diluted 10× with pre-warmed fresh medium and aliquoted into a 384-well plate before adding compounds. The starting concentration of the compound was either 300 or 1000 µM, and following dilution in two-fold intervals, the plate was incubated at 37 °C for 18 h without agitation. To measure cell viability, we used the resazurin-based assay as described previously [
40]. To each well, 12 μL of 10× AlamarBlue solution (resazurin solution, ThermoFisher, Waltham, MA, USA) was added, and a 384-well plate incubated at 37 °C for 1 h. The 384-well plates enabled us to use small final volumes of 20 µL conserving compound stock and slowing down evaporation due to the depth of the wells. Fluorescence was measured using a POLARstar Omega microplate-reader (BMG Labtech, Offenburg, Germany) with excitation filter set to 544 nm and emission filter to 590 nm. Cells exposed to only the equivalent concentration of DMSO were used as negative control. Bleed-through of fluorescence from resorufin between wells in the microtiter plate fluorescence reader, was measured and found to be <1% between adjacent wells. The 384-well plates were used to avoid this fluorescence bleed-through, achieved by skipping a well in-between bacterial cultures and compound dilutions. To check for quenching of fluorescence by any of the investigated compounds, grown bacterial cultures were mixed after 1 h incubation with resazurin and the compound of interest at the highest concentration to be assayed, and the measured fluorescence compared with samples without compound added. All tests of compound activity were performed in three independent replicates for more accurate determination of the half (EC
50) and 90% maximal effective concentration (EC
90).
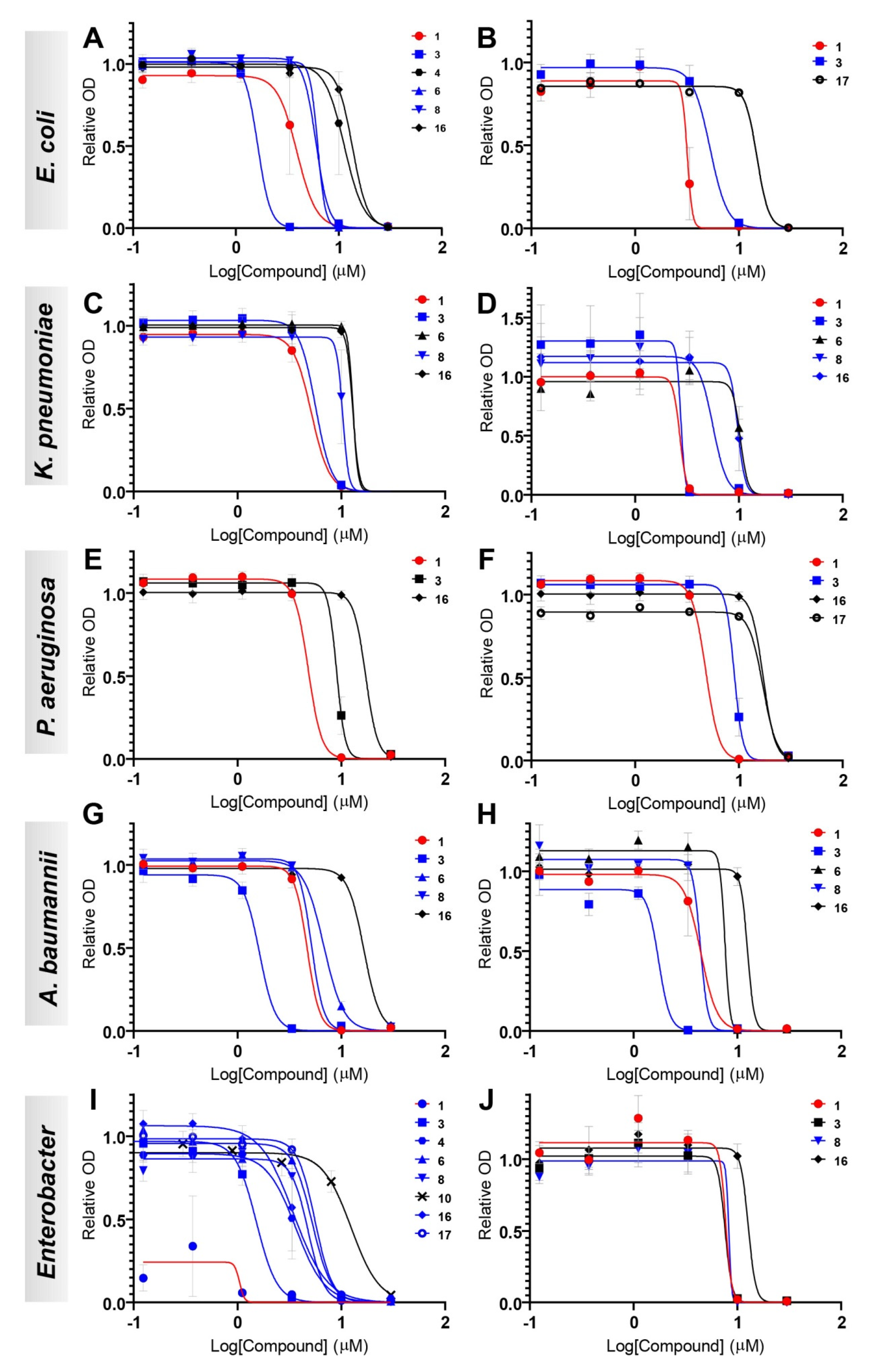
The antibacterial activities of eight compounds (1, 3, 4, 6, 8, 10, 16, and 17) were tested against 10 different isolates of Gram-negative bacteria from human and other sources, two E. coli (CCUG #67180 and CCUG #17620/ATCC #25922, control strain), two K. pneumoniae (CCUG #58547 and CCUG #225T), two P. aeruginosa (CCUG #17619 and CCUG #59347), two A. baumannii (CCUG #57035 and CCUG #57250), E. cloacae (CCUG #6323T), and E. hormaechei (CCUG #58962) in a 96-well plate format. The compounds to test were 3-fold diluted in six steps in cation-adjusted Mueller-Hinton (ca-MH) broth to final concentrations spanning from 30 to 0.12 µM, i.e., a 1667–416,667 times dilution from the 50 mM stocks. Bacterial mass from the ten isolates grown overnight on horse blood or Mueller–Hinton agar plates was suspended in ca-MH and adjusted to a final inoculum cell density of ~5 × 105 CFU/mL. For all assays, Biolog redox dye A diluted 100× from the stock solution was used for measurements of all ten isolates (up to a total volume of 120 µL per well). All plates included one well per isolate with only inoculum and dye but no test compound (i.e., positive control) and one well per isolate with inoculum, dye and DMSO diluted 1667×, corresponding to the DMSO concentration in the wells with the highest concentration of test compound. All measurements were performed using the Omnilog microplate reader (Biolog, Hayward, CA, USA) where the 96-well plates were read at 15 min intervals for a total of 24 h at 37 °C. Each compound was run in triplicate on three independent assay plates for more accurate determination of the half (EC50) and 90% maximal effective concentration (EC90).
Non-linear regression dose–response inhibition following a log (agonist) vs. response–Find ECanything was performed using GraphPad Prism version 9.2.0 for Windows, GraphPad Software, San Diego, CA, USA,
www.graphpad.com (accessed on 20 July 2022).
4.14. Cytotoxicity Assays
The cytotoxicity levels of all compounds were evaluated against human Michigan Cancer Foundation-7 (MCF-7) and Hepatoma G2 cell line (HepG2) cell lines. MCF-7 is an extensively characterized breast cancer cell line isolated in 1970 [
41], while HepG2 cells were derived from a hepatoma in 1975 [
42]. Both cell lines grow robustly during in vitro culture and have been widely used for cytotoxicity testing. Cells of hepatic origin, such as HepG2, are of particular relevance for toxicity studies as many drugs accumulate in the liver during metabolic conversion [
41,
42]. Cells were grown in Dulbecco’s Modified Eagle Medium supplemented with 10% fetal calf serum and kept in exponential growth, as previously reported [
43]. Before the assay, cells were reseeded into 96-well microtiter plates at a density allowing continued exponential growth and let to settle for 24 h. The compounds were added from a stock solution in DMSO, for a final concentration of 0.3%
v/
v of the solvent in the culture medium. After 24 h of incubation in presence of the compound, cell viability was assayed using PrestoBlue Cell Viability Reagent (resazurin-based solution, ThermoFisher, Waltham, MA, USA) according to the manufacturer’s instructions. A POLARstar Omega microplate-reader (BMG Labtech, Offenburg, Germany) was used to measure resorufin fluorescence at 544 nm excitation/590 nm emissions. Each assay contained a DMSO control at the equivalent starting concentration, positive control (uninhibited cell growth) and negative control (cell medium only). Survival was expressed as percentage of the solvent-only control. EC
50 values for each compound were calculated from three independent replicate experiments using 2-fold dilution intervals. Non-linear regression dose–response inhibition following a log(agonist) vs. response–Find ECanything was performed using GraphPad Prism version 9.2.0 for Windows, GraphPad Software, San Diego, California USA,
www.graphpad.com. The Selectivity Index (SI) for each compound and bacterial strain was calculated as the ratio between the mean of the EC
50 values for the two human cell lines and the EC
50 for the bacterial strain in question ((EC
50 MCF-7 + EC
50 HepG2)/2)/EC
50 bacterial strain). The higher the value, the more selective is the compound against the different bacterial strain.
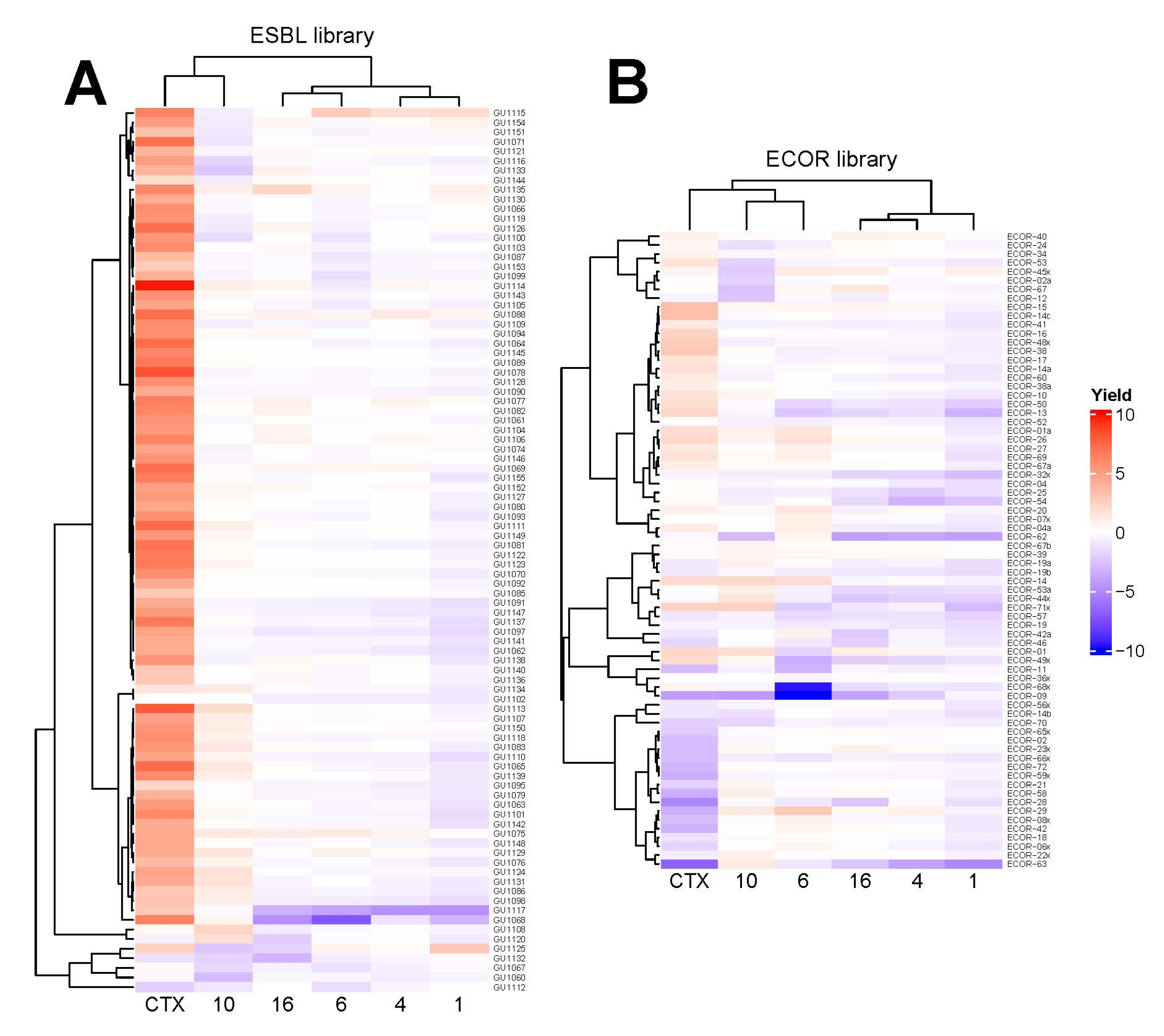
4.15. High-Resolution Microbial Phenomics (Scan-o-Matic) Assays
E. coli colonies were deposited as initially isogenic populations at initial population sizes of ~100,000 cells, with 1536 colonies deposited in systematic colony arrays on each plate on top of a solid matrix composed of LB medium supplemented with a sublethal concentration (60 µM) of five different compounds,
1,
4,
6,
10, and
16, and a known antimicrobial, cefotaxime (CTX; 2 µg/mL), using automated pinning by robot. The compound concentrations were empirically chosen to strongly but not completely inhibit colony growth on agar of the more sensitive strains in the collections, in order to enable quantitation of the difference in growth yield between more and less resistant strains [
44,
45]. Of these colonies, 384 were identical controls used to correct for spatial bias between and within plates. For CTX and
4,
10, and
16, each lineage was cultivated as six biological replicates on different plates. For
1, nine biological replicates were done. Population expansion for each colony was followed by measuring cell numbers at 10 min intervals using the Scan-o-Matic framework, version 2.0 [
44] with an
E. coli calibration curve [
45]. From each colony growth curve, the total cell yield after 8 h was extracted (growth yield). Experiments included automated transmissive scanning and signal calibration in 10 min intervals, as described [
44]. The absolute population yields were log(2) transformed and normalized to the corresponding measures of adjacent controls (fourth position
E. coli CCUG #17620/ATCC #25922 strain) on each plate, while data for missing or mis-quantified colonies were discarded. The relative growth yield of each strain (total n = 164) of the screened ECOR (n = 72) and ESBL (n = 92) libraries on the tested compounds was normalized to their corresponding growth without compound and the resulting ratios were clustered and visualized using heatmaps constructed using the R package
ComplexHeatmap v. 2.8.0 [
27].


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}