
RecA and Specialized Error-Prone DNA Polymerases Are Not Required for Mutagenesis and Antibiotic Resistance Induced by Fluoroquinolones in Pseudomonas aeruginosa


Abstract
:1. Introduction
2. Results and Discussion
2.1. Validation of the P. aeruginosa RecA-Deficient Mutant as an SOS Response- and Homologous Recombination-Null Strain
2.2. RecA Affects Intrinsic Resistance Only towards Fluoroquinolones and Genotoxic Agents
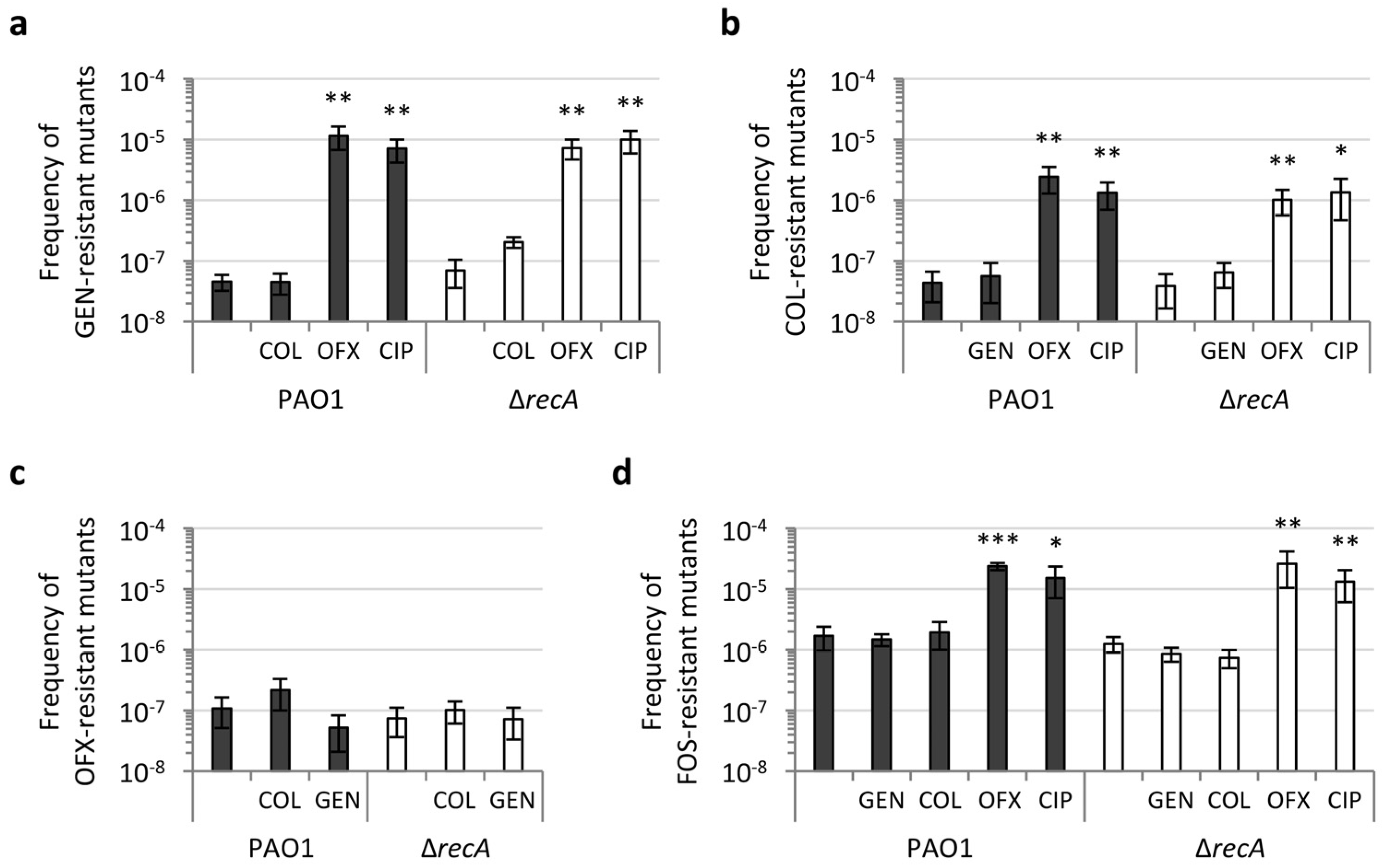
2.3. RecA Is Not Required for Antibiotic-Induced Mutagenesis and Acquisition of Antibiotic Resistance
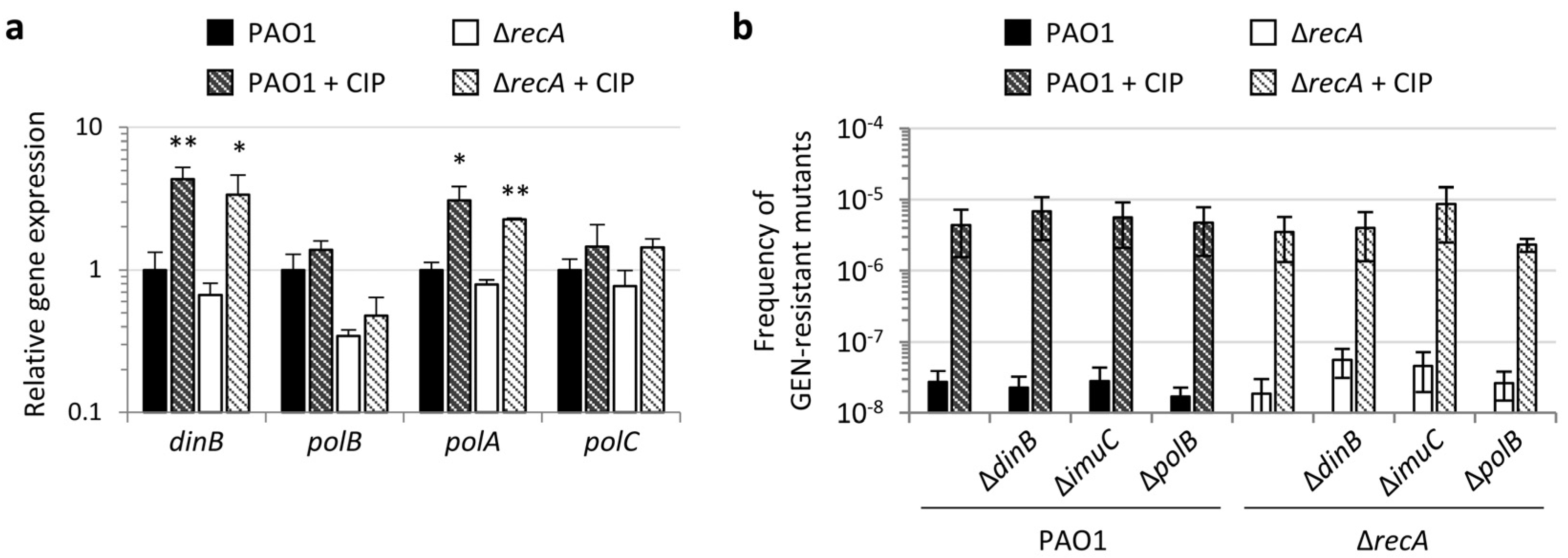
2.4. Specialized and Error-Prone DNA Polymerases Are not Involved in Fluoroquinolone-Induced Antibiotic Resistance
3. Conclusions
4. Materials and Methods
4.1. Bacterial Strains and Growth Media
4.2. Generation of the Complementing Construct Mini-CTX1recA and recA Deletion Mutants
4.3. Growth and Conjugation Assays
4.4. Gene Expression Analysis by qRT-PCR
4.5. MIC Assays
4.6. Selection and Frequency of Antibiotic-Resistant Mutants
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Foster, P.L. Adaptive mutation: Implications for evolution. BioEssays 2000, 22, 1067–1074. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, S.M. Evolving responsively: Adaptive mutation. Nat. Rev. Genet. 2001, 2, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Revitt-Mills, S.A.; Robinson, A. Antibiotic-Induced Mutagenesis: Under the Microscope. Front. Microbiol. 2020, 11, 585175. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, J.; Couce, A.; Rodríguez-Beltrán, J.; Rodríguez-Rojas, A. Antimicrobials as promoters of genetic variation. Curr. Opin. Microbiol. 2012, 15, 561–569. [Google Scholar] [CrossRef]
- Blázquez, J.; Rodríguez-Beltrán, J.; Matic, I. Antibiotic-Induced Genetic Variation: How It Arises and How It Can Be Prevented. Annu. Rev. Microbiol. 2018, 72, 209–230. [Google Scholar] [CrossRef]
- McKenzie, G.J.; Harris, R.S.; Lee, P.L.; Rosenberg, S.M. The SOS response regulates adaptive mutation. Proc. Natl. Acad. Sci. USA 2000, 97, 6646–6651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baharoglu, Z.; Mazel, D. SOS, the formidable strategy of bacteria against aggressions. FEMS Microbiol. Rev. 2014, 38, 1126–1145. [Google Scholar] [CrossRef] [Green Version]
- Maslowska, K.H.; Makiela-Dzbenska, K.; Fijalkowska, I.J. The SOS system: A complex and tightly regulated response to DNA damage. Environ. Mol. Mutagen. 2019, 60, 368–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janion, C. Inducible SOS Response System of DNA Repair and Mutagenesis in Escherichia coli. Int. J. Biol. Sci. 2008, 4, 338–344. [Google Scholar] [CrossRef] [Green Version]
- Kivisaar, M. Mechanisms of stationary-phase mutagenesis in bacteria: Mutational processes in pseudomonads. FEMS Microbiol. Lett. 2010, 312, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, M.F.; Woodgate, R. Translesion DNA Polymerases. Cold Spring Harb. Perspect. Biol. 2013, 5, a010363. [Google Scholar] [CrossRef]
- Henrikus, S.S.; van Oijen, A.; Robinson, A. Specialised DNA polymerases in Escherichia coli: Roles within multiple pathways. Curr. Genet. 2018, 64, 1189–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thi, T.D.; López, E.; Rodríguez-Rojas, A.; Rodríguez-Beltrán, J.; Couce, A.; Guelfo, J.R.; Castañeda-García, A.; Blázquez, J. Effect of recA inactivation on mutagenesis of Escherichia coli exposed to sublethal concentrations of antimicrobials. J. Antimicrob. Chemother. 2011, 66, 531–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baharoglu, Z.; Mazel, D. Vibrio cholerae Triggers SOS and Mutagenesis in Response to a Wide Range of Antibiotics: A Route towards Multiresistance. Antimicrob. Agents Chemother. 2011, 55, 2438–2441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlopoulou, A. RecA a universal drug target in pathogenic bacteria. Front. Biosci. 2018, 23, 36–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanyon-Hogg, T. Targeting the bacterial SOS response for new antimicrobial agents: Drug targets, molecular mechanisms and inhibitors. Future Med. Chem. 2021, 13, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Selwood, T.; Larsen, B.J.; Mo, C.Y.; Culyba, M.J.; Hostetler, Z.M.; Kohli, R.M.; Reitz, A.B.; Baugh, S.D.P. Advancement of the 5-Amino-1-(Carbamoylmethyl)-1H-1,2,3-Triazole-4-Carboxamide Scaffold to Disarm the Bacterial SOS Response. Front. Microbiol. 2018, 9, 2961. [Google Scholar] [CrossRef]
- Lyczak, J.B.; Cannon, C.L.; Pier, G.B. Establishment of Pseudomonas aeruginosa infection: Lessons from a versatile opportunist. Microbes Infect. 2000, 2, 1051–1060. [Google Scholar] [CrossRef]
- Driscoll, J.A.; Brody, S.L.; Kollef, M.H. The Epidemiology, Pathogenesis and Treatment of Pseudomonas aeruginosa Infections. Drugs 2007, 67, 351–368. [Google Scholar] [CrossRef]
- Poole, K. Pseudomonas Aeruginosa: Resistance to the Max. Front. Microbiol. 2011, 2, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, J. Pseudomonas aeruginosa in cystic fibrosis: Pathogenesis and persistence. Paediatr. Respir. Rev. 2002, 3, 128–134. [Google Scholar] [CrossRef]
- Folkesson, A.; Jelsbak, L.; Yang, L.; Johansen, H.K.; Ciofu, O.; Høiby, N.; Molin, S. Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: An evolutionary perspective. Nat. Rev. Genet. 2012, 10, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Cirz, R.T.; O’Neill, B.M.; Hammond, J.A.; Head, S.R.; Romesberg, F.E. Defining the Pseudomonas aeruginosa SOS Response and Its Role in the Global Response to the Antibiotic Ciprofloxacin. J. Bacteriol. 2006, 188, 7101–7110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penterman, J.; Singh, P.K.; Walker, G.C. Biological Cost of Pyocin Production during the SOS Response in Pseudomonas aeruginosa. J. Bacteriol. 2014, 196, 3351–3359. [Google Scholar] [CrossRef] [Green Version]
- Breidenstein, E.B.M.; Bains, M.; Hancock, R.E.W. Involvement of the Lon Protease in the SOS Response Triggered by Ciprofloxacin in Pseudomonas aeruginosa PAO1. Antimicrob. Agents Chemother. 2012, 56, 2879–2887. [Google Scholar] [CrossRef] [Green Version]
- Valencia, E.Y.; Esposito, F.; Spira, B.; Blázquez, J.; Galhardo, R.S. Ciprofloxacin-Mediated Mutagenesis Is Suppressed by Subinhibitory Concentrations of Amikacin in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2017, 61, e02107-16. [Google Scholar] [CrossRef] [Green Version]
- Torres-Barceló, C.; Kojadinovic, M.; Moxon, R.; MacLean, C. The SOS response increases bacterial fitness, but not evolvability, under a sublethal dose of antibiotic. Proc. R. Soc. B Biol. Sci. 2015, 282, 20150885. [Google Scholar] [CrossRef] [Green Version]
- Tanimoto, K.; Tomita, H.; Fujimoto, S.; Okuzumi, K.; Ike, Y. Fluoroquinolone Enhances the Mutation Frequency for Meropenem-Selected Carbapenem Resistance in Pseudomonas aeruginosa, but Use of the High-Potency Drug Doripenem Inhibits Mutant Formation. Antimicrob. Agents Chemother. 2008, 52, 3795–3800. [Google Scholar] [CrossRef] [Green Version]
- Boles, B.R.; Singh, P.K. Endogenous oxidative stress produces diversity and adaptability in biofilm communities. Proc. Natl. Acad. Sci. USA 2008, 105, 12503–12508. [Google Scholar] [CrossRef] [Green Version]
- Nair, C.G.; Chao, C.; Ryall, B.; Williams, H.D. Sub-lethal concentrations of antibiotics increase mutation frequency in the cystic fibrosis pathogen Pseudomonas aeruginosa. Lett. Appl. Microbiol. 2013, 56, 149–154. [Google Scholar] [CrossRef]
- Migliorini, L.B.; Brüggemann, H.; de Sales, R.O.; Koga, P.C.M.; de Souza, A.V.; Martino, M.D.V.; Galhardo, R.S.; Severino, P. Mutagenesis Induced by Sub-Lethal Doses of Ciprofloxacin: Genotypic and Phenotypic Differences Between the Pseudomonas aeruginosa Strain PA14 and Clinical Isolates. Front. Microbiol. 2019, 10, 1553. [Google Scholar] [CrossRef] [Green Version]
- Boles, B.R.; Thoendel, M.; Singh, P.K. Self-generated diversity produces "insurance effects" in biofilm communities. Proc. Natl. Acad. Sci. USA 2004, 101, 16630–16635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, J.; Kowalczykowski, S.C. RecA: Regulation and Mechanism of a Molecular Search Engine. Trends Biochem. Sci. 2016, 41, 491–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lusetti, S.L.; Cox, M.M. The Bacterial RecA Protein and the Recombinational DNA Repair of Stalled Replication Forks. Annu. Rev. Biochem. 2002, 71, 71–100. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.J.; Margulies, A.D. Isolation and Characterization of Recombination-Deficient Mutants of Escherichia coli K12. Proc. Natl. Acad. Sci. USA 1965, 53, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Hoang, T.T.; Karkhoff-Schweizer, R.R.; Kutchma, A.J.; Schweizer, H.P. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: Application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 1998, 212, 77–86. [Google Scholar] [CrossRef]
- Hoang, T.T.; Kutchma, A.J.; Becher, A.; Schweizer, H.P. Integration-Proficient Plasmids for Pseudomonas aeruginosa: Site-Specific Integration and Use for Engineering of Reporter and Expression Strains. Plasmid 2000, 43, 59–72. [Google Scholar] [CrossRef]
- Frangipani, E.; Visaggio, D.; Heeb, S.; Kaever, V.; Cámara, M.; Visca, P.; Imperi, F. The Gac/Rsm and cyclic-di-GMP signalling networks coordinately regulate iron uptake in Pseudomonas aeruginosa. Environ. Microbiol. 2014, 16, 676–688. [Google Scholar] [CrossRef]
- Blázquez, J.; Gómez-Gómez, J.-M.; Oliver, A.; Juan, C.; Kapur, V.; Martín, S. PBP3 inhibition elicits adaptive responses in Pseudomonas aeruginosa. Mol. Microbiol. 2006, 62, 84–99. [Google Scholar] [CrossRef]
- Luján, A.M.; Moyano, A.J.; Martino, R.A.; Feliziani, S.; Urretavizcaya, M.; Smania, A.M. ImuB and ImuC contribute to UV-induced mutagenesis as part of the SOS regulon in Pseudomonas aeruginosa. Environ. Mol. Mutagen. 2019, 60, 594–601. [Google Scholar] [CrossRef]
- Rella, M.; Haas, D. Resistance of Pseudomonas aeruginosa PAO to nalidixic acid and low levels of beta-lactam antibiotics: Mapping of chromosomal genes. Antimicrob. Agents Chemother. 1982, 22, 242–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drlica, K.; Malik, M.; Kerns, R.J.; Zhao, X. Quinolone-Mediated Bacterial Death. Antimicrob. Agents Chemother. 2008, 52, 385–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalczykowski, S.C. An Overview of the Molecular Mechanisms of Recombinational DNA Repair. Cold Spring Harb. Perspect. Biol. 2015, 7, a016410. [Google Scholar] [CrossRef] [Green Version]
- Kwan, B.W.; Chowdhury, N.; Wood, T.K. Combatting bacterial infections by killing persister cells with mitomycin C. Environ. Microbiol. 2015, 17, 4406–4414. [Google Scholar] [CrossRef] [PubMed]
- Van, T.T.; Minejima, E.; Chiu, C.A.; Butler-Wu, S.M. Don’t Get Wound Up: Revised Fluoroquinolone Breakpoints for Enterobacteriaceae and Pseudomonas aeruginosa. J. Clin. Microbiol. 2019, 57, e02072-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hocquet, D.; Bertrand, X. Metronidazole increases the emergence of ciprofloxacin- and amikacin-resistant Pseudomonas aeruginosa by inducing the SOS response. J. Antimicrob. Chemother. 2014, 69, 852–854. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Jiang, Q.; Woodgate, R.; Cox, M.M.; Goodman, M.F. A new model for SOS-induced mutagenesis: How RecA protein activates DNA polymerase V. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 171–184. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Di Luca, M.; Tkhilaishvili, T.; Trampuz, A.; Moreno, M.G. Synergistic Activity of Fosfomycin, Ciprofloxacin, and Gentamicin Against Escherichia coli and Pseudomonas aeruginosa Biofilms. Front. Microbiol. 2019, 10, 2522. [Google Scholar] [CrossRef] [Green Version]
- Sabnis, A.; Hagart, K.L.; Klöckner, A.; Becce, M.; Evans, L.E.; Furniss, R.C.D.; Mavridou, D.; Murphy, R.; Stevens, M.M.; Davies, J.C.; et al. Colistin kills bacteria by targeting lipopolysaccharide in the cytoplasmic membrane. eLife 2021, 10, e65836. [Google Scholar] [CrossRef]
- Mo, C.Y.; Manning, S.A.; Roggiani, M.; Culyba, M.J.; Samuels, A.N.; Sniegowski, P.D.; Goulian, M.; Kohli, R.M. Systematically Altering Bacterial SOS Activity under Stress Reveals Therapeutic Strategies for Potentiating Antibiotics. mSphere 2016, 1, e00163-16. [Google Scholar] [CrossRef] [Green Version]
- Tegova, R.; Tover, A.; Tarassova, K.; Tark, M.; Kivisaar, M. Involvement of Error-Prone DNA Polymerase IV in Stationary-Phase Mutagenesis in Pseudomonas putida. J. Bacteriol. 2004, 186, 2735–2744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, L.H.; Rockel, A.; Lu, H.; Wozniak, D.J.; Sutton, M.D. Role of Pseudomonas aeruginosa dinB-Encoded DNA Polymerase IV in Mutagenesis. J. Bacteriol. 2006, 188, 8573–8585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jatsenko, T.; Sidorenko, J.; Saumaa, S.; Kivisaar, M. DNA Polymerases ImuC and DinB Are Involved in DNA Alkylation Damage Tolerance in Pseudomonas aeruginosa and Pseudomonas putida. PLoS ONE 2017, 12, e0170719. [Google Scholar] [CrossRef] [Green Version]
- Sanders, L.H.; Devadoss, B.; Raja, G.V.; O’Connor, J.; Su, S.; Wozniak, D.J.; Hassett, D.J.; Berdis, A.J.; Sutton, M.D. Epistatic Roles for Pseudomonas aeruginosa MutS and DinB (DNA Pol IV) in Coping with Reactive Oxygen Species-Induced DNA Damage. PLoS ONE 2011, 6, e18824. [Google Scholar] [CrossRef]
- Jacobs, M.A.; Alwood, A.; Thaipisuttikul, I.; Spencer, D.; Haugen, E.; Ernst, S.; Will, O.; Kaul, R.; Raymond, C.; Levy, R.; et al. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2003, 100, 14339–14344. [Google Scholar] [CrossRef] [Green Version]
- Liberati, N.T.; Urbach, J.M.; Miyata, S.; Lee, D.G.; Drenkard, E.; Wu, G.; Villanueva, J.; Wei, T.; Ausubel, F.M. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc. Natl. Acad. Sci. USA 2006, 103, 2833–2838. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.A.; Gallagher, L.A.; Thongdee, M.; Staudinger, B.J.; Lippman, S.; Singh, P.K.; Manoil, C. General and condition-specific essential functions of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2015, 112, 5189–5194. [Google Scholar] [CrossRef] [Green Version]
- Wigle, T.J.; Sexton, J.Z.; Gromova, A.V.; Hadimani, M.B.; Hughes, M.A.; Smith, G.R.; Yeh, L.-A.; Singleton, S.F. Inhibitors of RecA Activity Discovered by High-Throughput Screening: Cell-Permeable Small Molecules Attenuate the SOS Response in Escherichia coli. J. Biomol. Screen. 2009, 14, 1092–1101. [Google Scholar] [CrossRef] [Green Version]
- Sexton, J.Z.; Wigle, T.J.; He, Q.; Hughes, M.A.; Smith, G.R.; Singleton, S.F.; Williams, A.L.; Yeh, L.-A. Novel Inhibitors of E. coli RecA ATPase Activity. Curr. Chem. Genom. 2010, 4, 34–42. [Google Scholar] [CrossRef]
- Peterson, E.J.; Janzen, W.P.; Kireev, D.; Singleton, S.F. High-Throughput Screening for RecA Inhibitors Using a Transcreener Adenosine 5′-O-Diphosphate Assay. ASSAY Drug Dev. Technol. 2012, 10, 260–268. [Google Scholar] [CrossRef] [Green Version]
- Alam, K.; Alhhazmi, A.; DeCoteau, J.F.; Luo, Y.; Geyer, C.R. RecA Inhibitors Potentiate Antibiotic Activity and Block Evolution of Antibiotic Resistance. Cell Chem. Biol. 2016, 23, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Song, L.Y.; Goff, M.; Davidian, C.; Mao, Z.; London, M.; Lam, K.; Yung, M.; Miller, J.H. Mutational Consequences of Ciprofloxacin in Escherichia coli. Antimicrob. Agents Chemother. 2016, 60, 6165–6172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Lo Sciuto, A.; Spinnato, M.C.; Pasqua, M.; Imperi, F. Generation of stable and unmarked conditional mutants in Pseudomonas aeruginosa. Methods Mol. Biol. 2022; in press. [Google Scholar]
- Scala, R.; Di Matteo, A.; Coluccia, A.; Lo Sciuto, A.; Federici, L.; Travaglini-Allocatelli, C.; Visca, P.; Silvestri, R.; Imperi, F. Mutational analysis of the essential lipopolysaccharide-transport protein LptH of Pseudomonas aeruginosa to uncover critical oligomerization sites. Sci. Rep. 2020, 10, 11276. [Google Scholar] [CrossRef] [PubMed]
- Spinnato, M.C.; Lo Sciuto, A.; Mercolino, J.; Lucidi, M.; Leoni, L.; Rampioni, G.; Visca, P.; Imperi, F. Effect of a Defective Clamp Loader Complex of DNA Polymerase III on Growth and SOS Response in Pseudomonas aeruginosa. Microorganisms 2022, 10, 423. [Google Scholar] [CrossRef]
- Simon, R.; Priefer, U.B.; Pühler, A. A Broad Host Range Mobilization System for In Vivo Genetic Engineering: Transposon Mutagenesis in Gram Negative Bacteria. Bio/technology 1983, 1, 784–791. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Strain | MIC (µg/mL) | |||||||
|---|---|---|---|---|---|---|---|---|
| CIP 1 | OFX 1 | NAL 1 | GEN 1 | TOB 1 | MER 1 | COL 1 | MMC 1 | |
| PAO1 | 0.125 | 1 | 250 | 0.5 | 0.25 | 0.5 | 1 | 4 |
| ΔrecA | 0.031 | 0.25 | 125 | 0.5 | 0.25 | 0.5 | 1 | 1 |
| ΔrecA recA+ | 0.125 | 1 | 250 | 0.5 | 0.25 | 0.5 | 1 | 4 |
| PAO1 CIPR-1 | 2 | n.t. 2 | n.t. | n.t. | n.t. | n.t. | n.t. | n.t. |
| PAO1 CIPR-1 ΔrecA | 1 | n.t. | n.t. | n.t. | n.t. | n.t. | n.t. | n.t. |
| PAO1 CIPR-2 | 8 | n.t. | n.t. | n.t. | n.t. | n.t. | n.t. | n.t. |
| PAO1 CIPR-2 ΔrecA | 2 | n.t. | n.t. | n.t. | n.t. | n.t. | n.t. | n.t. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mercolino, J.; Lo Sciuto, A.; Spinnato, M.C.; Rampioni, G.; Imperi, F. RecA and Specialized Error-Prone DNA Polymerases Are Not Required for Mutagenesis and Antibiotic Resistance Induced by Fluoroquinolones in Pseudomonas aeruginosa. Antibiotics 2022, 11, 325. https://doi.org/10.3390/antibiotics11030325
Mercolino J, Lo Sciuto A, Spinnato MC, Rampioni G, Imperi F. RecA and Specialized Error-Prone DNA Polymerases Are Not Required for Mutagenesis and Antibiotic Resistance Induced by Fluoroquinolones in Pseudomonas aeruginosa. Antibiotics. 2022; 11(3):325. https://doi.org/10.3390/antibiotics11030325
Chicago/Turabian StyleMercolino, Jessica, Alessandra Lo Sciuto, Maria Concetta Spinnato, Giordano Rampioni, and Francesco Imperi. 2022. "RecA and Specialized Error-Prone DNA Polymerases Are Not Required for Mutagenesis and Antibiotic Resistance Induced by Fluoroquinolones in Pseudomonas aeruginosa" Antibiotics 11, no. 3: 325. https://doi.org/10.3390/antibiotics11030325
APA StyleMercolino, J., Lo Sciuto, A., Spinnato, M. C., Rampioni, G., & Imperi, F. (2022). RecA and Specialized Error-Prone DNA Polymerases Are Not Required for Mutagenesis and Antibiotic Resistance Induced by Fluoroquinolones in Pseudomonas aeruginosa. Antibiotics, 11(3), 325. https://doi.org/10.3390/antibiotics11030325