
Performance of Novel Antimicrobial Protein Bg_9562 and In Silico Predictions on Its Properties with Reference to Its Antimicrobial Efficiency against Rhizoctonia solani


Abstract
:1. Introduction
2. Results
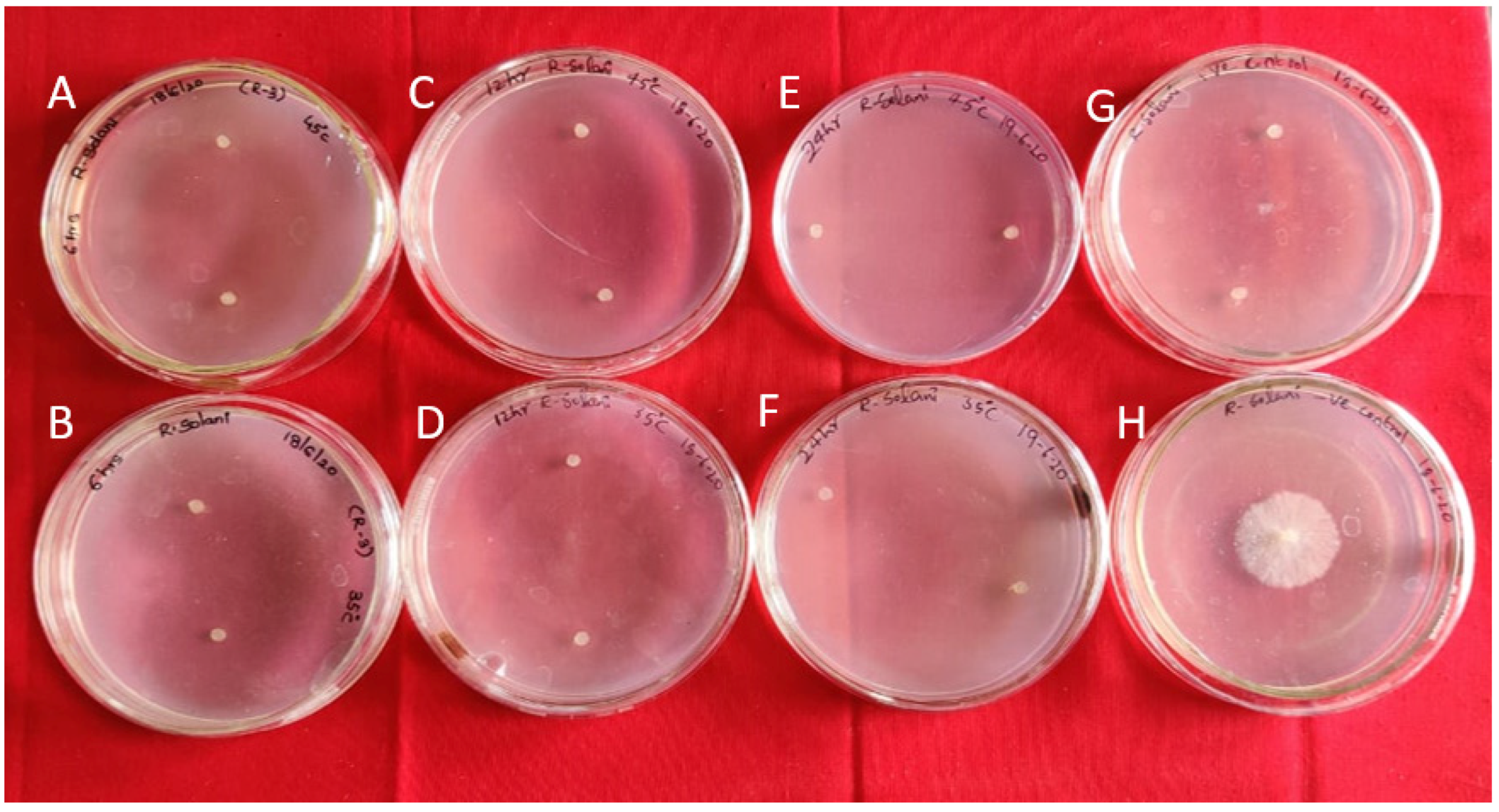
2.1. Antifungal Efficacy of Bg_9562 on RS
2.2. In Vitro Compatibility of Bioagents, Trichoderma Asperellum TAIK1 and Bacillus Subtilis BIK3 with Bg_9562
2.3. Assessment of Bg_9562 Protein Activity under Field Conditions
2.4. Amino Acid Composition in Bg_9562
2.5. Physiochemical Properties of Bg_9562 Determined Using ProtParam Tool
2.6. Physiochemical Properties of Bg_9562 Determined Using Protscale
2.7. Bg_9562 Properties Determined Using APD
2.8. Propensity of Crystallization
2.9. Sub-Program Sorting Using PSORT Tool
2.10. Secondary Structure of Bg_9562
2.11. 3D Structure Modeling of Bg_9562
2.12. Functional Analysis of Conserved and Disordered Regions of Bg_9562
3. Discussion
4. Materials and Methods
4.1. Preparation of Pure Bg_9562 Protein
4.2. Broad-Spectrum Bioactivity Assay of Bg_9562 on Fungal and Bacterial Growth
4.3. Efficacy of Bg_9562 Protein under Field Conditions
4.4. Sequence Retrieval
4.5. Prediction of Physiochemical Properties of Bg_9562 Protein
4.6. Prediction of Functional Properties of Bg_9562 Protein
4.7. Secondary Structure Prediction
4.8. 3D Structure Prediction
4.9. 3D Structure Validation
4.10. Prediction of Disordered Regions in Bg_9562
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Den Herder, G.; Van Isterdael, G.; Beeckman, T.; De Smet, I. The roots of a new green revolution. Trends Plant Sci. 2010, 15, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Thambugala, K.M.; Daranagama, D.A.; Phillips, A.J.; Kannangara, S.D.; Promputtha, I. Fungi vs. fungi in biocontrol: An overview of fungal antagonists applied against fungal plant pathogens. Front. Cell. Infect. Microbiol. 2020, 10, 718. [Google Scholar] [CrossRef] [PubMed]
- Jangir, M.; Sharma, S.; Sharma, S. Development of next-generation formulation against Fusarium oxysporum and unraveling bioactive antifungal metabolites of biocontrol agents. Sci. Rep. 2021, 11, 22895. [Google Scholar] [CrossRef] [PubMed]
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 2020, 11, 582779. [Google Scholar] [CrossRef] [PubMed]
- Shai, Y. Molecular recognition between membrane-spanning polypeptides. Trends Biochem. Sci. 1995, 20, 460–464. [Google Scholar] [CrossRef]
- Balkovec, J. Lipopeptide antifungal agents. Expert Opin. Investig. Drugs 1994, 3, 65–82. [Google Scholar] [CrossRef]
- Debono, M.; Gordee, R.S. Antibiotics that inhibit fungal cell wall development. Annu. Rev. Microbiol. 1994, 48, 471–497. [Google Scholar] [CrossRef]
- Singh, H.B. Management of plant pathogens with microorganisms. Proc. Indian Natl. Sci. Acad. 2014, 80, 443–454. [Google Scholar] [CrossRef]
- World Rice Production 2019/2020 World Agricultural Production. Available online: http://www.worldagriculturalproduction.com/crops/rice.aspx (accessed on 11 April 2020).
- Gao, J.P.; Chao, D.Y.; Lin, H.X. Understanding abiotic stress tolerance mechanisms, recent studies on stress response in rice. J. Integr. Plant Biol. 2007, 49, 742–750. [Google Scholar] [CrossRef]
- Gangopadhyay, S.; Chakrabarti, N.K. Sheath blight of rice. Rev. Plant Pathol. 1982, 61, 451–460. [Google Scholar]
- Carling, D.; Baird, R.; Gitaitis, R.; Brainard, K.; Kuninaga, S. Characterization of AG-13, a newly reported anastomosis group of Rhizoctonia solani. Phytopathology 2002, 92, 893–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carling, D.; Kuninaga, S.; Brainard, K. Hyphal anastomosis reactions, rDNA-internal transcribed spacer sequences, and virulence levels among subsets of Rhizoctonia solani anastomosis group-2 (AG-2) and AG-BI. Phytopathology 2002, 92, 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, P.; Mazumdar, P.; Harikrishna, J.A.; Babu, S. Sheath blight of rice: A review and identification of priorities for future research. Planta 2019, 250, 1387–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berlin, A.; Källström, H.N.; Lindgren, A.; Olson, Å. Scientific evidence for sustainable plant disease protection strategies for the main arable crops in Sweden. A systematic map protocol. Environ. Evid. 2018, 7, 31. [Google Scholar] [CrossRef] [Green Version]
- Köhl, J.; Kolnaar, R.; Ravensberg, W.J. Mode of action of microbial biological control agents against plant diseases: Relevance beyond efficacy. Front. Plant Sci. 2019, 10, 845. [Google Scholar] [CrossRef] [Green Version]
- Swain, D.M.; Yadav, S.K.; Tyagi, I.; Kumar, R.; Kumar, R.; Ghosh, S.; Das, J.; Jha, G. A prophage tail-like protein is deployed by Burkholderia bacteria to feed on fungi. Nat. Commun. 2017, 8, 404. [Google Scholar] [CrossRef] [Green Version]
- Kannan, C.; Mishra, D.; Rekha, G.; Maruthi, P.; Shaik, H.; Sundaram, R.M. Diversity analysis of antagonistic microbes against bacterial leaf and fungal sheathblight diseases of rice. Egypt. J. Biol. Pest Control 2021, 31, 115. [Google Scholar] [CrossRef]
- Boman, H.G. Antibacterial peptides: Basic facts and emerging concepts. J. Intern. Med. 2003, 254, 197–215. [Google Scholar] [CrossRef]
- Molla, K.A.; Karmakar, S.; Molla, J.; Bajaj, P.; Varshney, R.K.; Datta, S.K.; Datta, K. Understanding sheath blight resistance in rice: The road behind and the road ahead. Plant Biotechnol. J. 2020, 18, 895–915. [Google Scholar] [CrossRef]
- Khammanee, N.; Qiu, Y.; Kungskulniti, N.; Bignert, A.; Meng, Y.; Zhu, Z.; Teffera, Z.L. Presence and Health Risks of Obsolete and Emerging Pesticides in Paddy Rice and Soil from Thailand and China. Int. J. Environ. Res. Public Health 2020, 17, 3786. [Google Scholar] [CrossRef]
- Chatterjee, S.; Gangopadhyay, C.; Bandyopadhyay, P.; Bhowmick, M.; Roy, S.; Majumder, A.; Gathala, M.; Tanwar, R.; Singh, S.; Birah, A.; et al. Input-based assessment on integrated pest management for transplanted rice (Oryza sativa) in India. Crop Prot. 2021, 141, 105444. [Google Scholar] [CrossRef]
- Selitrennikoff, C.P. Antifungal proteins. Appl. Environ. Microbiol. 2001, 67, 2883–2894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, S.; Badri, J.; Prakasam, V.; Bhadana, V.P.; Eswari, K.B.; Laha, G.S.; Priyanka, C.; Aku, R.; Ram, T. Identification and agro-morphological characterization of rice genotypes resistant to sheath blight. Australas. Plant Pathol. 2016, 45, 145–153. [Google Scholar] [CrossRef]
- Shai, Y.; Fox, J.; Caratsch, C.; Shih, Y.L.; Edwards, C.; Lazarovici, P. Sequencing and synthesis of pardaxin, a polypeptide from the Red Sea Moses sole with ionophore activity. FEBS Lett. 1988, 242, 161–166. [Google Scholar] [CrossRef] [Green Version]
- Oren, Z.; Hong, J.; Shai, Y. A repertoire of novel antibacterial diastereomeric peptides with selective cytolytic activity. J. Biol. Chem. 1997, 272, 14643–14649. [Google Scholar] [CrossRef] [Green Version]
- Johansson, J.; Gudmundsson, G.H.; Rottenberg, M.E.; Berndt, K.D.; Agerberth, B. Conformation-dependent antibacterial activity of the naturally occurring human peptide LL-37. J. Biol. Chem. 1988, 273, 3718–3724. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Bai, X.; Luan, N.; Yao, H.; Zhang, Z.; Liu, W.; Chen, Y.; Yan, X.; Rong, M.; Lai, R.; et al. A Designed Tryptophan- and Lysine/Arginine-Rich Antimicrobial Peptide with Therapeutic Potential for Clinical Antibiotic-Resistant Candida albicans Vaginitis. J. Med. Chem. 2016, 59, 1791–1799. [Google Scholar] [CrossRef]
- Godballe, T.; Mojsoska, B.; Nielsen, H.M.; Jenssen, H. Antimicrobial activity of GN peptides and their mode of action. Biopolymers 2016, 106, 172–183. [Google Scholar] [CrossRef] [Green Version]
- Deslouches, B.; Hasek, M.L.; Craigo, J.K.; Steckbeck, J.D.; Montelaro, R.C. Comparative functional properties of engineered cationic antimicrobial peptides consisting exclusively of tryptophan and either lysine or arginine. J. Med. Microbiol. 2016, 65, 554–565. [Google Scholar] [CrossRef]
- Hussain, S.; Khaliq, A.; Ali, B.; Hussain, H.A.; Qadir, T.; Hussain, S. Temperature Extremes: Impact on Rice Growth and Development. In Plant Abiotic Stress Tolerance; Hasanuzzaman, M., Hakeem, K., Nahar, K., Alharby, H., Eds.; Springer: Cham, Switzerland, 2019. [Google Scholar] [CrossRef]
- Kumar, S.; Tsai, C.J.; Nussinov, R. Factors enhancing protein thermostability. Protein Eng. 2000, 13, 179–191. [Google Scholar] [CrossRef] [Green Version]
- Haki, G.D.; Rakshit, S.K. Developments in industrially important thermostable enzymes: A review. Bioresour. Technol. 2003, 89, 17–34. [Google Scholar] [CrossRef]
- Holdbrook, D.A.; Singh, S.; Choong, Y.K.; Petrlova, J.; Malmsten, M.; Bond, P.J.; Verma, N.K.; Schmidtchen, A.; Saravanan, R. Influence of pH on the activity of thrombin-derived antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 2018, 1860, 2374–2384. [Google Scholar] [CrossRef] [PubMed]
- Mao, A.H.; Crick, S.L.; Vitalis, A.; Chicoine, C.L.; Pappu, R.V. Net charge per residue modulates conformational ensembles of intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 8183–8188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, M. Antifungal properties of haem peroxidase from Acorus calamus. Ann. Bot. 2006, 98, 1145–1153. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Haney, E.F.; Vogel, H.J. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 2011, 29, 464–472. [Google Scholar] [CrossRef]
- Hancock, R.E. Peptide antibiotics. Lancet 1997, 349, 418–422. [Google Scholar] [CrossRef]
- Hwang, P.M.; Vogel, H.J. Structure-function relationships of antimicrobial peptides. Biochem. Cell Biol. 1988, 76, 235–246. [Google Scholar] [CrossRef]
- Kim, H.; Jang, J.H.; Kim, S.C.; Cho, J.H. De novo generation of short antimicrobial peptides with enhanced stability and cell specificity. J. Antimicrob. Chemother. 2014, 69, 121–132. [Google Scholar] [CrossRef]
- Anjana, R.; Vaishnavi, M.K.; Sherlin, D.; Kumar, S.P.; Naveen, K.; Kanth, P.S.; Sekar, K. Aromatic-aromatic interactions in structures of proteins and protein-DNA complexes: A study based on orientation and distance. Bioinformation 2012, 8, 1220–1224. [Google Scholar] [CrossRef]
- Butterfield, S.M.; Patel, P.R.; Waters, M.L. Contribution of aromatic interactions to alpha-helix stability. J. Am. Chem. Soc. 2002, 124, 9751–9755. [Google Scholar] [CrossRef]
- Meyer, E.A.; Castellano, R.K.; Diederich, F. Interactions with aromatic rings in chemical and biological recognition. Angew. Chem. 2003, 42, 1210–1250. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Phillips, R.S.; Li, J. Editorial: Aromatic Amino Acid Metabolism. Front. Mol. Biosci. 2012, 6, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, L.J.; Fiebig, K.M.; Schwalbe, H.; Dobson, C.M. The concept of a random coil. Residual structure in peptides and denatured proteins. Fold. Des. 1996, 1, R95–R106. [Google Scholar] [CrossRef] [Green Version]
- González-Faune, P.; Sánchez-Arévalo, I.; Sarkar, S.; Majhi, K.; Bandopadhyay, R.; Cabrera-Barjas, G.; Gómez, A.; Banerjee, A. Computational Study on Temperature Driven Structure–Function Relationship of Polysaccharide Producing Bacterial Glycosyl Transferase Enzyme. Polymers 2021, 13, 1771. [Google Scholar] [CrossRef]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef] [Green Version]
- Torrent, M.; Andreu, D.; Nogués, V.M.; Boix, E. Connecting peptide physicochemical and antimicrobial properties by a rational prediction model. PLoS ONE 2011, 6, e16968. [Google Scholar] [CrossRef]
- Narayana, J.L.; Chen, J.Y. Antimicrobial peptides: Possible anti-infective agents. Peptides 2015, 72, 88–94. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, B.; Li, B.; Wang, C.; Luo, Y. Preparation and identification of peptides and their zinc complexes with antimicrobial activities from silver carp (Hypophthalmichthys molitrix) protein hydrolysates. Food Res. Int. 2014, 64, 91–98. [Google Scholar] [CrossRef]
- Jezowska-Bojczuk, M.; Stokowa-Sołtys, K. Peptides having antimicrobial activity and their complexes with transition metal ions. Eur. J. Med. Chem. 2018, 143, 997–1009. [Google Scholar] [CrossRef]
- Chen, Y.; Guarnieri, M.T.; Vasil, A.I.; Vasil, M.L.; Mant, C.T.; Hodges, R.S. Role of peptide hydrophobicity in the mechanism of action of a-helical antimicrobial peptides. Antimicrob. Agents Chemother. 2007, 51, 1398–1406. [Google Scholar] [CrossRef] [Green Version]
- Radzicka, A.; Wolfenden, R. Comparing the polarities of the amino acids: Side-chain distribution coefficients between the vapor phase, cyclohexane, 1-octanol, and neutral aqueous solution. Biochemistry 1988, 27, 1664–1670. [Google Scholar] [CrossRef]
- Jiang, Z.Y.; Xu, L.L.; Lu, M.C.; Chen, Z.Y.; Yuan, Z.W.; Xu, X.L.; Guo, X.K.; Zhang, X.J.; Sun, H.P.; You, Q.D. Structure-Activity and Structure-Property Relationship and Exploratory in Vivo Evaluation of the Nanomolar Keap1-Nrf2 Protein-Protein Interaction Inhibitor. J. Med. Chem. 2015, 58, 6410–6421. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Smeekens, J.M.; Wu, R. Systematic study of the dynamics and half-lives of newly synthesized proteins in human cells. Chem. Sci. 2016, 7, 1393–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirski, T.; Lidia, M.; Nakonieczna, A.; Gryko, R. Bacteriophages, phage endolysins and antimicrobial peptides—The possibilities for their common use to combat infections and in the design of new drugs. Ann. Agric. Environ. Med. 2019, 26, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, P.; Zheng, C.; Huang, Y.P. Characterization of maltocin P28, a novel phage tail-like bacteriocin from Stenotrophomonas maltophilia. Appl. Environ. Microbiol. 2018, 79, 5593–5600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hockett, K.L.; Renner, T.; Baltrus, D.A. Independent Co-Option of a Tailed Bacteriophage into a Killing Complex in Pseudomonas. mBio 2015, 6, e00452. [Google Scholar] [CrossRef] [Green Version]
- Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu. Rev. Biochem. 2014, 83, 553–584. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Linking folding and binding. Curr. Opin. Struct. Biol. 2009, 19, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Miyatake, H.; Yokota, N.; Matsuyama, S.; Tokuda, H.; Miki, K. Crystal structures of bacterial lipoprotein localization factors, LolA and LolB. EMBO J. 2003, 22, 3199–3209. [Google Scholar] [CrossRef] [Green Version]
- Ramamourthy, G.; Park, J.; Seo, C.; JVogel, H.; Park, Y. Antifungal and Antibiofilm Activities and the Mechanism of Action of Repeating Lysine-Tryptophan Peptides against Candida albicans. Microorganisms 2020, 8, 758. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; John, M.W., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Li, X.; Wang, Z. APD2: The updated antimicrobial peptide database and its application in peptide design. Nucleic Acids Res. 2009, 37, D933–D937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wimley, W.C.; White, S.H. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 1996, 3, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Chen, C.P.; Kernytsky, A.; Rost, B. Transmembrane helix predictions revisited. Protein Sci. A Publ. Protein Soc. 2002, 11, 2774–2791. [Google Scholar] [CrossRef] [Green Version]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Horton, P.; Nakai, K. Better Prediction of Protein Cellular Localization Sites with it k Nearest Neighbors Classifier. In Proceedings of the International Conference on Intelligent Systems for Molecular Biology, Halkidiki, Greece, 21–26 June 1997; Volume 5, pp. 147–152. [Google Scholar]
- Kurgan, L.A.; Razib, A.A.; Aghakhani, S.; Dick, S.; Mizianty, M.J.; Jahandideh, S. CRYSTALP2: Sequence-based protein crystallization propensity prediction. BMC Struct. Biol. 2009, 9, 50. [Google Scholar] [CrossRef] [Green Version]
- Mizianty, M.J.; Kurgan, L.A. Sequence-based prediction of protein crystallization, purification, and production propensity. Bioinformatics 2011, 27, i24–i33. [Google Scholar] [CrossRef]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. Con Surf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [Green Version]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef]
- Buchan, D.W.A.; Jones, D.T. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res. 2019, 47, W402–W407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Combet, C.; Blanchet, C.; Geourjon, C.; Deléage, G. NPS@: Network protein sequence analysis. Trends Biochem. Sci. 2000, 25, 147–150. [Google Scholar] [CrossRef]
- King, R.D.; Sternberg, M.J. Identification and application of the concepts important for accurate and reliable protein secondary structure prediction. Protein Sci. 1996, 5, 2298–2310. [Google Scholar] [CrossRef] [PubMed]
- Guermeur, Y.; Geourjon, C.; Gallinari, P.; Deléage, G. Improved performance in protein secondary structure prediction by inhomogeneous score combination. Bioinformatics 1999, 15, 413–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rost, B.; Sander, C. Prediction of protein secondary structure at better than 70% accuracy. J. Mol. Biol. 1993, 232, 584–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frishman, D.; Argos, P. Incorporation of non-local interactions in protein secondary structure prediction from the amino acid sequence. Protein Eng. 1996, 9, 133–142. [Google Scholar] [CrossRef]
- Geourjon, C.; Deléage, G. SOPMA: Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput. Appl. Biosci. CABIOS 1995, 11, 681–684. [Google Scholar] [CrossRef]
- Levin, J.M.; Robson, B.; Garnier, J. An algorithm for secondary structure determination in proteins based on sequence similarity. FEBS Lett. 1985, 205, 303–308. [Google Scholar] [CrossRef] [Green Version]
- Deléage, G.; Blanchet, C.; Geourjon, C. Protein structure prediction. Implications for the biologist. Biochimie 1997, 79, 681–686. [Google Scholar] [CrossRef]
- Biegert, A.; Mayer, C.; Remmert, M.; Söding, J.; Lupas, A.N. The MPI Bioinformatics Toolkit for protein sequence analysis. Nucleic Acids Res. 2006, 34, W335–W339. [Google Scholar] [CrossRef] [Green Version]
- Fiser, A.; Sali, A. Modeller: Generation and refinement of homology-based protein structure models. Methods Enzymol. 2003, 374, 461–491. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Sternberg, M.J. Protein structure prediction on the Web: A case study using the Phyre server. Nat. Protoc. 2009, 4, 363–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Källberg, M.; Wang, H.; Wang, S.; Peng, J.; Wang, Z.; Lu, H.; Xu, J. Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 2012, 7, 1511–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhang, Y. LOMETS: A local meta-threading-server for protein structure prediction. Nucleic Acids Res. 2007, 35, 3375–3382. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Faraggi, E.; Zhao, H.; Zhou, Y. Improving protein fold recognition and template-based modeling by employing probabilistic-based matching between predicted one-dimensional structural properties of query and corresponding native properties of templates. Bioinformatics 2011, 27, 2076–2082. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32, W526–W531. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Zhang, Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins 2012, 80, 1715–1735. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Mirdita, M.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. bioRxiv 2021, 8, 9939. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK—A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef]
- Yang, J.; Roy, A.; Zhang, Y. Protein–ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 2013, 29, 2588–2595. [Google Scholar] [CrossRef]
- Ishida, T.; Kinoshita, K. PrDOS: Prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 2007, 35, W460–W464. [Google Scholar] [CrossRef] [PubMed]
- Prilusky, J.; Feder, C.E.; Zeev-Ben-Mordehai, T.; Rydberg, E.; Man, O.; Beckman, J.S.; Silman, I.; Sussman, J.L. FoldIndex: A simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics 2005, 21, 3435–3438. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No | Parameters | Values/Scores |
|---|---|---|
| 1 | Number of amino acids | 111 |
| 2 | Molecular weight | 11,451.97 |
| 3 | Theoretical isoelectric point | 4.65 |
| 4 | Total number of negatively-charged residues (Asp + Glu) | 12 |
| 5 | Total number of positively-charged residues (Arg + Lys) | 8 |
| 6 | Total number of hydrophobic amino acids (Ile + Val + Lue + Phe + Cys + Met + Ala + Tyr) | 46 |
| 7 | Total number of atoms | 1612 |
| 8 | Formula | C491H813N137O166S5 |
| 9 | Instability index | 21.19 |
| 10 | Aliphatic index | 89.92 |
| 11 | Grand average of hydropathicity index (GRAVY) | −0.012 |
| 12 | Estimated half-life (mammalian reticulocytes, in vitro) | 30 h |
| 13 | Estimated half-life (yeast, in vitro) | >20 h |
| 14 | Estimated half-life (Escherichia coli, in vivo) | >10 h |
| 15 | Extinction coefficient | 0 M−1 cm−1 |
| S. No. | Parameter | Values |
|---|---|---|
| 1 | Protein binding potential (Boman index) | 1.27 kcal/mol |
| 2 | The Wimley–White whole-residue hydrophobicity of the peptide | 26.18 kcal/mol |
| 3 | APD defined total hydrophobic ratio of protein | 41% |
| 4 | Total net charge | −3.75 |
| 5 | Antimicrobial activity | Yes |
| Secondary Structure | DSC * | HNN * | MLRC * | PHD * | PREDATOR | Secondary Consensus |
|---|---|---|---|---|---|---|
| Alpha helix | 55.86% | 27.3% | 26.13% | 34.23% | 22.52% | 36.04% |
| Extended strand | 3.6% | 7.21% | 9.91% | 16.22% | 3.6% | 9.91% |
| Random coil | 40.54% | 65.77% | 63.96% | 49.55% | 73.87% | 52.25% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karnati, P.; Gonuguntala, R.; Barbadikar, K.M.; Mishra, D.; Jha, G.; Prakasham, V.; Chilumula, P.; Shaik, H.; Pesari, M.; Sundaram, R.M.; et al. Performance of Novel Antimicrobial Protein Bg_9562 and In Silico Predictions on Its Properties with Reference to Its Antimicrobial Efficiency against Rhizoctonia solani. Antibiotics 2022, 11, 363. https://doi.org/10.3390/antibiotics11030363
Karnati P, Gonuguntala R, Barbadikar KM, Mishra D, Jha G, Prakasham V, Chilumula P, Shaik H, Pesari M, Sundaram RM, et al. Performance of Novel Antimicrobial Protein Bg_9562 and In Silico Predictions on Its Properties with Reference to Its Antimicrobial Efficiency against Rhizoctonia solani. Antibiotics. 2022; 11(3):363. https://doi.org/10.3390/antibiotics11030363
Chicago/Turabian StyleKarnati, Pranathi, Rekha Gonuguntala, Kalyani M. Barbadikar, Divya Mishra, Gopaljee Jha, Vellaisamy Prakasham, Priyanka Chilumula, Hajira Shaik, Maruthi Pesari, Raman Meenakshi Sundaram, and et al. 2022. "Performance of Novel Antimicrobial Protein Bg_9562 and In Silico Predictions on Its Properties with Reference to Its Antimicrobial Efficiency against Rhizoctonia solani" Antibiotics 11, no. 3: 363. https://doi.org/10.3390/antibiotics11030363
APA StyleKarnati, P., Gonuguntala, R., Barbadikar, K. M., Mishra, D., Jha, G., Prakasham, V., Chilumula, P., Shaik, H., Pesari, M., Sundaram, R. M., & Chinnaswami, K. (2022). Performance of Novel Antimicrobial Protein Bg_9562 and In Silico Predictions on Its Properties with Reference to Its Antimicrobial Efficiency against Rhizoctonia solani. Antibiotics, 11(3), 363. https://doi.org/10.3390/antibiotics11030363