
Development of New Antimycobacterial Sulfonyl Hydrazones and 4-Methyl-1,2,3-thiadiazole-Based Hydrazone Derivatives

 ,
,  , ,
, , 
Abstract
:1. Introduction
2. Results
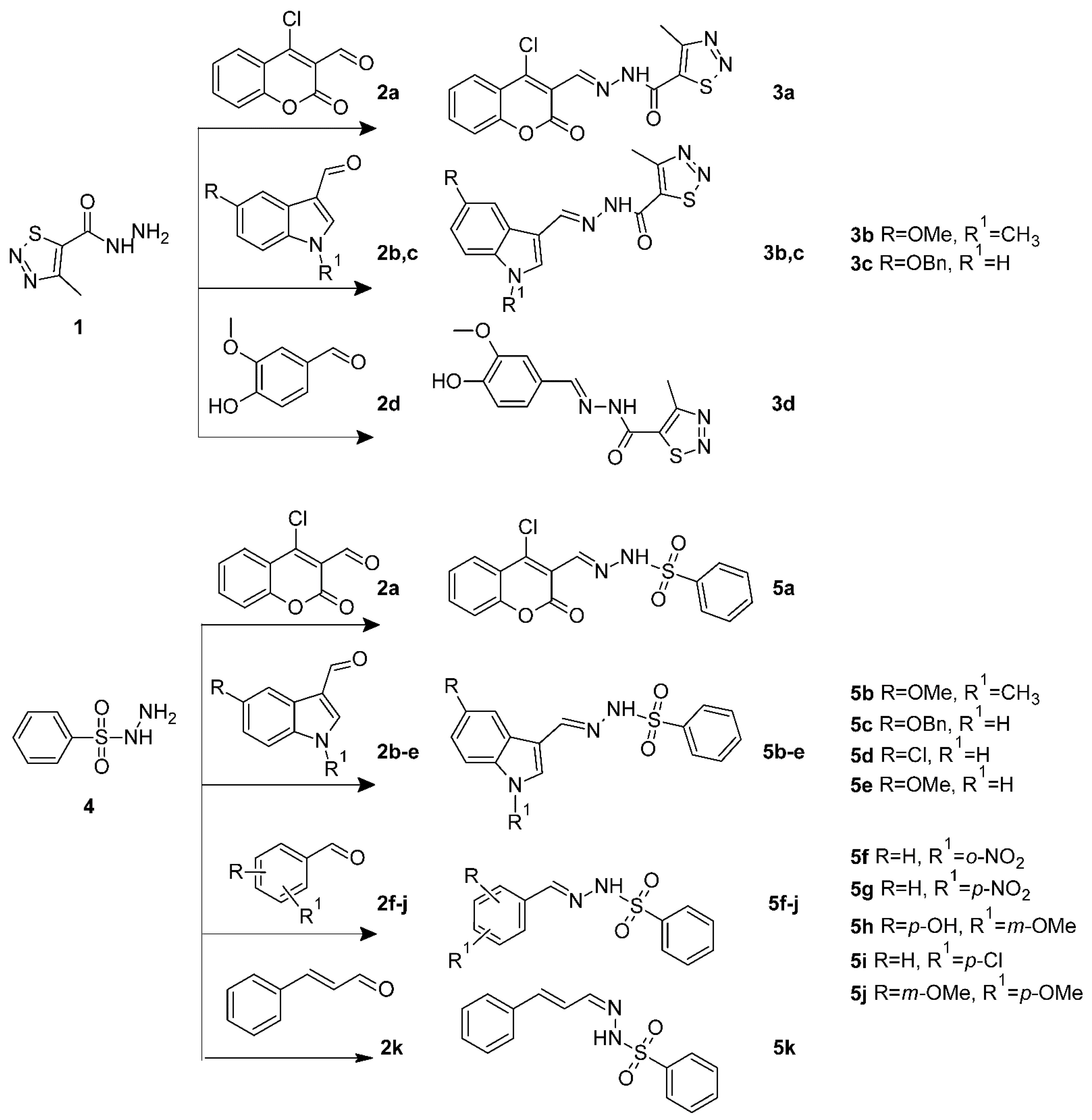
2.1. Chemistry
2.2. M. Tuberculosis Growth Inhibition and Cytotoxic Activity of Novel Compounds against Normal Cell Lines
2.3. ADME/Tox Screening Results
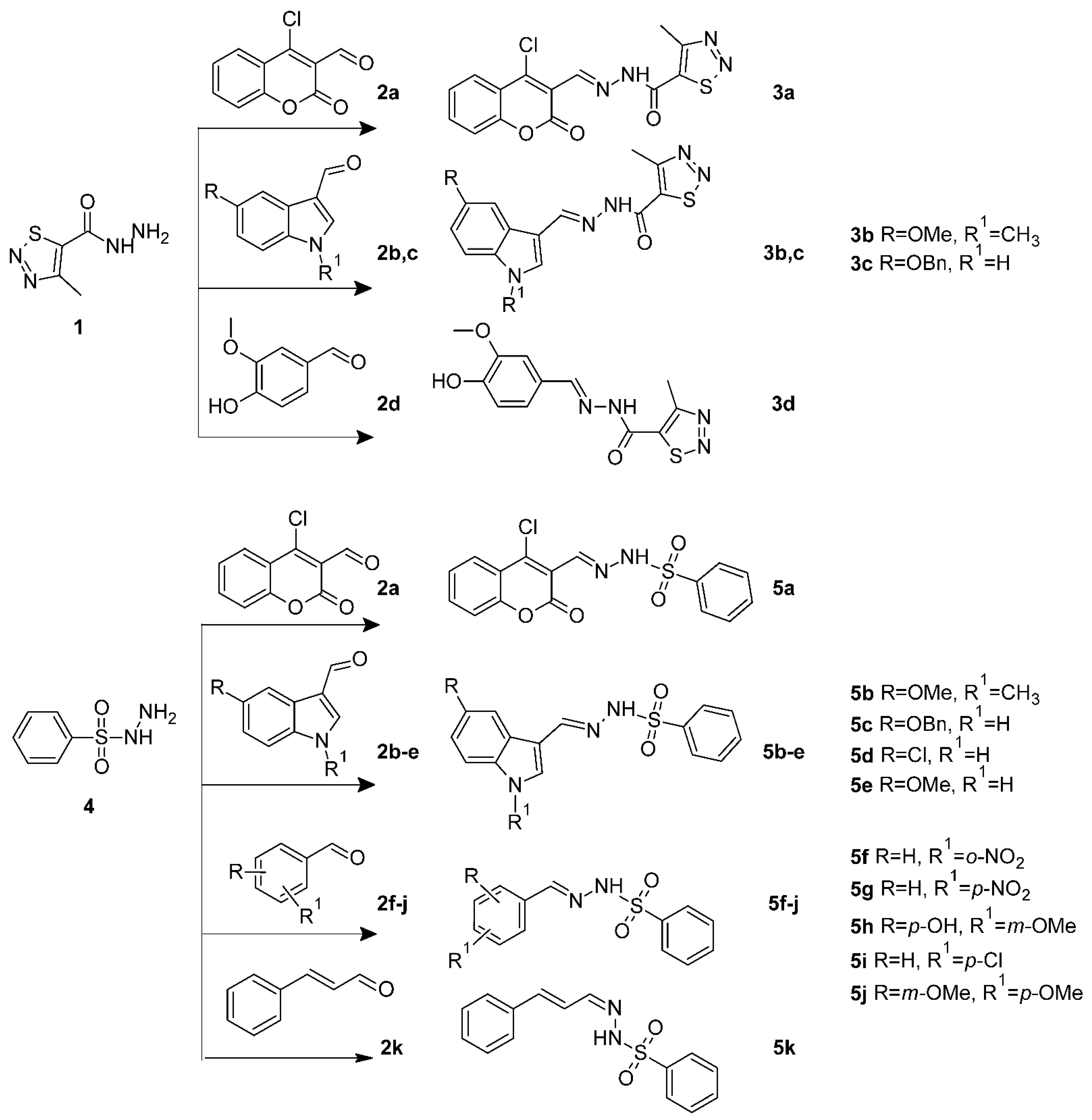
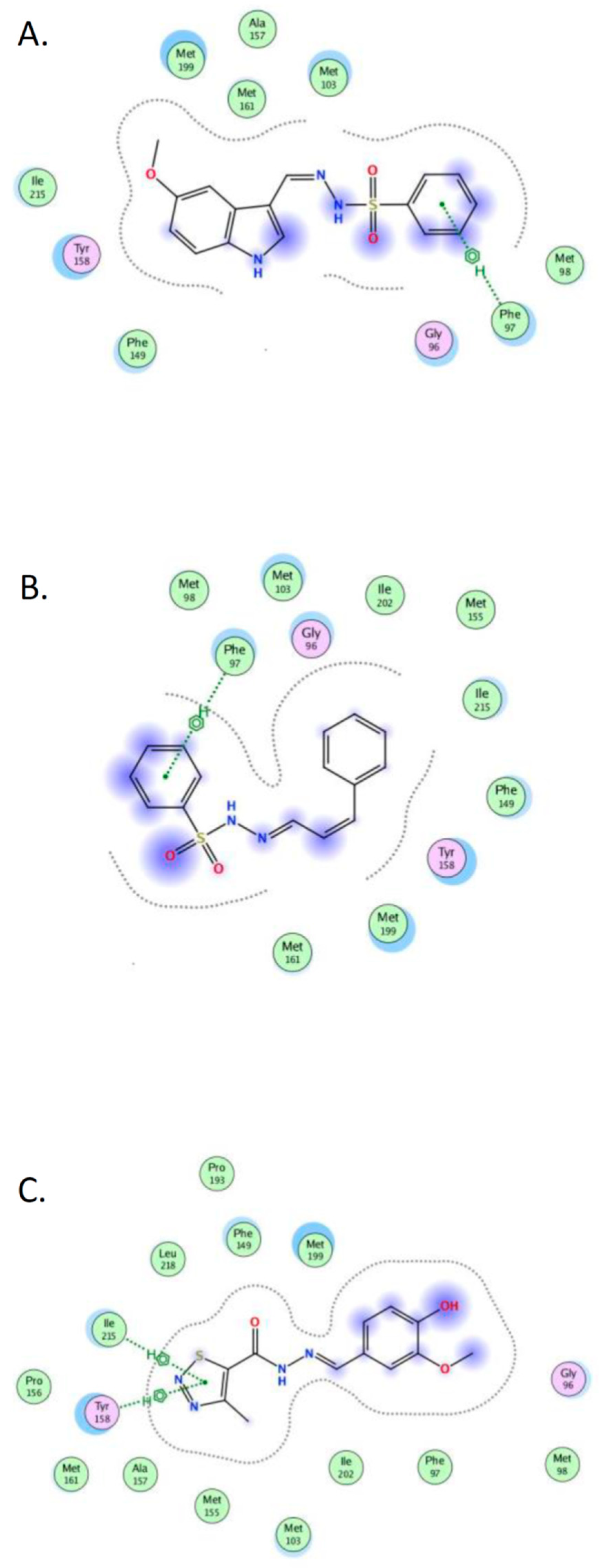
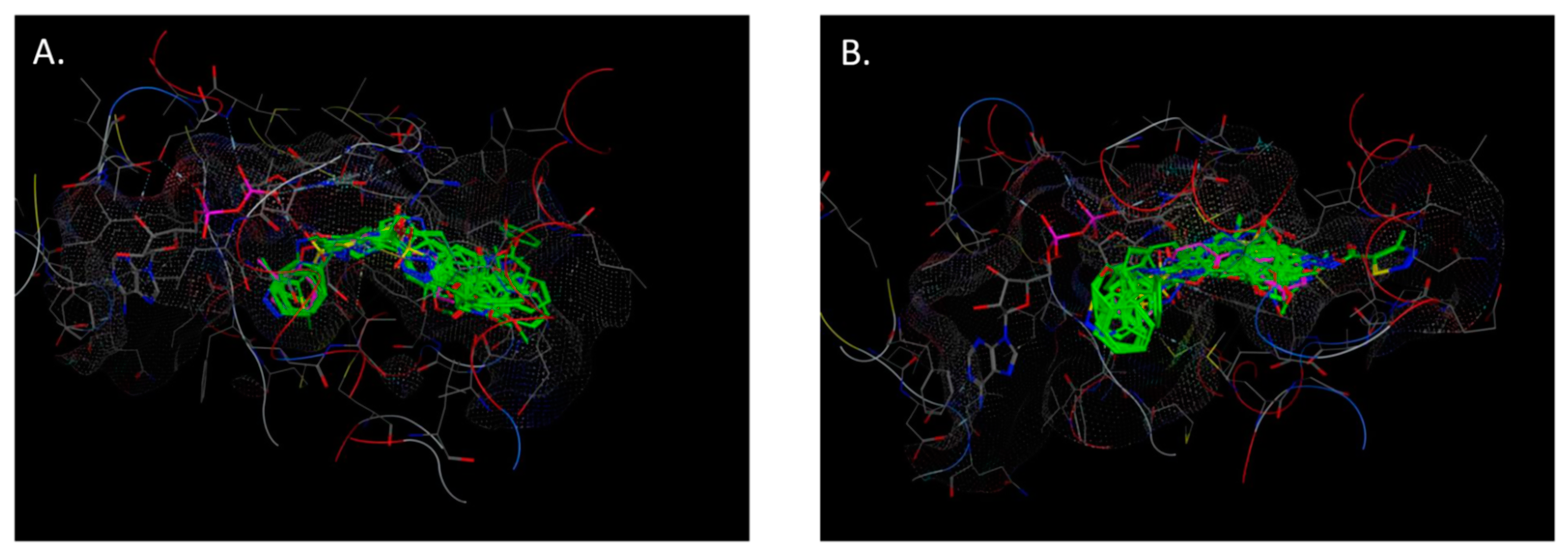
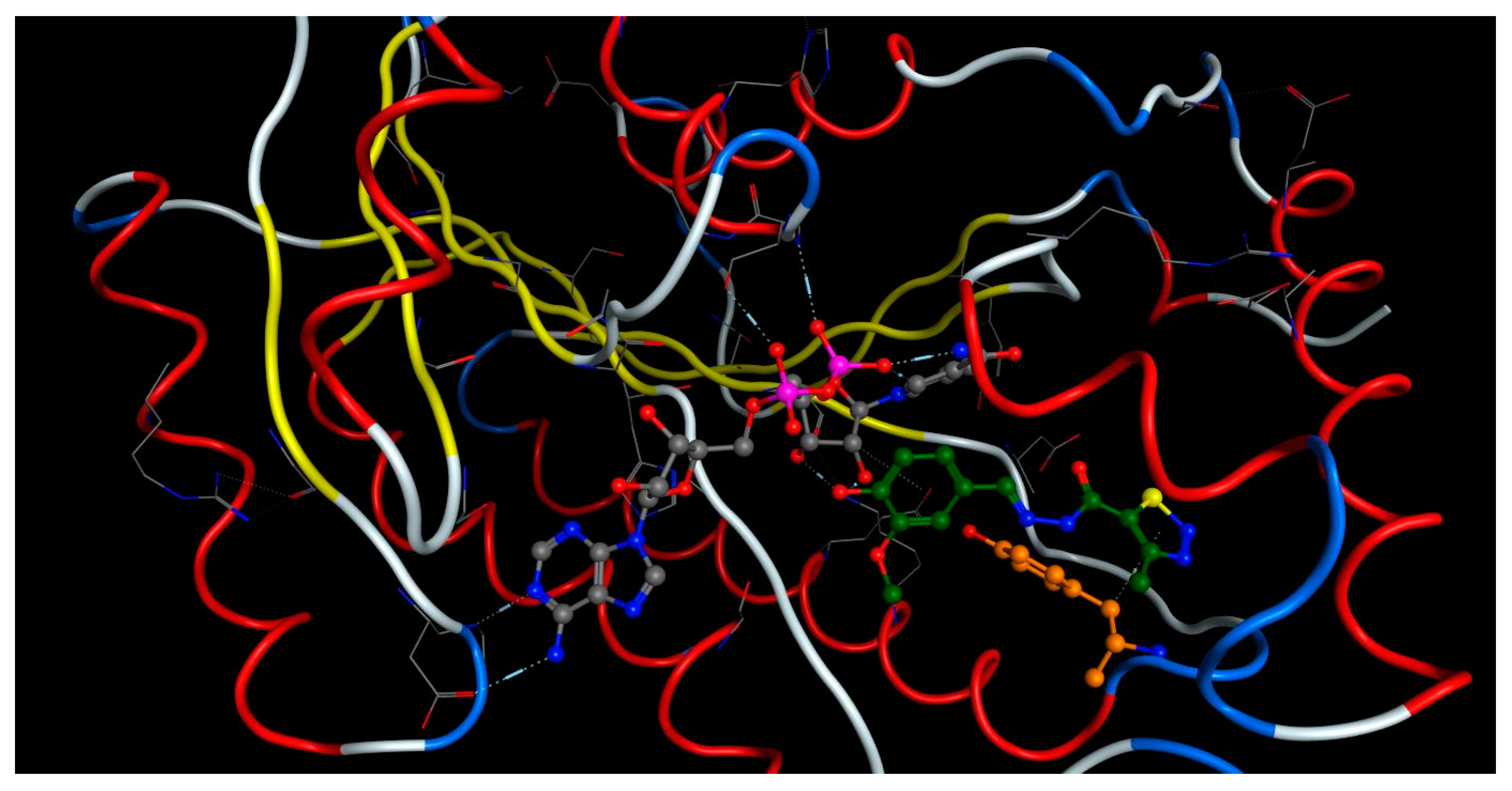
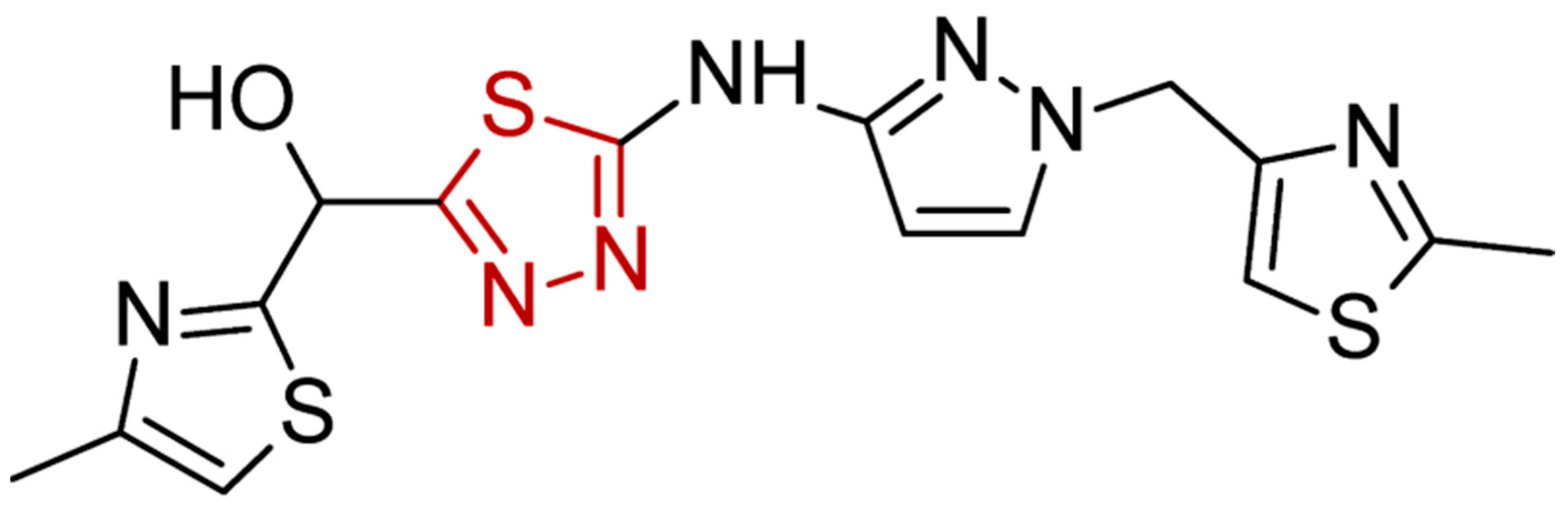
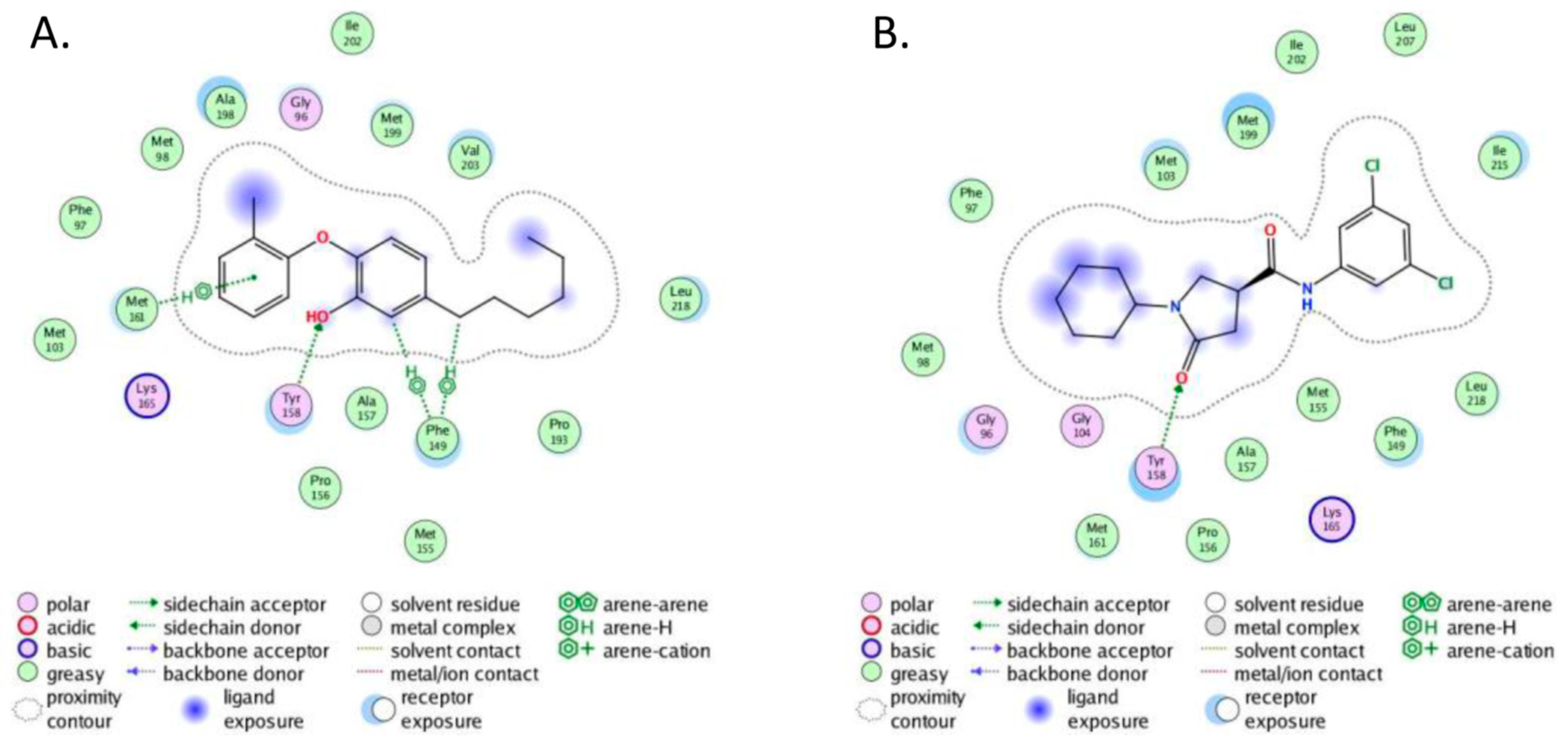
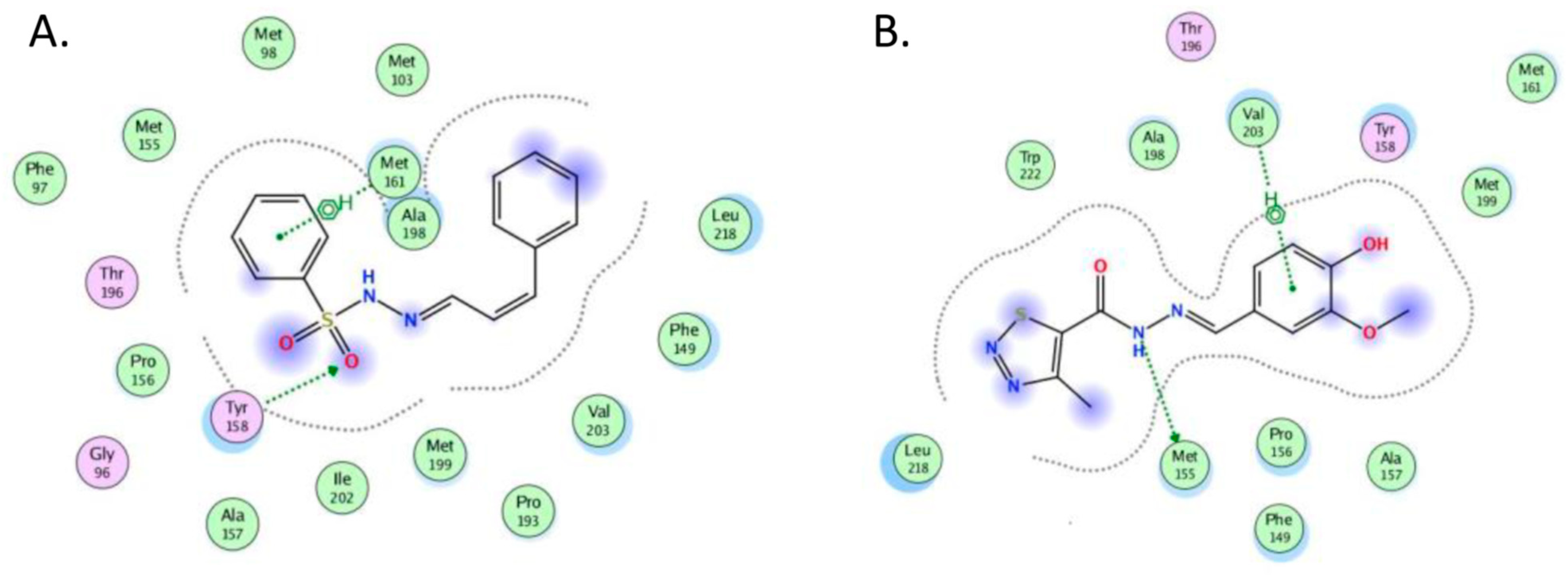
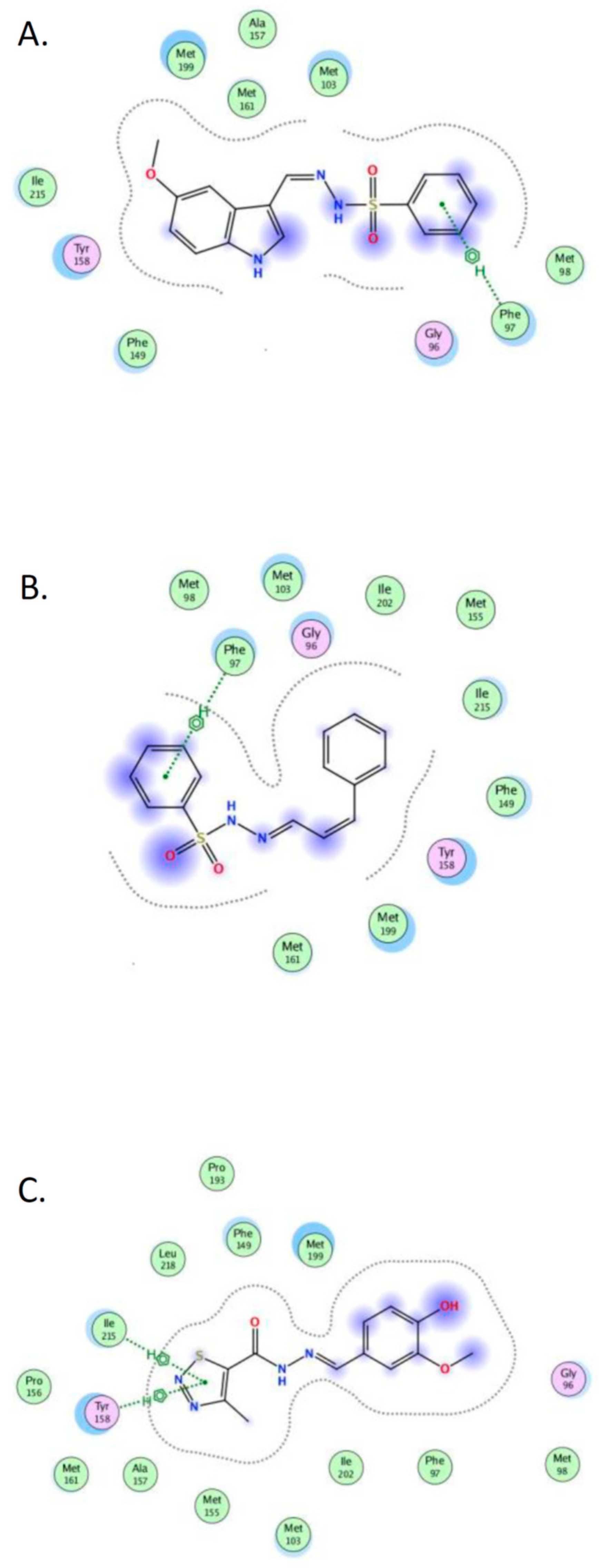
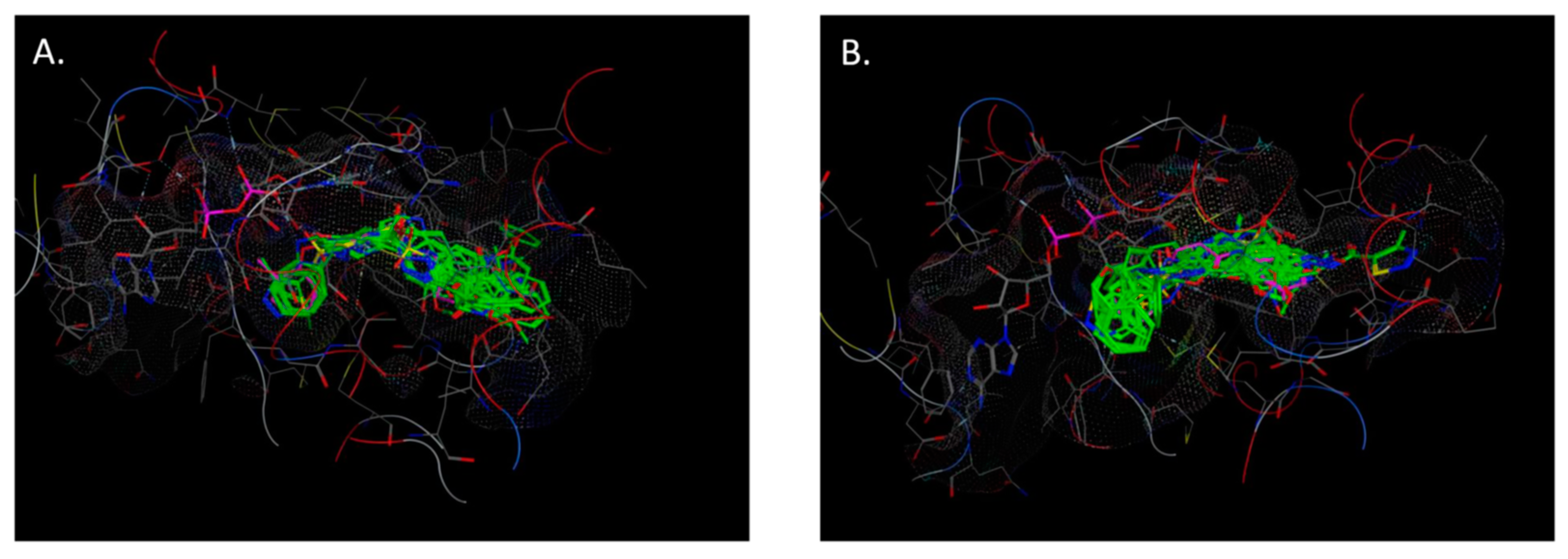
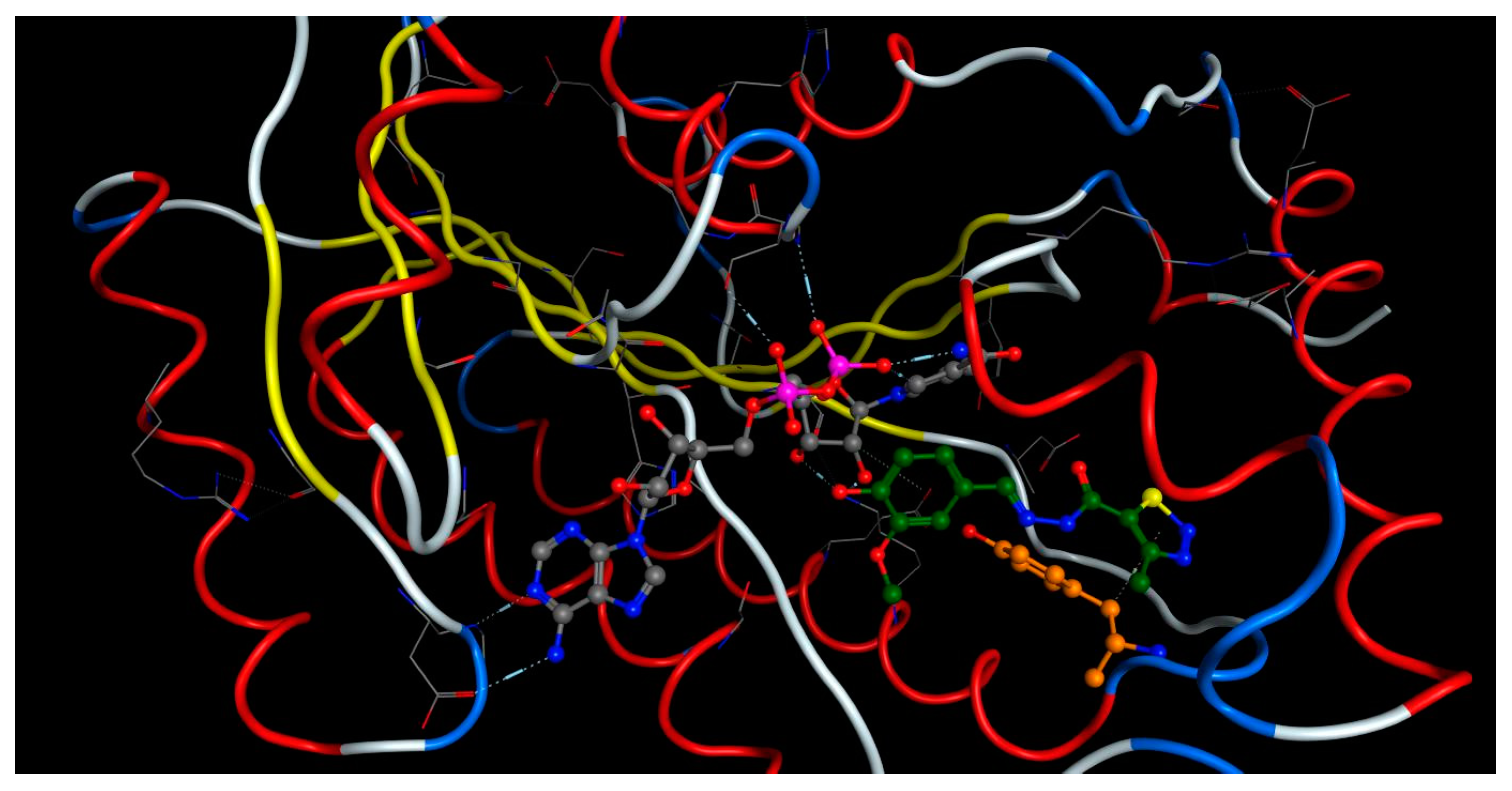
2.4. Molecular Docking
- (1)
- The crystal structure of M. tuberculosis InhA complexed with 5-hexyl-2-(2-methylphenoxy)phenol (TCU) with the co-factor nicotinamide adenine dinucleotide (NAD+) was extracted from the Protein Data Bank (http://www.rcsb.org/ (accessed on 20 April 2022), PDB ID 2X22);
- (2)
- The crystal structure of M. tuberculosis InhA complexed with (3S)-1-cyclohexyl-N-(3,5-dichlorophenyl)-5-oxopyrrolidine-3-carboxamide (ligand ID 641, further denoted as 641), also with a co-factor NAD+, extracted from PDB (PDB ID 4TZK).
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedure for the Synthesis of 3a–d
- N’-[(E)-(4-chloro-2-oxo-2H-1-benzopyran-3-yl)methylidene]-4-methyl-1,2,3-thiadiazole-5-carbohydrazide, 3a [19] Yield: 77%; m.p. 252–253 °C. HRMS (ESI) m/z: calcd: [M+H]+ 337.165902. Found: [M+H]+ 337.16517.
- N’-[(E)-(5-methoxy-1-methyl-1H-indol-3-yl)methylidene]-4-methyl-1,2,3-thiadiazole-5-carbohydrazide, 3b Yellow solid. Yield: 77%; m.p. 191–192 °C. 1H NMR (600 MHz, DMSO-d6) δ 11.98 (s, 1H, NH), 8.35 (s, 1H, CH=N), 7.90 (s, 1H, H-2), 7.67 (d, J = 2.4 Hz, 1H, H-4), 7.45 (d, J = 8.9 Hz, 1H, H-7), 6.93 (dd, J = 2.4, 8.9 Hz, 1H, H-6), 3.83 (s, 3H, OCH3), 3.82 (s, 3H, NCH3), 2.96 (s, 3H, CH3). 13C NMR (151 MHz, DMSO-d6) δ 162.63 (C=O), 158.95 (C-4′), 155.03 (C-5), 143.04 (CH=N), 136.41 (C-5′), 135.94 (C-2), 132.67 (C-7a), 124.53 (C-3a), 112.97 (C-6), 111.64 (C-7), 109.22 (C-3), 102.31 (C-4), 55.45 (OCH3), 33.14 (NCH3), 14.83 (CH3). HRMS (ESI) m/z: calcd: [M+H]+ 330.10192. Found: [M+H]+ 330.1009.
- N’-((E)-[5-(benzyloxy)-1H-indol-3-yl]methylidene)-4-methyl-1,2,3-thiadiazole-5-carbohydrazide, 3c Yellow solid. Yield: 76%; m.p. 229–230 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.02 (s, 1H, NH-indol), 11.73 (s, 1H, NH), 8.38 (s, 1H, CH=N), 7.92 (d, J = 2.3 Hz, 1H, H-2), 7.80 (d, J = 2.4 Hz, 1H, H-4), 7.49–7.47 (m, 2H, o-Ar), 7.42–7.39 (m, 3H, m-Ar and H-7), 7.33 (tt, J = 1.5, 7.4 Hz, 1H, o-Ar), 6.97 (dd, J = 2.4, 8.8 Hz, 1H, H-6), 5.15 (s, 2H, CH2), 2.97 (s, 3H, CH3). 13C NMR (151 MHz, DMSO-d6) δ 162.57 (C=O), 159.01 (C-5‘), 153.80 (C-5), 143.59 (CH=N), 137.27 (i-Ar), 136.61 (C-4′), 132.86 (C-2), 132.16 (C-7a), 128.40 (m-Ar), 128.10 (o-Ar), 127.75 (p-Ar), 124.00 (C-3), 113.64 (C-6), 113.12 (C-7), 110.50 (C3a), 103.69 (C-4), 69.84 (CH2), 14.79 (CH3). HRMS (ESI) m/z: calcd: [M+H]+ 392.117571. Found: [M+H]+ 392.1166
- N’-[(E)-(4-hydroxy-3-methoxyphenyl)methylidene]-4-methyl-1,2,3-thiadiazole-5-carbohydrazide, 3d Yellow solid. Yield: 80%; m.p. 229–230 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.25 (bs, NH), 9.75 (bs, 1H, OH), 8.08 (s, 1H, CH=N), 7.38 (d, J = 1.9 Hz, 1H, H-2), 7.22 (dd, J = 1.9, 8.2 Hz, 1H, H-6), 6.90 (d, J = 8.2 Hz, 1H, H-5), 3.89 (s, 3H, OMe), 2.97 (s, 3H, CH3). 13C NMR (151 MHz, DMSO-d6) δ 163.19 (C=O), 159.64 (C-5), 149.44 (C-3-Ar), 148.08 (C-4-Ar), 146.16 (CH=N), 135.40 (C-4), 124.69 (C-1-Ar), 122.32 (C-6-Ar), 115.80 (C-5-Ar), 110.15 (C-2-Ar), 55.51 (OCH3), 15.08 (CH3). HRMS (ESI) m/z: calcd: [M+H]+ 393.070287. Found: [M+H]+ 393.0694.
3.1.2. General Procedure for the Synthesis of 5a–k
- N’-[(E)-(4-chloro-2-oxo-2H-1-benzopyran-3-yl)-methylidene]benzenesulfonohydrazide,5a Yellow solid. Yield: 75%; m.p. 163–165 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.04 (s, 1H, NH), 7.98 (s, 1H, CH=N), 7.95 (dd, J = 1.5, 8.3 Hz, 1H, H-5), 7.91 (dd, J = 1.3, 8.4 Hz, 2H, H-o), 7.72 (ddd, J = 1.4, 7.4, 8.3 Hz, 1H, H-7), 7.69 (tt, J = 1.6, 7.4 Hz, 1H, H-p), 7.64 (tt, J = 1.6, 7.7 Hz, 2H, H-m), 7.47 (ddd, J = 1.2, 6.6, 7.8 Hz, 1H, H-6), 7.46 (d, J = 8.3 Hz, 1H, H-8). 13C NMR (151 MHz, DMSO-d6) δ 157.81 (C=O), 151.28 (C-8a), 144.57 (C-4), 140.18 (CH=N), 138.87 (C-i), 133.72 (C-7), 133.32 (C-p), 129.30 (C-m), 127.38 (C-o), 126.14 (C-5), 125.35 (C-6), 118.70 (C-3), 118.29 (C-4a), 116.58 (C-8). HRMS (ESI) m/z: calcd: [M+H]+ 363.020081. Found: [M+H]+ 363.02012.
- N’-[(E)-(5-methoxy-1-methyl-1H-indol-3-yl)methylidene]benzenesulfonohydrazide,5b Yellow solid. Yield: 83%; m.p. 190–191 °C. 1H NMR (600 MHz, DMSO-d6) δ 10.93 (s, 1H, NH), 8.05 (s, 1H, CH=N), 7.92 (dd, J = 1.5, 7.0 Hz, 2H, H-o), 7.64 (s, 1H, H-2), 7.63 (tt, J = 1.4, 7.4 Hz, 1H, H-p), 7.59 (tt, J = 1.7, 7.3 Hz, 2H, H-m), 7.44 (d, J = 2.5 Hz, 1H, H-4), 7.35 (d, J = 8.9 Hz, 1H, H-7), 6.85 (dd, J = 2.6, 8.9 Hz, 1H, H-6), 3.75 (s, 3H, OCH3), 3.73 (s, 3H, NCH3). 13C NMR (151 MHz, DMSO-d6) δ 154.68 (C-5), 145.35 (CH=N), 139.16 (C-i), 134.39 (C-2), 132.86 (C-p), 132.52 (C-7a), 129.09 (C-m), 127.34 (C-o), 124.87 (C-3a), 112.58 (C-6), 111.09 (C-7), 109.62 (C-3), 103.21 (C-4), 55.21 (OCH3), 32.94 (NCH3). HRMS (ESI) m/z: calcd: [M+H]+ 344.106338. Found: [M+H]+ 344.10625.
- N’-((E)-[5-(benzyloxy)-1H-indol-3-yl]methylidenebenzenesulfonohydrazide,5c Yellow solid. Yield: 87%; m.p. 208–209 °C. 1H NMR (600 MHz, DMSO-d6) δ 11.41 (s, 1H, NH-indol), 10.95 (s, 1H, NH), 8.08 (s, 1H, CH=N), 7.92 (d, J = 7.4 Hz, 2H, H-o), 7.68 (s, 1H, H-2), 7.61 (t, J = 6.8 Hz, 1H, H-p), 7.55–7.56 (m, 3H, H-m and H-4), 7.52 (d, J = 7.3 Hz, 2H, H-o), 7.42 (t, J = 6.9 Hz, 2H, H-m), 7.35 (t, J = 6.9 Hz, 1H, H-p), 7.29 (d, J = 8.5 Hz, 1H, H-7), 6.87 (d, J = 8.2 Hz, 1H, H-6), 5.03 (s, 2H, CH2). 13C NMR (151 MHz, DMSO-d6) δ 153.40 (C-5), 145.82 (CH=N), 139.22 (C-i), 137.38 (C-i), 132.83 (C-p), 132.01 (C-7a), 131.10 (C-2), 129.04 (C-m), 128.51 (C-m), 127.90 (C-o), 127.85 (C-p), 127.33 (C-o), 124.48 (C-3a), 113.07 (C-6), 112.56 (C-7), 110.77 (C-3), 104.81 (C-4), 69.66 (CH2). HRMS (ESI) m/z: calcd: [M+H]+ 406.121988. Found: [M+H]+ 406.12167.
- N’-[(E)-(5-chloro-1H-indol-3-yl)methylidene]benzenesulfonohydrazide,5d Yellow solid. Yield: 81%; m.p. 183–184 °C. 1H NMR (600 MHz, DMSO-d6) δ 11.70 (bs, 1H, NH-indol), 11.05 (s, 1H, NH), 8.08 (s, 1H, CH=N), 7.92 (d, J = 7.0 Hz, 2H, H-o), 7.89 (d, J = 2.0 Hz, 1H, H-4), 7.80 (d, J = 2.7 Hz, 1H, H-2), 7.66 (t, J = 7.2 Hz, 1H, H-p), 7.62 (t, J = 7.3 Hz, 2H, H-m), 7.41 (d, J = 8.6 Hz, 1H, H-7), 7.17 (dd, J = 2.1, 8.6 Hz, 1H, H-6). 13C NMR (151 MHz, DMSO-d6) δ 144.88 (CH=N), 138.98 (C-i), 135.36 (C-7a), 133.02 (C-p), 131.94 (C-2), 129.14 (C-m), 127.37 (C-o), 125.09 (C-3a), 125.02 (C-5), 122.56 (C-6), 120.73 (C-4), 113.47 (C-7), 110.68 (C-3). HRMS (ESI) m/z: calcd: [M+H]+ 334.041151. Found: [M+H]+ 334.04123.
- N’-[(E)-(5-methoxy-1H-indol-3-yl)methylidene]benzenesulfonohydrazide,5e Yellow solid. Yield: 90%; m.p. 174–175 °C. 1H NMR (600 MHz, DMSO-d6) δ 11.40 (d, J = 2.0 Hz, 1H, NH-indol), 10.94 (s, 1H, NH), 8.08 (s, 1H, CH=N), 7.93 (td, J = 1.6, 6.5 Hz, 2H, H-o), 7.67 (d, J = 2.8 Hz, 1H, H-2), 7.64 (tt, J = 1.8, 7.3 Hz, 1H, H-p), 7.60 (tt, J = 1.7, 7.1 Hz, 2H, H-m), 7.43 (d, J = 2.5 Hz, 1H, H-4), 7.28 (d, J = 8.8 Hz, 1H, H-7), 6.79 (dd, J = 2.6, 8.8 Hz, 1H, H-6), 3.74 (s, 3H, CH3). 13C NMR (151 MHz, DMSO-d6) δ 154.38 (C-5), 145.82 (CH), 139.17 (C-i), 132.85 (C-p), 131.79 (C-7a), 130.95 (C-2), 129.08 (C-m), 127.35 (C-o), 124.46 (C-3a), 112.65 (C-6), 112.55 (C-7), 110.77 (C-3), 103.03 (C-4), 55.15 (CH3). HRMS (ESI) m/z: calcd: [M+H]+ 330.090688. Found: [M+H]+ 330.09057.
- N’-[(E)-(2-nitrophenyl)methylidene]benzenesulfonohydrazide,5f Yellow solid. Yield: 79%; m.p. 148–149 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.00 (s, 1H, NH), 8.29 (s, 1H, CH=N), 8.02 (dd, J = 1.2, 8.2 Hz, 1H, H-3), 7.89 (td, J = 1.5, 6.6 Hz, 2H, H-o), 7.81 (dd, J = 1.5, 7.9 Hz, 1H, H-6), 7.75 (dt, J = 1.0, 7.6 Hz, 1H, H-5), 7.69 (tt, J = 1.6, 7.3 Hz, 1H, H-p), 7.62–7.56 (m, 3H, H-m and H-4). 13C NMR (151 MHz, DMSO-d6) δ 147.88 (CH=N), 142.71 (C-2), 138.92 (C-i), 133.82 (C-5), 133.30 (C-p), 130.79 (C-4), 129.43 (C-m), 128.00 (C-1), 127.90 (C-6), 127.14 (C-o), 124.71 (C-3). HRMS (ESI) m/z: calcd: [M+H]+ 306.054302. Found: [M+H]+ 306.0535.
- N’-[(E)-(4-nitrophenyl)methylidene]benzenesulfonohydrazide,5g Yellow solid. Yield: 92%; m.p. 166–167 °C. 1H NMR (600 MHz, DMSO-d6) δ 12.01 (s, 1H, NH), 8.23 (td, J = 2.1, 9.3 Hz, 2H, H-3 and H-5), 8.03 (s, 1H, CH=N), 7.90 (td, J = 1.5, 6.7 Hz, 2H, H-o), 7.83 (td, J = 1.4, 8.9 Hz, 2H, H-2 and H-6), 7.68 (tt, J = 1.6, 7.4 Hz, 1H, H-p), 7.62 (tt, J = 1.5, 7.5 Hz, 2H, H-m). 13C NMR (151 MHz, DMSO-d6) δ 147.90 (C-p), 144.62 (CH=N), 139.78 (C-i), 138.85 (C-i), 133.32 (C-p), 129.42 (C-m), 127.76 (C-2 and C-6), 127.15 (C-o), 124.09 (C-3 and C-05). HRMS (ESI) m/z: calcd: [M+H]+ 306.054302. Found: [M+H]+ 306.0535.
- N’-[(E)-(3-hydroxy-4-methoxyphenyl)methylidene]benzenesulfonohydrazide,5h Yellow solid. Yield: 75%; m.p. 129–13 °C. 1H NMR (600 MHz, DMSO-d6) δ 11.29 (s, 1H, NH), 9.25 (bs, 1H, OH), 7.86 (td, J = 1.6, 6.6 Hz, 2H, H-o), 7.77 (s, 1H, CH=N), 7.66 (tt, J = 1.7, 7.4 Hz, 1H, H-p), 7.60 (tt, J = 1.6, 11.6 Hz, 2H, H-m), 7.05 (d, J = 1.3 Hz, 1H, H-2), 6.89–6.92 (m, 2H, H-5 and H-6), 3.76 (s, 3H, OCH3). 13C NMR (151 MHz, DMSO-d6) δ 149.80 (C-4), 147.58 (CH=N), 146.76 (C-34), 139.13 (C-i), 133.01 (C-p), 129.24 (C-m), 127.16 (C-o), 126.46 (C-1), 120.14 (C-6), 111.87 (C-2), 111.75 (C-5), 55.56 (OCH3). HRMS (ESI) m/z: calcd: [M+H]+ 307.074703. Found: [M+H]+ 307.0738.
- N’-[(E)-(4-chlorophenyl)methylidene]benzenesulfonohydrazide,5i White solid. Yield: 80%; m.p. 161–163 °C. 1H NMR (600 MHz, DMSO-d6) δ 11.64 (s, 1H, NH), 7.91 (s, 1H, CH=N), 7.88 (td, J = 1.5, 6.6 Hz, 2H, H-o), 7.67 (tt, J = 1.6, 7.1 Hz, 1H, H-p), 7.61 (tt, J = 1.5, 7.2 Hz, 2H, H-m), 7.58 (td, J = 2.2, 9.1 Hz, 2H, H-2 and H-6), 7.45 (td, J = 2.2, 9.1 Hz, 2H, H-3 and H-5). 13C NMR (151 MHz, DMSO-d6) δ 145.90 (CH=N), 138.95 (C-i), 134.60 (C-4), 133.16 (C-p), 132.56 (C-1), 129.32 (C-m), 128.93 (C-3 and C-5), 128.44 (C-2 and C-6), 127.18 (C-o). HRMS (ESI) m/z: calcd: [M+H]+ 295.030252. Found: [M+H]+ 295.03044.
- N’-[(E)-(3,4-dimethoxyphenyl)methylidene]benzenesulfonohydrazide,5j White solid. Yield: 87%; m.p. 150–152 °C. 1H NMR (600 MHz, DMSO-d6) δ 11.33 (s, 1H, NH), 7.88 (td, J = 2.1, 7.7 Hz, 2H, H-o), 7.83 (s, 1H, CH=N), 7.66 (tt, J = 1.7, 11.1 Hz, 1H, H-p), 7.61 (tt, J = 1.6, 7.5 Hz, 2H, H-m), 7.12 (d, J = 1.9 Hz, 1H, H-2), 7.08 (dd, J = 1.9, 8.3 Hz, 1H, H-6), 6.95 (d, J = 8.4 Hz, 1H, H-5), 3.76 (s, 3H, OCH3), 3.76 (s, 3H, OCH3). 13C NMR (151 MHz, DMSO-d6) δ 150.66 (C-4), 148.90 (C-3), 147.45 (CH=N), 139.01 (C-i), 133.05 (C-p), 129.21 (C-m), 127.26 (C-o), 126.36 (s, 1C), 121.00 (C-6), 111.49 (C-5), 108.58 (C-2), 55.56 (OCH3), 55.42 (OCH3). HRMS (ESI) m/z: calcd: [M+H]+ 321.090353. Found: [M+H]+ 321.0895.
- N’-[(1E,2E)-3-phenylprop-2-en-1-ylidene]benzenesulfonohydrazid,5k White solid. Yield: 85%; m.p. 168–170 °C. 1H NMR (600 MHz, DMSO-d6) δ 11.50 (s, 1H, NH), 7.83–7.85 (m, 2H, H-o), 7.73 (d, J = 9.2 Hz, 1H, H-1), 7.66–7.68 (m, 1H, H-p), 7.60–7.63 (m, 2H, H-m), 7.54–7.56 (m, 2H, H-o), 7.33–7.36 (m, 2H, H-m), 7.28–7.31 (m, 1H, H-p), 6.95 (d, J = 16.1 Hz, 1H, H-3), 6.84 (dd, J = 9.2, 16.1 Hz, 1H, H-2). 13C NMR (151 MHz, DMSO-d6) δ 149.48 (C-1), 139.38 (C-3), 139.14 (C-i), 135.65 (C-i), 133.06 (C-p), 129.32 (C-m), 128.94 (C-p), 128.80 (C-m), 127.15 (C-o), 127.12 (C-o), 124.68 (C-2). HRMS (ESI) m/z: calcd: [M+H]+ 287.084874. Found: [M+H]+ 287.0842.
3.2. In Vitro Antimycobacterial Activity
3.3. In Vitro Cytotoxicity Screening
3.4. Statistical Methods
3.5. ADME/Tox Screening
3.6. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McQuaid, C.F.; Vassall, A.; Cohen, T.; Fiekert, K.; COVID/TB Modelling Working Group; White, R.G. The impact of COVID-19 on TB: A review of the data. Int. J. Tuberc. Lung Dis. 2021, 25, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Pavlovic, J.M.; Pesut, D.P.; Stosic, M.B. Influence of the COVID-19 pandemic on the incidence of tuberculosis and influenza. Rev. Inst. Med. Trop. São Paulo 2021, 63, e53. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.R.; Mello, F.C.D.Q.; D’Ambrosio, L.; Centis, R.; Dalcolmo, M.P.; Migliori, G.B. Tuberculosis and COVID-19, the new cursed duet: What differs between Brazil and Europe? J. Bras. Pneumol. 2021, 47, e20210044. [Google Scholar]
- Visca, D.; Ong, C.; Tiberi, S.; Centis, R.; D’Ambrosio, L.; Chen, B.; Mueller, J.; Duarte, R.; Dalcolmo, M.; Sotgiu, G.; et al. Tuberculosis and COVID-19 interaction: A review of biological, clinical and public health effects. Pulmonology 2021, 27, 151–165. [Google Scholar] [CrossRef]
- Shariq, M.; Sheikh, J.A.; Quadir, N.; Sharma, N.; Hasnain, S.E.; Ehtesham, N.Z. COVID-19 and tuberculosis: The double whammy of respiratory pathogens. Eur. Respir. Rev. 2022, 31, 210264. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, A.J.; Klinton, J.S.; Oga-Omenka, C.; Heitkamp, P.; Nyirenda, C.N.; Furin, J.; Pai, M. Tuberculosis in times of COVID-19. J. Epidemiol. Community Health 2021, 76, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Hegde, P.; Boshoff, H.I.; Rusman, Y.; Aragaw, W.W.; Salomon, C.E.; Dick, T.; Aldrich, C.C. Reinvestigation of the structure-activity relationships of isoniazid. Tuberculosis 2021, 129, 102100. [Google Scholar] [CrossRef] [PubMed]
- Shirude, P.S.; Madhavapeddi, P.; Naik, M.; Murugan, K.; Shinde, V.; Nandishaiah, R.; Bhat, J.; Kumar, A.; Hameed, S.; Holdgate, G.; et al. Methyl-thiazoles: A novel mode of inhibition with the potential to develop novel inhibitors targeting InhA in Mycobacterium tuberculosis. J. Med. Chem. 2013, 56, 8533–8542. [Google Scholar] [CrossRef]
- Hartkoorn, R.C.; Sala, C.; Neres, J.; Pojer, F.; Magnet, S.; Mukherjee, R.; Uplekar, S.; Boy-Röttger, S.; Altmann, K.H.; Cole, S.T. Towards a new tuberculosis drug: Pyridomycin–nature’s isoniazid. EMBO Mol. Med. 2012, 4, 1032–1042. [Google Scholar] [CrossRef]
- Kamsri, P.; Hanwarinroj, C.; Phusi, N.; Pornprom, T.; Chayajarus, K.; Punkvang, A.; Suttipanta, N.; Srimanote, P.; Suttisintong, K.; Songsiriritthigul, C.; et al. Discovery of new and potent inha inhibitors as antituberculosis agents: Structure-based virtual screening validated by biological assays and x-ray crystallography. J. Chem. Inf. Model. 2019, 60, 226–234. [Google Scholar] [CrossRef]
- Angula, K.; Legoabe, L.; Beteck, R. Chemical Classes Presenting Novel Antituberculosis Agents Currently in Different Phases of Drug Development: A 2010–2020 Review. Pharmaceuticals 2021, 14, 461. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Hoyos, M.; Perez-Herran, E.; Gulten, G.; Encinas, L.; Álvarez-Gómez, D.; Alvarez, E.; Ferrer-Bazaga, S.; García-Pérez, A.; Ortega, F.; Angulo-Barturen, I.; et al. Antitubercular drugs for an old target: GSK693 as a promising InhA direct inhibitor. EBioMedicine 2016, 8, 291–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, B.; Suresh, J.; Ahsan, M.J.; Mathew, G.E.; Usman, D.; Subramanyan, P.N.S.; Safna, K.F.; Maddela, S. Hydrazones as a privileged structural linker in antitubercular agents: A review. Infect. Disord. Drug Targets 2015, 15, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Angelova, V.T.; Valcheva, V.; Pencheva, T.; Voynikov, Y.; Vassilev, N.; Mihaylova, R.; Momekov, G.; Shivachev, B. Synthesis, antimycobacterial activity and docking study of 2-aroyl-[1] benzopyrano [4, 3-c] pyrazol-4 (1H)-one deriv-atives and related hydrazide-hydrazones. Bioorg. Med. Chem. Lett. 2017, 27, 2996–3002. [Google Scholar] [CrossRef] [PubMed]
- Ghiano, D.G.; Recio-Balsells, A.; Bortolotti, A.; Defelipe, L.A.; Turjanski, A.; Morbidoni, H.R.; Labadie, G.R. New one-pot synthesis of anti-tuberculosis compounds inspired on isoniazid. Eur. J. Med. Chem. 2020, 208, 112699. [Google Scholar] [CrossRef] [PubMed]
- Shtyrlin, N.V.; Khaziev, R.M.; Shtyrlin, V.G.; Gilyazetdinov, E.M.; Agafonova, M.N.; Usachev, K.S.; Islamov, D.R.; Klimovitskii, A.E.; Vinogradova, T.I.; Dogonadze, M.Z. Isonicotinoyl hydrazones of pyridoxine derivatives: Synthesis and antimycobacterial activity. Med. Chem. Res. 2021, 30, 952–963. [Google Scholar] [CrossRef]
- Lalavani, N.H.; Gandhi, H.R.; Bhensdadia, K.A.; Patel, R.K.; Baluja, S.H. Synthesis, pharmacokinetic and molecular docking studies of new benzohydrazide derivatives possessing antitubercular activity against Mycobacterium tuberculosis H37Rv. J. Mol. Struct. 2022, 1250, 131884. [Google Scholar] [CrossRef]
- Bonnett, S.A.; Ollinger, J.; Chandrasekera, S.; Florio, S.; O’Malley, T.; Files, M.; Jee, J.-A.; Ahn, J.; Casey, A.; Ovechkina, Y. A target-based whole cell screen approach to identify potential inhibitors of Mycobacterium tuberculosis signal peptidase. ACS Infect. Dis. 2016, 2, 893–902. [Google Scholar] [CrossRef]
- Angelova, V.T.; Valcheva, V.; Vassilev, N.G.; Buyukliev, R.; Momekov, G.; Dimitrov, I.; Saso, L.; Djukic, M.; Shivachev, B. Antimycobacterial activity of novel hydrazide-hydrazone derivatives with 2 H -chromene and coumarin scaffold. Bioorg. Med. Chem. Lett. 2017, 27, 223–227. [Google Scholar] [CrossRef]
- Angelova, V.T.; Pencheva, T.; Vassilev, N.; Simeonova, R.; Momekov, G.; Valcheva, V. New indole and indazole derivatives as potential antimycobacterial agents. Med. Chem. Res. 2019, 28, 485–497. [Google Scholar] [CrossRef]
- Hu, Y.-Q.; Zhang, S.; Zhao, F.; Gao, C.; Feng, L.-S.; Lv, Z.-S.; Xu, Z.; Wu, X. Isoniazid derivatives and their antitubercular activity. Eur. J. Med. Chem. 2017, 133, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Vavříková, E.; Polanc, S.; Kočevar, M.; Horváti, K.; Bősze, S.; Stolaříková, J.; Vávrová, K.; Vinšová, J. New fluorine-containing hydrazones active against MDR-tuberculosis. Eur. J. Med. Chem. 2011, 46, 4937–4945. [Google Scholar] [CrossRef] [PubMed]
- Vergara, F.M.; Lima, C.H.d.S.; Maria das Graças, M.d.O.; Candéa, A.L.; Lourenço, M.C.; Ferreira, M.d.L.; Kaiser, C.R.; de Souza, M.V. Synthesis and antimycobacterial activity of N′-[(E)-(monosubstituted-benzylidene)]-2-pyrazinecarbohydrazide derivatives. Eur. J. Med. Chem. 2009, 44, 4954–4959. [Google Scholar] [CrossRef] [PubMed]
- Šink, R.; Sosič, I.; Živec, M.; Fernandez-Menendez, R.; Turk, S.; Pajk, S.; Alvarez-Gomez, D.; Lopez-Roman, E.M.; Gonzales-Cortez, C.; Rullas-Triconado, J.; et al. Design, synthesis, and evaluation of new thiadiazole-based direct inhibitors of enoyl acyl carrier protein reductase (InhA) for the treatment of tuberculosis. J. Med. Chem. 2015, 58, 613–624. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, K.N.; Chiaradia, L.D.; Martins, P.G.A.; Mascarello, A.; Cordeiro, M.N.S.; Guido, R.V.C.; Andricopulo, A.D.; Yunes, R.A.; Nunes, R.J.; Vernal, J.; et al. Sulfonyl-hydrazones of cyclic imides derivatives as potent inhibitors of the Mycobacterium tuberculosis protein tyrosine phosphatase B (PtpB). MedChemComm 2011, 2, 500–504. [Google Scholar] [CrossRef]
- Mascarello, A.; Mori, M.; Chiaradia-Delatorre, L.D.; Menegatti, A.C.O.; Monache, F.D.; Ferrari, F.; Yunes, R.A.; Nunes, R.J.; Terenzi, H.; Botta, B.; et al. Discovery of Mycobacterium tuberculosis protein tyrosine phosphatase B (PtpB) inhibitors from natural products. PLoS ONE 2013, 8, e77081. [Google Scholar] [CrossRef]
- Ghiya, S.; Joshi, Y.C. Synthesis and antimicrobial evaluation of hydrazones derived from 4-methylbenzenesulfonohydrazide in aqueous medium. Med. Chem. Res. 2016, 25, 970–976. [Google Scholar] [CrossRef]
- Siemann, S.; Evanoff, D.P.; Marrone, L.; Clarke, A.J.; Viswanatha, T.; Dmitrienko, G.I. N -Arylsulfonyl Hydrazones as Inhibitors of IMP-1 Metallo-β-Lactamase. Antimicrob. Agents Chemother. 2002, 46, 2450–2457. [Google Scholar] [CrossRef] [Green Version]
- Popiołek, Ł. The bioactivity of benzenesulfonyl hydrazones: A short review. Biomed. Pharmacother. 2021, 141, 111851. [Google Scholar] [CrossRef]
- Özdemir, Ü.Ö.; Arslan, F.; Hamurcu, F. Synthesis, characterization, antibacterial activities and carbonic anhydrase enzyme inhibitor effects of new arylsulfonylhydrazone and their Ni(II), Co(II) complexes. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2010, 75, 121–126. [Google Scholar] [CrossRef]
- Aslan, H.G.; Özcan, S.; Karacan, N. The antibacterial activity of some sulfonamides and sulfonyl hydrazones, and 2D-QSAR study of a series of sulfonyl hydrazones. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2012, 98, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Alsaeedi, H.S.; Aljaber, N.A.; Ara, I. Synthesis and investigation of antimicrobial activity of some nifuroxazide analogues. Asian J. Chem. 2015, 27, 3639. [Google Scholar] [CrossRef]
- Segretti, N.D.; Serafim, R.A.; Segretti, M.C.; Miyata, M.; Coelho, F.R.; Augusto, O.; Ferreira, E.I. New anti-bacterial agents: Hybrid bioisoster derivatives as potential E. coli FabH inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 3988–3993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, M.; Poojary, B.; Kumar, S.M.; Hussain, M.M.; Pai, N.; Revanasiddappa, B.; Kullaiah, B. Structural, crystallographic, Hirshfeld surface, thermal and antimicrobial evaluation of new sulfonyl hydrazones. J. Mol. Struct. 2018, 1159, 55–66. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, X.; Zhou, T. Synthesis and antibacterial activity of benzenesulfonylhydrazone derivatives of methyl dehydroabietate. Russ. J. Gen. Chem. 2019, 89, 819–823. [Google Scholar] [CrossRef]
- Shaaban, M.M.; Ragab, H.M.; Akaji, K.; McGeary, R.P.; Bekhit, A.-E.A.; Hussein, W.M.; Kurz, J.L.; Elwakil, B.H.; Bekhit, S.A.; Ibrahim, T.M.; et al. Design, synthesis, biological evaluation and in silico studies of certain aryl sulfonyl hydrazones conjugated with 1,3-diaryl pyrazoles as potent metallo-β-lactamase inhibitors. Bioorg. Chem. 2020, 105, 104386. [Google Scholar] [CrossRef]
- Zhang, D.; Ma, Y.; Liu, Y.; Liu, Z.P. Synthesis of Sulfonylhydrazone-and Acylhydrazone-Substituted 8-Ethoxy-3-nitro-2H-chromenes as Potent Antiproliferative and Apoptosis Inducing Agents. Arch. Pharm. 2014, 347, 576–588. [Google Scholar] [CrossRef]
- Alaoui, S.; Dufies, M.; Driowya, M.; Demange, L.; Bougrin, K.; Robert, G.; Auberger, P.; Pagès, G.; Benhida, R. Synthesis and anti-cancer activities of new sulfonamides 4-substituted-triazolyl nucleosides. Bioorg. Med. Chem. Lett. 2017, 27, 1989–1992. [Google Scholar] [CrossRef]
- Rajput, J.D.; Bagul, S.D.; Bendre, R.S. Synthesis, biological activities and molecular docking simulation of hydrazone scaffolds of carvacrol, thymol and eugenol. Res. Chem. Intermed. 2017, 43, 6601–6616. [Google Scholar] [CrossRef]
- Korcz, M.; Sączewski, F.; Bednarski, P.J.; Kornicka, A. Synthesis, Structure, Chemical Stability, and In Vitro Cytotoxic Properties of Novel Quinoline-3-Carbaldehyde Hydrazones Bearing a 1,2,4-Triazole or Benzotriazole Moiety. Molecules 2018, 23, 1497. [Google Scholar] [CrossRef] [Green Version]
- Wei, D.; Pan, Y.; Wang, H.; Xu, W.; Chen, C.; Zheng, J.; Cai, D. Synthesis of substituted aromatic heterocyclic sulfonyl hydrazone compounds and in vitro anti-hepatoma activity: Preliminary results. Eur. Rev. Med. Pharm. Sci. 2018, 22, 4720–4729. [Google Scholar]
- Xie, Z.; Song, Y.; Xu, L.; Guo, Y.; Zhang, M.; Li, L.; Chen, K.; Liu, X. Rapid Synthesis of N-Tosylhydrazones under Solvent-Free Conditions and Their Potential Application Against Human Triple-Negative Breast Cancer. ChemistryOpen 2018, 7, 977–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Govindaiah, P.; Dumala, N.; Mattan, I.; Grover, P.; Prakash, M.J. Design, synthesis, biological and in silico evaluation of coumarin-hydrazone derivatives as tubulin targeted antiproliferative agents. Bioorg. Chem. 2019, 91, 103143. [Google Scholar] [CrossRef] [PubMed]
- Popiołek, Ł.; Gawrońska Grzywacz, M.; Berecka Rycerz, A.; Paruch, K.; Piątkowska Chmiel, I.; Natorska Chomicka, D.; Herbet, M.; Gumieniczek, A.; Dudka, J.; Wujec, M. New benzenesulphonohydrazide derivatives as potential an-titumour agents. Oncol. Lett. 2020, 20, 136. [Google Scholar] [CrossRef]
- Celebioglu, H.U.; Erden, Y.; Hamurcu, F.; Taslimi, P.; Şentürk, O.S.; Özmen, Ü.Ö.; Tuzun, B.; Gulçin, I. Cytotoxic effects, carbonic anhydrase isoenzymes, α-glycosidase and acetylcholinesterase inhibitory properties, and molecular docking studies of heteroatom-containing sulfonyl hydrazone derivatives. J. Biomol. Struct. Dyn. 2021, 39, 5539–5550. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Yang, J.-Q.; Luo, S.-H.; Mei, W.-J.; Lin, J.-Y.; Zhan, J.-Q.; Wang, Z.-Y. Synthesis of N-2(5H)-furanonyl sulfonyl hydrazone derivatives and their biological evaluation in vitro and in vivo activity against MCF-7 breast cancer cells. Bioorg. Chem. 2021, 107, 104518. [Google Scholar] [CrossRef] [PubMed]
- Heimpel, H.; Raghavachar, A. Hematological side effects of co-trimoxazole. Infection 1987, 15, S248–S253. [Google Scholar] [CrossRef]
- Loncle, C.; Brunel, J.M.; Vidal, N.; Dherbomez, M.; Letourneux, Y. Synthesis and antifungal activity of cho-lesterol-hydrazone derivatives. Eur. J. Med. Chem. 2004, 39, 1067–1071. [Google Scholar] [CrossRef]
- Backes, G.L.; Neumann, D.M.; Jursic, B.S. Synthesis and antifungal activity of substituted salicyl aldehyde hydrazones, hy-drazides and sulfohydrazides. Bioorg. Med. Chem. 2014, 22, 4629–4636. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, X.; Zhi, X.; Xiao, X.; Yang, C.; Xu, H. Synthesis and insecticidal activity of novel hydrazone compounds derived from a naturally occurring lignan podophyllotoxin against Mythimna separata (Walker). Bioorg. Med. Chem. Lett. 2014, 24, 2621–2624. [Google Scholar] [CrossRef]
- Gao, Z.; Lv, M.; Li, Q.; Xu, H. Synthesis of heterocycle-attached methylidenebenzenesulfonohydrazones as antifungal agents. Bioorg. Med. Chem. Lett. 2015, 25, 5092–5096. [Google Scholar] [CrossRef]
- Özdemir, Ü.Ö.; Altuntaş, A.; Gündüzalp, A.B.; Arslan, F.; Hamurcu, F. New aromatic/heteroaromatic pro-panesulfonylhydrazone compounds: Synthesis, physical properties and inhibition studies against carbonic anhydrase II (CAII) enzyme. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 128, 452–460. [Google Scholar] [CrossRef]
- De Oliveira, K.N.; Costa, P.; Santin, J.R.; Mazzambani, L.; Bürger, C.; Mora, C.; Nunes, R.J.; De Souza, M.M. Synthesis and antidepressant-like activity evaluation of sulphonamides and sulphonyl-hydrazones. Bioorg. Med. Chem. 2011, 19, 4295–4306. [Google Scholar] [CrossRef] [Green Version]
- Abid, S.M.A.; Younus, H.A.; Al-Rashida, M.; Arshad, Z.; Maryum, T.; Gilani, M.A.; Alharthi, A.I.; Iqbal, J. Sulfonyl hydrazones derived from 3-formylchromone as non-selective inhibitors of MAO-A and MAO-B: Synthesis, molecular modelling and in-silico ADME Evaluation. Bioorg. Chem. 2017, 75, 291–302. [Google Scholar] [CrossRef]
- Wang, H.; Ren, S.-X.; He, Z.-Y.; Wang, D.-L.; Yan, X.-N.; Feng, J.-T.; Zhang, X. Synthesis, antifungal activities and qualitative structure activity relationship of carabrone hydrazone derivatives as potential antifungal agents. Int. J. Mol. Sci. 2014, 15, 4257–4272. [Google Scholar] [CrossRef] [Green Version]
- Qu, H.; Lv, M.; Yu, X.; Lian, X.; Xu, H. Discovery of some piperine-based phenylsulfonylhydrazone derivatives as potent botanically narcotic agents. Sci. Rep. 2015, 5, 13077. [Google Scholar] [CrossRef] [Green Version]
- Karaman, N.; Oruç-Emre, E.E.; Sıcak, Y.; Çatıkkaş, B.; Karaküçük-İyidoğan, A.; Öztürk, M. Microwave-assisted synthesis of new sulfonyl hydrazones, screening of biological activities and investigation of structure–activity relationship. Med. Chem. Res. 2016, 25, 1590–1607. [Google Scholar] [CrossRef]
- Murtaza, S.; Shamim, S.; Kousar, N.; Tahir, M.N.; Sirajuddin, M.; Rana, U.A. Synthesis, biological investigation, calf thymus DNA binding and docking studies of the sulfonyl hydrazides and their derivatives. J. Mol. Struct. 2016, 1107, 99–108. [Google Scholar] [CrossRef]
- Fernandes, T.B.; Cunha, M.R.; Sakata, R.P.; Candido, T.M.; Baby, A.R.; Tavares, M.T.; Barbosa, E.G.; Almeida, W.P.; Parise-Filho, R. Synthesis, Molecular Modeling, and Evaluation of Novel Sulfonylhydrazones as Acetylcholinesterase Inhibitors for Alzheimer’s Disease. Arch. Pharm. 2017, 350, 1700163. [Google Scholar] [CrossRef]
- Queen, A.; Khan, P.; Idrees, D.; Azam, A.; Hassan, I. Biological evaluation of p-toluene sulphonylhydrazone as carbonic anhydrase IX inhibitors: An approach to fight hypoxia-induced tumors. Int. J. Biol. Macromol. 2018, 106, 840–850. [Google Scholar] [CrossRef]
- da Costa Nunes, I.K.; de Souza, E.T.; Martins, I.R.R.; Barbosa, G.; Junior, M.O.D.M.; Medeiros, M.D.M.; Silva, S.W.D.; Balliano, T.L.; da Silva, B.A.; Silva, P.M.R.; et al. Discovery of sulfonyl hydrazone derivative as a new selective PDE4A and PDE4D inhibitor by lead-optimization approach on the prototype LASSBio-448: In vitro and in vivo preclinical studies. Eur. J. Med. Chem. 2020, 204, 112492. [Google Scholar] [CrossRef]
- Younus, H.A.; Hameed, A.; Mahmood, A.; Khan, M.S.; Saeed, M.; Batool, F.; Asari, A.; Mohamad, H.; Pelletier, J.; Sévigny, J. Sulfonylhydrazones: Design, synthesis and investigation of ectonucleotidase (ALP & e5′ NT) inhibition ac-tivities. Bioorg. Chem. 2020, 100, 103827. [Google Scholar]
- Angelova, V.T.; Simeonova, R. Effects of a new 1,2,3-thiadiazole containing hydrazone antimycobacterial agent on serum and liver biochemical parameters in female mice. Drug Chem. Toxicol. 2019, 45, 113–119. [Google Scholar] [CrossRef]
- Sakaeda, T.; Okamura, N.; Nagata, S.; Yagami, T.; Horinouchi, M.; Okumura, K.; Yamashita, F.; Hashida, M. Molecular and pharmacokinetic properties of 222 commercially available oral drugs in humans. Biol. Pharm. Bull. 2001, 24, 935–940. [Google Scholar] [CrossRef] [Green Version]
- Moriguchi, I.; Hirono, S.; Nakagome, I.; Hirano, H. Comparison of reliability of log P values for drugs calculated by several methods. Chem. Pharm. Bull. 1994, 42, 976–978. [Google Scholar] [CrossRef] [Green Version]
- Doğan, Ş.D.; Gündüz, M.G.; Doğan, H.; Krishna, V.S.; Lherbet, C.; Sriram, D. Design and synthesis of thio-urea-based derivatives as Mycobacterium tuberculosis growth and enoyl acyl carrier protein reductase (InhA) inhibitors. Eur. J. Med. Chem. 2020, 199, 112402. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Desta, Z.; Soukhova, N.V.; Flockhart, D.A. Inhibition of cytochrome P450 (CYP450) isoforms by isoniazid: Potent inhibition of CYP2C19 and CYP3A. Antimicrob. Agents Chemother. 2001, 45, 382–392. [Google Scholar] [CrossRef] [Green Version]
- Hollenberg, P.F. Characteristics and common properties of inhibitors, inducers, and activators of CYP enzymes. Drug Metab. Rev. 2002, 34, 17–35. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Deodhar, M.; Al Rihani, S.; Arwood, M.; Darakjian, L.; Dow, P.; Turgeon, J.; Michaud, V. Mechanisms of CYP450 inhibition: Understanding drug-drug interactions due to mechanism-based inhibition in clinical practice. Pharmaeutics 2020, 12, 846. [Google Scholar] [CrossRef]
- Banerjee, P.; Dehnbostel, F.O.; Preissner, R. Prediction is a balancing act: Importance of sampling methods to balance sensitivity and specificity of predictive models based on imbalanced chemical data sets. Front. Chem. 2018, 6, 362. [Google Scholar] [CrossRef]
- Doğan, H.; Doğan, Ş.D.; Gündüz, M.G.; Krishna, V.S.; Lherbet, C.; Sriram, D.; Şahin, O.; Sarıpınar, E. Discovery of hydrazone containing thiadiazoles as Mycobacterium tuberculosis growth and enoyl acyl carrier protein reductase (InhA) inhibitors. Eur. J. Med. Chem. 2020, 188, 112035. [Google Scholar] [CrossRef]
- Menendez, C.; Gau, S.; Lherbet, C.; Rodriguez, F.; Inard, C.; Pasca, M.R.; Baltas, M. Synthesis and biological activities of triazole derivatives as inhibitors of InhA and antituberculosis agents. Eur. J. Med. Chem. 2011, 46, 5524–5531. [Google Scholar] [CrossRef]
- Parikh, S.; Moynihan, D.P.; Xiao, G.; Tonge, P.J. Roles of tyrosine 158 and lysine 165 in the catalytic mechanism of InhA, the enoyl-ACP reductase from Mycobacterium tuberculosis. Biochemistry 1999, 38, 13623–13634. [Google Scholar] [CrossRef]
- Rozwarski, D.A.; Vilchèze, C.; Sugantino, M.; Bittman, R.; Sacchettini, J.C. Crystal structure of the Mycobacterium tuberculosis enoyl-ACP reductase, InhA, in complex with NAD+ and a C16 fatty acyl substrate. J. Biol. Chem. 1999, 274, 15582–15589. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.M.; Shawon, J.; Halim, M.A. Multiple receptor conformers based molecular docking study of fluorine enhanced ethionamide with mycobacterium enoyl ACP reductase (InhA). J. Mol. Graph. Model. 2017, 77, 386–398. [Google Scholar] [CrossRef]
- Schön, T.; Werngren, J.; Machado, D.; Borroni, E.; Wijkander, M.; Lina, G.; Mouton, J.; Matuschek, E.; Kahlmeter, G.; Giske, C. Antimicrobial susceptibility testing of Mycobacterium tuberculosis complex isolates–the EUCAST broth microdilution reference method for MIC determination. Clin. Microbiol. Infect. 2020, 26, 1488–1492. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Konstantinov, S.M.; Eibl, H.; Berger, M.R. BCR-ABL influences the antileukaemic efficacy of alkylphospho-cholines. Br. J. Haematol. 1999, 107, 365–374. [Google Scholar] [CrossRef]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Cryst. 2002, 40, 82–92. [Google Scholar]
- Zoete, V.; Daina, A.; Bovigny, C.; Michielin, O. SwissSimilarity: A Web Tool for Low to Ultra High Throughput Ligand-Based Virtual Screening. J. Chem. Inf. Model. 2016, 56, 1398–1404. [Google Scholar] [CrossRef]
- Fox, T.; Kriegl, J.M. Machine learning techniques for in silico modeling of drug metabolism. Curr. Top. Med. Chem. 2006, 6, 1579–1591. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | Formula | MIC a (µM) | IC50 (µM) b CCL-1 | IC50 (µM) c HEK-293 | SI d CCL-1 | SI d HEK-293 |
|---|---|---|---|---|---|---|
| 3a | 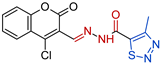 | 0.3914 | 32.9 ± 6.2 | 51.2 ± 4.7 | 84 | 130 |
| 3b | 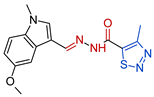 | 0.3294 | 818.3 ± 23.7 | 361 ± 11.3 | 2487 | 1244 |
| 3c | 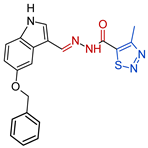 | 0.1744 | 390.8 ± 12.0 | 713.5 ± 18.5 | 2242 | 4093 |
| 3d |  | 0.0730 | 256.7 ± 13.3 | 217.5 ± 17.2 | 3516 | 2979 |
| 5a | 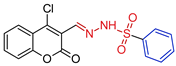 | 0.1814 | 48.0 ± 5.9 | 83.9 ± 4.0 | 267 | 463 |
| 5b | 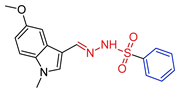 | 0.2027 | 2.9 ± 0.3 | 16.2 ± 2.7 | 14 | 82 |
| 5c |  | 0.3434 | 321.4 ± 16.2 | 32.3 ± 1.5 | 945 | 95 |
| 5d |  | 0.1669 | 13.8 ± 0.9 | 16.9 ± 3.4 | 11 | 97 |
| 5e |  | 0.1647 | 5.1 ± 1.1 | 15.6 ± 4.1 | 31 | 95 |
| 5f | 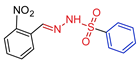 | 0.3053 | 72.4 ± 5.8 | 100.1 ± 2.1 | 237 | 327 |
| 5g | 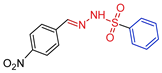 | 0.0763 | 191 ± 13.2 | 138.3 ± 7.4 | 1812 | 1819 |
| 5h | 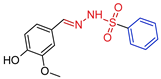 | 0.3203 | 223 ± 11.5 | 150.2 ± 5.1 | 469 | 696 |
| 5i | 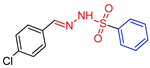 | 0.1473 | 131.4 ± 11.4 | 100.1 ± 9.3 | 892 | 679 |
| 5j | 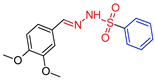 | 0.3210 | 83.5 ± 7.0 | 47.4 ± 6.8 | 146 | 260 |
| 5k | 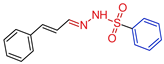 | 0.0716 | 240 ± 9.6 | 158.5 ± 11.2 | 3380 | 2216 |
| INH | 0.0343 | - | - | - | - |
| Scheme | MW 1 (g/mol) | TPSA 2 (Å2) | HBA 3 | HBD 4 | Rotatable Bonds | Moriguchi’s LogP | Water Solubility |
|---|---|---|---|---|---|---|---|
| 3a | 348.76430 | 125.69 | 6 | 1 | 4 | 1.37 | Poorly soluble |
| 3b | 329.37890 | 109.64 | 5 | 1 | 5 | 0.67 | Moderately soluble |
| 3c | 391.44628 | 120.50 | 5 | 2 | 7 | 1.64 | Moderately soluble |
| 3d | 292.31368 | 124.94 | 6 | 2 | 5 | 0.03 | Soluble |
| 5a | 362.78754 | 97.12 | 5 | 1 | 4 | 2.15 | Poorly soluble |
| 5b | 343.40018 | 81.07 | 4 | 1 | 5 | 1.44 | Moderately soluble |
| 5c | 405.46950 | 91.93 | 4 | 2 | 7 | 2.37 | Poorly soluble |
| 5d | 333.79268 | 82.70 | 3 | 2 | 4 | 2.01 | Poorly soluble |
| 5e | 329.37360 | 91.93 | 4 | 2 | 5 | 1.20 | Moderately soluble |
| 5f | 305.30914 | 112.73 | 5 | 1 | 5 | 1.86 | Moderately soluble |
| 5g | 305.30914 | 112.73 | 5 | 1 | 5 | 1.86 | Moderately soluble |
| 5h | 306.33696 | 96.37 | 5 | 2 | 5 | 1.23 | Moderately soluble |
| 5i | 294.75664 | 66.91 | 3 | 1 | 4 | 2.61 | Moderately soluble |
| 5j | 320.36354 | 85.37 | 5 | 1 | 6 | 1.49 | Moderately soluble |
| 5k | 286.34886 | 66.91 | 3 | 1 | 5 | 2.52 | Moderately soluble |
| INH | 137.14 | 68.01 | 3 | 2 | 2 | −0.47 | Soluble |
| EMB | 204.31 | 64.52 | 4 | 4 | 9 | 0.18 | Soluble |
| Pharmacokinetics | Drug Likeness | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | GI Absorbtion | BBB Perm. | P-gp Substrate | CYP1A2 Inhib. | CYP2C19 Inhib. | CYP2C9 Inhib. | CYP2D6 Inhib. | CYP3A4 Inhib. | Log Kp (cm/s) | Lipinski | Ghose | Veber | Bio. Score |
| 3a | high | no | no | yes | yes | no | no | no | −6.56 | yes | yes | yes | 0.55 |
| 3b | high | no | no | yes | yes | yes | no | yes | −6.80 | yes | yes | yes | 0.55 |
| 3c | high | no | no | yes | yes | yes | no | yes | −6.08 | yes | yes | yes | 0.55 |
| 3d | high | no | no | no | no | no | no | no | −6.88 | yes | yes | yes | 0.55 |
| 5a | high | no | no | no | yes | no | no | no | −6.34 | yes | yes | yes | 0.55 |
| 5b | high | no | no | no | yes | yes | no | yes | −6.57 | yes | yes | yes | 0.55 |
| 5c | high | no | no | yes | yes | yes | yes | yes | −5.86 | yes | yes | yes | 0.55 |
| 5d | high | no | no | yes | yes | yes | no | yes | −6.01 | yes | yes | yes | 0.55 |
| 5e | high | no | no | yes | yes | yes | no | no | −6.45 | yes | yes | yes | 0.55 |
| 5f | high | no | yes | yes | no | no | no | no | −6.49 | yes | yes | yes | 0.55 |
| 5g | high | no | yes | yes | no | no | no | no | −6.49 | yes | yes | yes | 0.55 |
| 5h | high | no | no | no | no | no | no | no | −6.67 | yes | yes | yes | 0.55 |
| 5i | high | yes | no | yes | yes | yes | no | no | −5.86 | yes | yes | yes | 0.55 |
| 5j | high | no | no | yes | yes | yes | no | no | −6.51 | yes | yes | yes | 0.55 |
| 5k | high | yes | no | yes | yes | yes | no | no | −5.95 | yes | yes | yes | 0.55 |
| INH | high | no | no | no | no | no | no | no | −7.63 | yes | no:3 viol. | yes | 0.55 |
| EMB | high | no | no | no | no | no | no | no | −7.60 | yes | yes | yes | 0.55 |
| No. | Compound | Oral Toxicity Class | Predicted LD50 (mg/kg) | Organ Toxicity (Hepatotoxicity) | Carcinogenicity | Immunotoxicity | Mutagenicity | Cytotoxicity | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pr | Prob | Pr | Prob | Pr | Prob | Pr | Prob | Pr | Prob | ||||
| 1 | 3a | III | 187 | A | 0.61 | I | 0.55 | I | 0.97 | A | 0.52 | I | 0.69 |
| 2 | 3b | IV | 1120 | A | 0.51 | I | 0.56 | A | 0.78 | A | 0.54 | I | 0.67 |
| 3 | 3c | V | 4920 | A | 0.63 | I | 0.52 | I | 0.82 | A | 0.55 | I | 0.65 |
| 4 | 3d | V | 4920 | A | 0.60 | A | 0.56 | I | 0.55 | A | 0.56 | I | 0.64 |
| 5 | 5a | IV | 500 | I | 0.53 | I | 0.60 | I | 0.99 | I | 0.68 | I | 0.71 |
| 7 | 5b | IV | 500 | I | 0.50 | I | 0.51 | I | 0.92 | I | 0.50 | I | 0.72 |
| 8 | 5c | IV | 500 | I | 0.50 | I | 0.53 | I | 0.99 | I | 0.55 | I | 0.78 |
| 9 | 5d | IV | 500 | I | 0.55 | I | 0.58 | I | 0.99 | I | 0.68 | I | 0.77 |
| 10 | 5e | IV | 500 | I | 0.50 | I | 0.50 | I | 0.94 | I | 0.53 | I | 0.81 |
| 11 | 5f | IV | 500 | I | 0.50 | A | 0.51 | I | 0.99 | I | 0.55 | I | 0.79 |
| 12 | 5g | IV | 500 | A | 0.52 | A | 0.57 | I | 0.99 | I | 0.63 | I | 0.73 |
| 13 | 5h | IV | 500 | I | 0.53 | A | 0.55 | I | 0.95 | I | 0.59 | I | 0.87 |
| 14 | 5i | IV | 500 | I | 0.63 | I | 0.60 | I | 0.99 | I | 0.66 | I | 0.70 |
| 15 | 5j | IV | 500 | I | 0.51 | A | 0.54 | I | 0.99 | I | 0.55 | I | 0.88 |
| 16 | 5k | III | 283 | I | 0.59 | A | 0.51 | I | 0.99 | I | 0.59 | I | 0.69 |
| INH | III | 133 | A | 0.94 | A | 0.98 | I | 0.99 | I | 0.63 | I | 0.81 | |
| EMB | IV | 998 | A | 0.63 | I | 0.56 | I | 0.99 | I | 0.95 | I | 0.72 | |
| Compound | 2X22 E_Score1 * (kcal/mol) | 4TZK E_Score1 * (kcal/mol) |
|---|---|---|
| 3a | −10.93 (13) | −11.56 (14) |
| 3b | −10.02 (15) | −11.10 (15) |
| 3c | −11.09 (9) | −11.62 (13) |
| 3d | −12.19 (2) | −12.38 (6) |
| 5a | −11.06 (10) | −12.15 (9) |
| 5b | −10.99 (12) | −12.30 (7) |
| 5c | −10.73 (14) | −12.66 (4) |
| 5d | −11.60 (5) | −12.19 (8) |
| 5e | −12.03 (3) | −14.67 (1) |
| 5f | −11.29 (7) | −11.88 (10) |
| 5g | −11.98 (4) | −12.80 (3) |
| 5h | −11.43 (6) | −12.49 (5) |
| 5i | −11.18 (8) | −11.87 (11) |
| 5j | −11.06 (11) | −11.75 (12) |
| 5k | −12.36 (1) | −12.83 (2) |
| INH | −9.18 (16) | −8.51 (16) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angelova, V.T.; Pencheva, T.; Vassilev, N.; K-Yovkova, E.; Mihaylova, R.; Petrov, B.; Valcheva, V. Development of New Antimycobacterial Sulfonyl Hydrazones and 4-Methyl-1,2,3-thiadiazole-Based Hydrazone Derivatives. Antibiotics 2022, 11, 562. https://doi.org/10.3390/antibiotics11050562
Angelova VT, Pencheva T, Vassilev N, K-Yovkova E, Mihaylova R, Petrov B, Valcheva V. Development of New Antimycobacterial Sulfonyl Hydrazones and 4-Methyl-1,2,3-thiadiazole-Based Hydrazone Derivatives. Antibiotics. 2022; 11(5):562. https://doi.org/10.3390/antibiotics11050562
Chicago/Turabian StyleAngelova, Violina T., Tania Pencheva, Nikolay Vassilev, Elena K-Yovkova, Rositsa Mihaylova, Boris Petrov, and Violeta Valcheva. 2022. "Development of New Antimycobacterial Sulfonyl Hydrazones and 4-Methyl-1,2,3-thiadiazole-Based Hydrazone Derivatives" Antibiotics 11, no. 5: 562. https://doi.org/10.3390/antibiotics11050562
APA StyleAngelova, V. T., Pencheva, T., Vassilev, N., K-Yovkova, E., Mihaylova, R., Petrov, B., & Valcheva, V. (2022). Development of New Antimycobacterial Sulfonyl Hydrazones and 4-Methyl-1,2,3-thiadiazole-Based Hydrazone Derivatives. Antibiotics, 11(5), 562. https://doi.org/10.3390/antibiotics11050562