
A Universal LC-MS/MS Method for Simultaneous Detection of Antibiotic Residues in Animal and Environmental Samples



Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
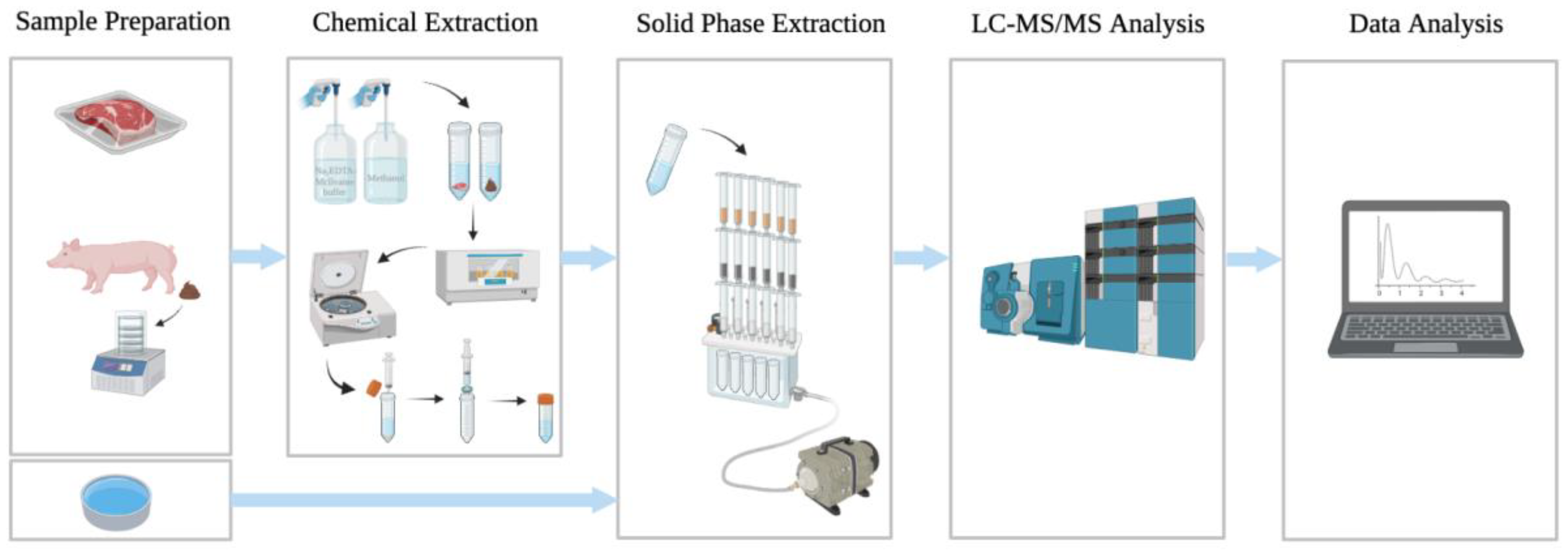
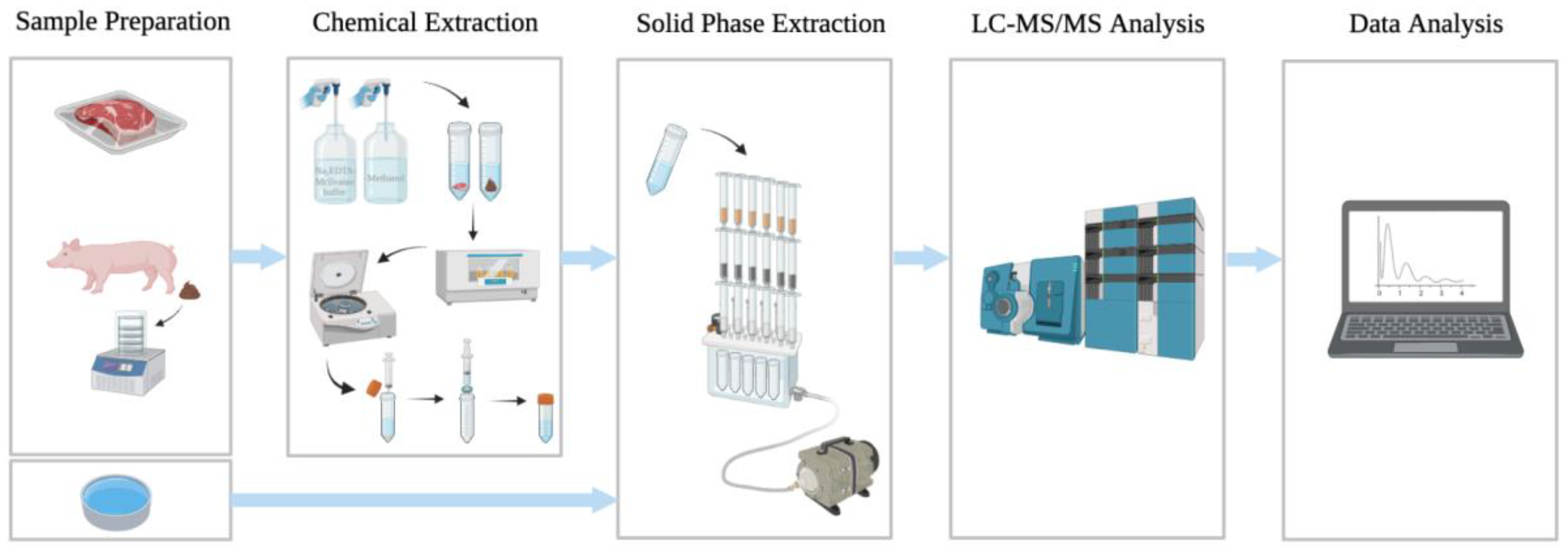
2.2. Sample Preparation
2.3. Chemical Extraction
2.4. Solid-Phase Extraction (SPE)
2.5. LC-MS/MS Analysis
2.6. Data Analysis
3. Results
3.1. Limit of Detection
3.2. Solid-Phase Extraction
3.3. Chemical Extraction
3.4. Sensitivity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Antimicrobial Resistance. 2020. Available online: https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance (accessed on 31 March 2021).
- Ventola, C.L. The antibiotic resistance crisis: Part 1: Causes and threats. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- World Health Organization. WHO, FAO, and OIE Unite in the Fight Against Antimicrobial Resistance; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- European Commission. A European One Health Action Plan against Antimicrobial Resistance (AMR); European Commission: Brussels, Belgium, 2017. [Google Scholar]
- World Health Organization. Antimicrobial Resistance: Global Report on Surveillance; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Lopes, R.; Augusti, D.; Oliveira, A.; Oliveira, F.; Vargas, E.; Augusti, R. Development and validation of a methodology to qualitatively screening veterinary drugs in porcine muscle via an innovative extraction/clean-up procedure and LC-MS/MS analysis. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2011, 28, 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Abd El-Aty, A.M.; Kim, S.K.; Choi, J.M.; Park, D.H.; Yoo, K.H.; Kang, Y.S.; Jeon, J.S.; Hacimuftuoglu, A.; Shim, J.H.; et al. Development and validation of a solid-phase extraction method coupled with LC-MS/MS for the simultaneous determination of 16 antibiotic residues in duck meat. Biomed. Chromatogr. 2019, 33, e4501. [Google Scholar] [CrossRef] [PubMed]
- Dubreil, E.; Gautier, S.; Fourmond, M.-P.; Bessiral, M.; Gaugain, M.; Verdon, E.; Pessel, D. Validation approach for a fast and simple targeted screening method for 75 antibiotics in meat and aquaculture products using LC-MS/MS. Food Addit. Contam. Part A 2017, 34, 453–468. [Google Scholar] [CrossRef] [PubMed]
- Alija, G.; Hajrulai-Musliu, Z.; Uzunov, R. Development and validation of confirmatory LC–MS/MS method for multi-residue analysis of antibiotic drugs in bovine milk. SN Appl. Sci. 2020, 2, 1563. [Google Scholar] [CrossRef]
- Gaugain-Juhel, M.; Delepine, B.; Gautier, S.; Fourmond, M.P.; Gaudin, V.; Hurtaud-Pessel, D.; Verdon, E.; Sanders, P. Validation of a liquid chromatography-tandem mass spectrometry screening method to monitor 58 antibiotics in milk: A qualitative approach. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2009, 26, 1459–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufmann, A.; Roth, S.; Ryser, B.; Widmer, M.; Guggisberg, D. Quantitative LC/MS-MS determination of sulfonamides and some other antibiotics in honey. J. AOAC Int. 2002, 85, 853–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mokh, S.; El Khatib, M.; Koubar, M.; Daher, Z.; Al Iskandarani, M. Innovative SPE-LC-MS/MS technique for the assessment of 63 pharmaceuticals and the detection of antibiotic-resistant-bacteria: A case study natural water sources in Lebanon. Sci. Total Environ. 2017, 609, 830–841. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Song, W.; Lin, H.; Wang, W.; Du, L.; Xing, W. Antibiotics and antibiotic resistance genes in global lakes: A review and meta-analysis. Environ. Int. 2018, 116, 60–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Zhang, H.; Luo, Y.; Song, J. Occurrence and assessment of veterinary antibiotics in swine manures: A case study in East China. Chin. Sci. Bull. 2012, 57, 606–614. [Google Scholar] [CrossRef] [Green Version]
- Duelge, K.J.; Nishshanka, U.; De Alwis, H.G. An LC-MS/MS method for the determination of antibiotic residues in distillers grains. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1053, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Barreca, S.; Forni, C.; Colzani, L.; Clerici, L.; Daverio, D.; Dellavedova, P. Study on the stability of antibiotics, pesticides and drugs in water by using a straightforward procedure applying HPLC-mass spectrometric determination for analytical purposes. Separations 2021, 8, 179. [Google Scholar] [CrossRef]
- Okerman, L.; Van Hende, J.; De Zutter, L. Stability of frozen stock solutions of beta-lactam antibiotics, cephalosporins, tetracyclines and quinolones used in antibiotic residue screening and antibiotic susceptibility testing. Anal. Chim. Acta 2007, 586, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, B.J.; Elbers, I.J.; Stolker, A.A. Determination of the stability of antibiotics in matrix and reference solutions using a straightforward procedure applying mass spectrometric detection. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2011, 28, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Rossi, R.; Saluti, G.; Moretti, S.; Diamanti, I.; Giusepponi, D.; Galarini, R. Multiclass methods for the analysis of antibiotic residues in milk by liquid chromatography coupled to mass spectrometry: A review. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2018, 35, 241–257. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, X.Q.; Li, H.M.; Zhang, Q.H.; Gao, Y.; Li, X.J. Antibiotic residues in honey: A review on analytical methods by liquid chromatography tandem mass spectrometry. TrAC Trends Anal. Chem. 2019, 110, 344–356. [Google Scholar] [CrossRef]
- Andrade-Eiroa, A.; Canle, M.; Leroy-Cancellieri, V.; Cerdà, V. Solid-phase extraction of organic compounds: A critical review. part ii. TrAC Trends Anal. Chem. 2016, 80, 655–667. [Google Scholar] [CrossRef]
- Pico, Y.; Fernandez, M.; Ruiz, M.J.; Font, G. Current trends in solid-phase-based extraction techniques for the determination of pesticides in food and environment. J. Biochem. Biophys. Methods 2007, 70, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Seifrtova, M.; Novakova, L.; Lino, C.; Pena, A.; Solich, P. An overview of analytical methodologies for the determination of antibiotics in environmental waters. Anal. Chim. Acta 2009, 649, 158–179. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Classes | Antibiotics |
|---|---|
| Aminoglycosides | Gentamicin |
| Kanamycin sulfate | |
| Neomycin trisulfate salt hydrate | |
| Spectinomycin hydrochloride pentahydrate | |
| Streptomycin sulfate salt | |
| Amphenicols | Chloramphenicol |
| Florfenicol | |
| Antifolate | Trimethoprim |
| Carbapenems | Meropenem |
| Cephalosporins | Cefalexin |
| Cefquinome sulfate | |
| Ceftazidime | |
| Ceftiofur sodium | |
| Cefuroxime | |
| Fluoroquinolones | Ciprofloxacin |
| Enrofloxacin | |
| Levofloxacin | |
| Norfloxacin | |
| Ofloxacin | |
| Glycopeptides | Vancomycin |
| Lincosamides | Clindamycin phosphate |
| Lincomycin hydrochloride | |
| Macrolides | Erythromycin |
| Tilmicosin | |
| Tylosin tartrate salt | |
| Tylvalosin | |
| Nitroimidazole | Metronidazole |
| Penicillins | Amoxicillin |
| Ampicillin | |
| Penicillin G sodium salt | |
| Pleuromutilins | Tiamulin |
| Polymyxins | Colistin A |
| Colistin B | |
| Quinoxaline 1,4-di-N-oxides (QdNOs) | Mequindox |
| Sulfonamides | Sulfachloropyridazine |
| Sulfadiazine | |
| Sulfadimidine | |
| Sulfamethoxazole | |
| Sulfamonomethoxine | |
| Tetracyclines | Chlortetracycline hydrochloride |
| Doxycycline | |
| Oxytetracycline | |
| Tetracycline |
| Antibiotics | Retention Time (min) | Transition 1 (m/z) | Transition 2 (m/z) | Limit of Detection (ppb) |
|---|---|---|---|---|
| Amoxicillin | 0.83 | 366.1 > 348.9 | 366.1 > 208 | 8.51 |
| Ampicillin | 1.78 | 350 > 191.9 | 350 > 160 | 0.49 |
| Cefalexin | 1.79 | 348 > 158 | 348 > 174 | 0.9 |
| Cefquinome sulfate | 2.33 | 529 > 396 | 529 > 134 | 1.11 |
| Ceftazidime | 2.14 | 547.1 > 467.8 | 547.1 > 396 | 3.44 |
| Ceftiofur sodium | 3.53 | 524 > 241 | 524 > 285 | 0.31 |
| Cefuroxime | 2.57 | 447 > 385.7 | 447 > 342 | 4.1 |
| Chloramphenicol | 2.56 | 323.1 > 274.9 | 323.1 > 304.8 | 5.41 |
| Chlortetracycline hydrochloride | 2.66 | 479 > 444 | 479 > 462 | 4.06 |
| Ciprofloxacin | 2.35 | 332.1 > 313.9 | 332.1 > 231.1 | 0.4 |
| Clindamycin phosphate | 3.16 | 505.1 > 457 | 505.1 > 487.1 | 0.55 |
| Colistin A | 1.95 | 585.6 > 535.5 | 585.6 > 576.4 | 862.53 |
| Colistin B | 1.77 | 578.5 > 528.4 | 578.5 > 569.5 | 793.3 |
| Doxycycline | 2.94 | 445.1 > 428 | 445.1 > 267 | 0.49 |
| Enrofloxacin | 2.58 | 360 > 316 | 360 > 245 | 0.4 |
| Erythromycin | 3.83 | 734.3 > 576.3 | 734.3 > 157.9 | 14.52 |
| Florfenicol | 2.26 | 358 > 340 | 358 > 241 | 13.21 |
| Gentamicin | 2.79 | 500.1 > 456 | 500.1 > 227.1 | 320 |
| Kanamycin sulfate | 0.27 | 485 > 324 | 485 > 163 | 6.54 |
| Levofloxacin | 2.36 | 362.1 > 318.2 | 362.1 > 261.1 | 0.47 |
| Lincomycin hydrochloride | 1.43 | 407 > 126 | 407 > 359 | 0.45 |
| Mequindox | 2.18 | 219 > 143 | 219 > 185 | 1.7 |
| Meropenem | 1.79 | 384.1 > 340.1 | 384.1 > 297.7 | 1.76 |
| Metronidazole | 1.06 | 172 > 128.2 | 172 > 82.1 | 0.46 |
| Neomycin trisulfate salt hydrate | 0.26 | 615 > 293 | 615 > 161 | 345.18 |
| Norfloxacin | 2.3 | 320 > 302 | 320 > 231.2 | 0.54 |
| Ofloxacin | 2.35 | 362 > 318 | 362 > 261 | 0.43 |
| Oxytetracycline | 2.02 | 461 > 426 | 461 > 444 | 1.98 |
| Penicillin G sodium salt | 2.36 | 335 > 160 | 335 > 176 | 10.79 |
| Spectinomycin hydrochloride pentahydrate | 0.3 | 333 > 189 | 333 > 140 | 2.52 |
| Streptomycin sulfate salt | 4.13 | 582 > 174 | 582 > 156 | 425.98 |
| Sulfachloropyridazine | 2.3 | 285 > 156 | 285 > 108 | 0.6 |
| Sulfadiazine | 1.45 | 251 > 156 | 251 > 92 | 0.45 |
| Sulfadimidine | 2.08 | 279 > 186 | 279 > 156 | 0.36 |
| Sulfamethoxazole | 2.4 | 254.1 > 155.8 | 254.1 > 108.2 | 0.43 |
| Sulfamonomethoxine | 2.38 | 281 > 156 | 281 > 126 | 0.62 |
| Tetracycline | 2.14 | 445 > 410 | 445 > 269 | 0.49 |
| Tiamulin | 4.12 | 494 > 192 | 494 > 119 | 0.59 |
| Tilmicosin | 3.38 | 869.4 > 696 | 869.4 > 174 | 5.73 |
| Trimethoprim | 1.92 | 291.1 > 230 | 291.1 > 260.9 | 0.41 |
| Tylosin tartrate salt | 4.13 | 916.3 > 772 | 916.3 > 174 | 11.63 |
| Tylvalosin | 5.2 | 1042.3 > 814 | 1042.3 > 174 | 64.27 |
| Vancomycin | 1.95 | 726 > 144 | 725 > 144 | 26.63 |
| Antibiotics | SPE Recovery | Mainly Retained |
|---|---|---|
| Amoxicillin | Poor | MAX |
| Ampicillin | Poor | MAX |
| Cefalexin | Good | MCX |
| Cefquinome sulfate | Poor | MAX/HLB |
| Ceftazidime | Good | MAX |
| Ceftiofur sodium | Good | MAX |
| Cefuroxime | Good | MAX |
| Chloramphenicol | Good | MAX |
| Chlortetracycline hydrochloride | Satisfactory | MAX/HLB |
| Ciprofloxacin | Good | MCX |
| Clindamycin phosphate | Good | MAX |
| Colistin A | Satisfactory | MCX |
| Colistin B | Satisfactory | MCX |
| Doxycycline | Satisfactory | MAX/HLB |
| Enrofloxacin | Satisfactory | MCX |
| Erythromycin | Good | HLB |
| Florfenicol | Good | MAX |
| Gentamicin | Satisfactory | MAX |
| Kanamycin sulfate | Good | MCX |
| Levofloxacin | Good | MCX |
| Lincomycin hydrochloride | Good | MCX |
| Mequindox | Satisfactory | MAX/HLB |
| Meropenem | Poor | MAX/HLB |
| Metronidazole | Good | MCX |
| Neomycin trisulfate salt hydrate | Good | MCX |
| Norfloxacin | Good | MCX |
| Ofloxacin | Good | MCX |
| Oxytetracycline | Satisfactory | MCX |
| Penicillin G sodium salt | Good | MAX |
| Spectinomycin hydrochloride pentahydrate | Satisfactory | MCX |
| Streptomycin sulfate salt | Good | HLB |
| Sulfachloropyridazine | Good | MAX |
| Sulfadiazine | Good | MAX/HLB/MCX |
| Sulfadimidine | Good | MAX/HLB/MCX |
| Sulfamethoxazole | Good | MAX |
| Sulfamonomethoxine | Good | MAX |
| Tetracycline | Good | MCX |
| Tiamulin | Poor | MCX |
| Tilmicosin | Satisfactory | HLB |
| Trimethoprim | Good | MCX |
| Tylosin tartrate salt | Good | HLB |
| Tylvalosin | Good | HLB |
| Vancomycin | Good | MCX |
| Antibiotics | Water Sample | Fecal Sample | Meat Sample |
|---|---|---|---|
| Amoxicillin | N.D. | N.D. | N.D. |
| Ampicillin | Detected | Detected | Detected |
| Cefalexin | Detected | Detected | Detected |
| Cefquinome sulfate | N.D. | N.D. | N.D. |
| Ceftazidime | N.D. | N.D. | N.D. |
| Ceftiofur sodium | Detected | Detected | Detected |
| Cefuroxime | N.D. | N.D. | N.D. |
| Chloramphenicol | Detected | Detected | Detected |
| Chlortetracycline hydrochloride | Detected | Detected | Detected |
| Ciprofloxacin | N.D. | N.D. | N.D. |
| Clindamycin phosphate | Detected | Detected | Detected |
| Colistin A | N.D. | N.D. | N.D. |
| Colistin B | N.D. | N.D. | N.D. |
| Doxycycline | Detected | Detected | Detected |
| Enrofloxacin | Detected | Detected | Detected |
| Erythromycin | Detected | Detected | Detected |
| Florfenicol | Detected | Detected | Detected |
| Gentamicin | N.D. | N.D. | N.D. |
| Kanamycin sulfate mixture of kanamycin A (main component) and kanamycin B and C | N.D. | N.D. | N.D. |
| Levofloxacin | Detected | Detected | Detected |
| Lincomycin hydrochloride | Detected | Detected | Detected |
| Mequindox | Detected | Detected | Detected |
| Meropenem | N.D. | N.D. | N.D. |
| Metronidazole | Detected | Detected | Detected |
| Neomycin trisulfate salt hydrate | N.D. | N.D. | N.D. |
| Norfloxacin | N.D. | N.D. | N.D. |
| Ofloxacin | Detected | Detected | Detected |
| Oxytetracycline | Detected | Detected | Detected |
| Penicillin G sodium salt | Detected | Detected | Detected |
| Spectinomycin hydrochloride pentahydrate | Detected | Detected | Detected |
| Streptomycin sulfate salt | Detected | Detected | Detected |
| Sulfachloropyridazine | Detected | Detected | Detected |
| Sulfadiazine | Detected | Detected | Detected |
| Sulfadimidine | Detected | Detected | Detected |
| Sulfamethoxazole | Detected | Detected | Detected |
| Sulfamonomethoxine | Detected | Detected | Detected |
| Tetracycline | Detected | Detected | Detected |
| Tiamulin | Detected | Detected | Detected |
| Tilmicosin | Detected | Detected | Detected |
| Trimethoprim | Detected | Detected | Detected |
| Tylosin tartrate salt | Detected | Detected | Detected |
| Tylvalosin | Detected | Detected | Detected |
| Vancomycin | N.D. | N.D. | N.D. |
| Antibiotics | Sensitivity | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 10 μg Spiked-in | 5 μg Spiked-in | 1 μg Spiked-in | |||||||
| Water Sample | Fecal Sample | Meat Sample | Water Sample | Fecal Sample | Meat Sample | Water Sample | Fecal Sample | Meat Sample | |
| Amoxicillin | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Ampicillin | 100 | 100 | 100 | 100 | 100 | 100 | 66.7 | 0 | 33.3 |
| Cefalexin | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Cefquinome sulfate | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Ceftazidime | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Ceftiofur sodium | 100 | 100 | 100 | 100 | 100 | 100 | 66.7 | 66.7 | 0 |
| Cefuroxime | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Chloramphenicol | 100 | 100 | 100 | 100 | 100 | 100 | 0 | 0 | 66.7 |
| Chlortetracycline hydrochloride | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 66.7 |
| Ciprofloxacin | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Clindamycin phosphate | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Colistin A | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Colistin B | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Doxycycline | 100 | 100 | 100 | 100 | 100 | 100 | 0 | 33.3 | 0 |
| Enrofloxacin | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Erythromycin | 100 | 100 | 100 | 100 | 33.3 | 100 | 33.3 | 0 | 33.3 |
| Florfenicol | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Gentamicin | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Kanamycin sulfate | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Levofloxacin | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Lincomycin hydrochloride | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Mequindox | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Meropenem | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Metronidazole | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 33.3 | 66.7 |
| Neomycin trisulfate salt hydrate | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Norfloxacin | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Ofloxacin | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Oxytetracycline | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Penicillin G sodium salt | 100 | 100 | 100 | 100 | 33.3 | 100 | 0 | 0 | 0 |
| Spectinomycin hydrochloride pentahydrate | 100 | 66.7 | 100 | 66.7 | 66.7 | 100 | 0 | 0 | 0 |
| Streptomycin sulfate salt | 66.7 | 100 | 100 | 33.3 | 0 | 66.7 | 0 | 0 | 0 |
| Sulfachloropyridazine | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Sulfadiazine | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Sulfadimidine | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Sulfamethoxazole | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Sulfamonomethoxine | 100 | 100 | 66.7 | 100 | 100 | 100 | 100 | 100 | 100 |
| Tetracycline | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 66.7 |
| Tiamulin | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Tilmicosin | 66.7 | 100 | 100 | 66.7 | 100 | 100 | 0 | 33.3 | 33.3 |
| Trimethoprim | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Tylosin tartrate salt | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 66.7 |
| Tylvalosin | 100 | 100 | 100 | 100 | 100 | 100 | 33.3 | 100 | 100 |
| Vancomycin | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Antibiotics Group | Sensitivity | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 10 μg Spiked-in | 5 μg Spiked-in | 1 μg Spiked-in | |||||||
| Water Sample | Fecal Sample | Meat Sample | Water Sample | Fecal Sample | Meat Sample | Water Sample | Fecal Sample | Meat Sample | |
| Aminoglycosides | 33.3 | 33.3 | 40 | 20 | 13.3 | 33.3 | 0 | 0 | 0 |
| Amphenicols | 100 | 100 | 100 | 100 | 100 | 100 | 50 | 50 | 83.3 |
| Antifolate | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Carbapenems | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Cephalosporins | 40 | 40 | 40 | 40 | 40 | 40 | 33.3 | 33.3 | 20 |
| Fluoroquinolones | 60 | 60 | 60 | 60 | 60 | 60 | 60 | 60 | 60 |
| Glycopeptides | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Lincosamides | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Macrolides | 91.7 | 100 | 100 | 91.7 | 83.3 | 100 | 41.7 | 58.3 | 58.3 |
| Nitroimidazole | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 33.3 | 66.7 |
| Penicillins | 66.7 | 66.7 | 66.7 | 66.7 | 44.4 | 66.7 | 22.2 | 0 | 11.1 |
| Pleuromutilins | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Polymyxins | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Quinoxaline 1,4-di-N-oxides (QdNOs) | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Sulfonamides | 100 | 100 | 93.3 | 100 | 100 | 100 | 100 | 100 | 100 |
| Tetracyclines | 100 | 100 | 100 | 100 | 100 | 100 | 75 | 83.3 | 58.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, C.-L.; Wai, H.K.-F.; Wu, P.; Lai, S.-W.; Chan, O.S.-K.; Tun, H.M. A Universal LC-MS/MS Method for Simultaneous Detection of Antibiotic Residues in Animal and Environmental Samples. Antibiotics 2022, 11, 845. https://doi.org/10.3390/antibiotics11070845
Chan C-L, Wai HK-F, Wu P, Lai S-W, Chan OS-K, Tun HM. A Universal LC-MS/MS Method for Simultaneous Detection of Antibiotic Residues in Animal and Environmental Samples. Antibiotics. 2022; 11(7):845. https://doi.org/10.3390/antibiotics11070845
Chicago/Turabian StyleChan, Chak-Lun, Hogan Kok-Fung Wai, Peng Wu, Siu-Wai Lai, Olivia Sinn-Kay Chan, and Hein M. Tun. 2022. "A Universal LC-MS/MS Method for Simultaneous Detection of Antibiotic Residues in Animal and Environmental Samples" Antibiotics 11, no. 7: 845. https://doi.org/10.3390/antibiotics11070845
APA StyleChan, C.-L., Wai, H. K.-F., Wu, P., Lai, S.-W., Chan, O. S.-K., & Tun, H. M. (2022). A Universal LC-MS/MS Method for Simultaneous Detection of Antibiotic Residues in Animal and Environmental Samples. Antibiotics, 11(7), 845. https://doi.org/10.3390/antibiotics11070845