
Emergence of a Novel Lineage and Wide Spread of a blaCTX-M-15/IncHI2/ST1 Plasmid among Nosocomial Enterobacter in Guadeloupe
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Clinical Incidence of Hospital Acquired ESBL-Producing Enterobacteriaceae at the UHCG between 2018 and 2019
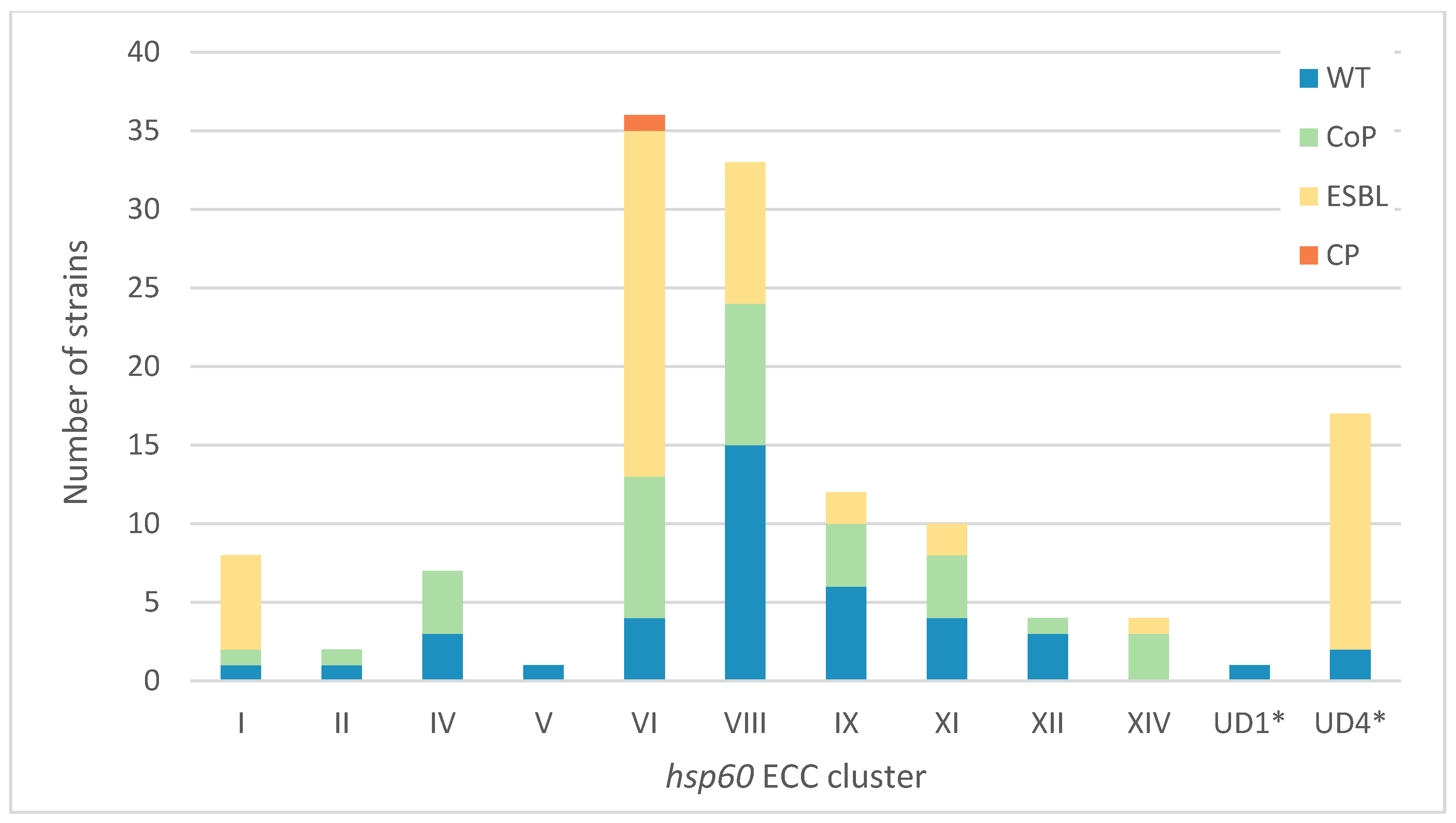
2.2. Enterobacter hsp60 Distribution and Antibiotic Resistance Patterns at the UHCG
2.3. Specificities of ESBL-Producing ECC Populations
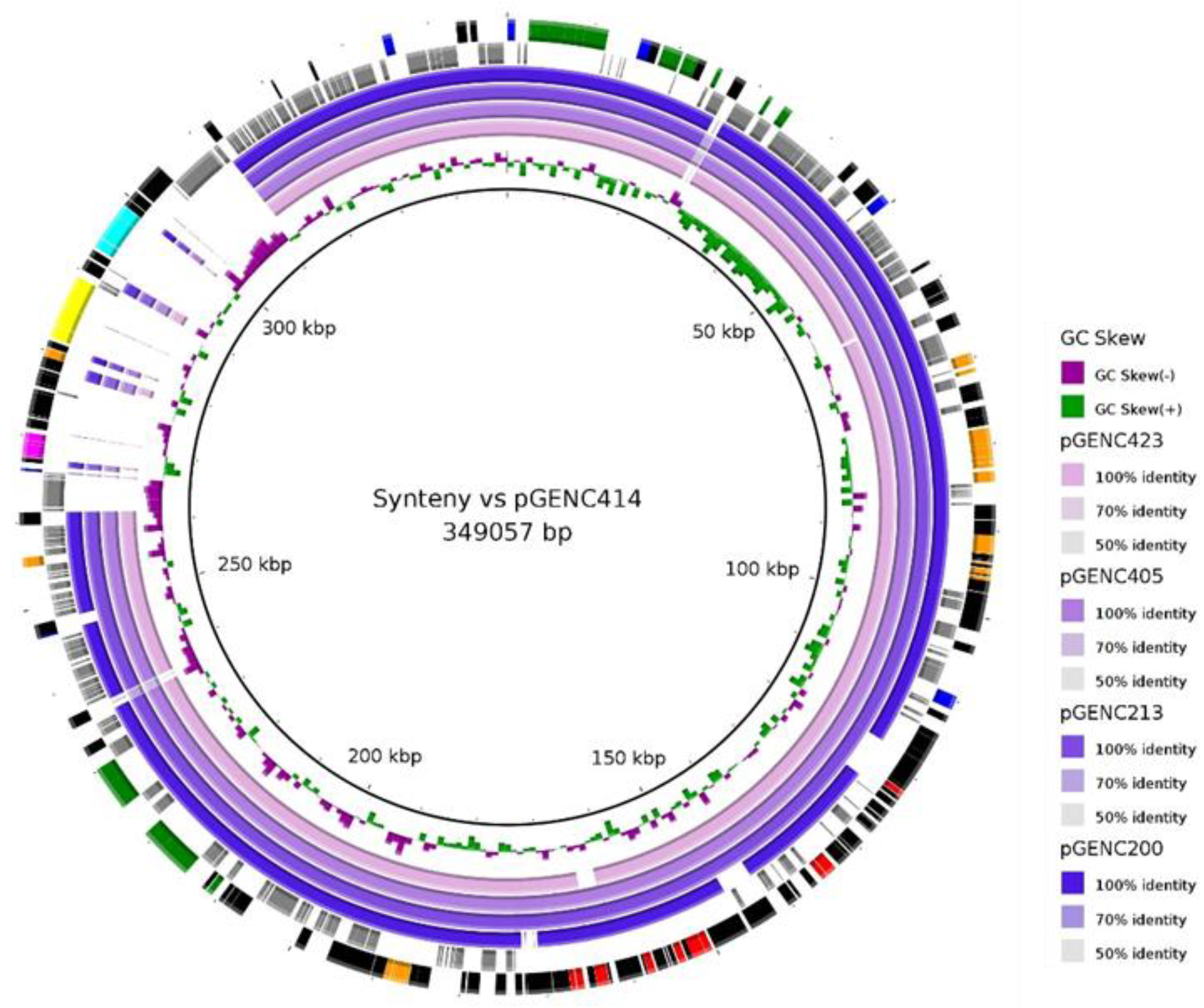
2.4. Sequence Analyses and Synteny of Major blaCTXM-15/IncHI2/ST1 Plasmids
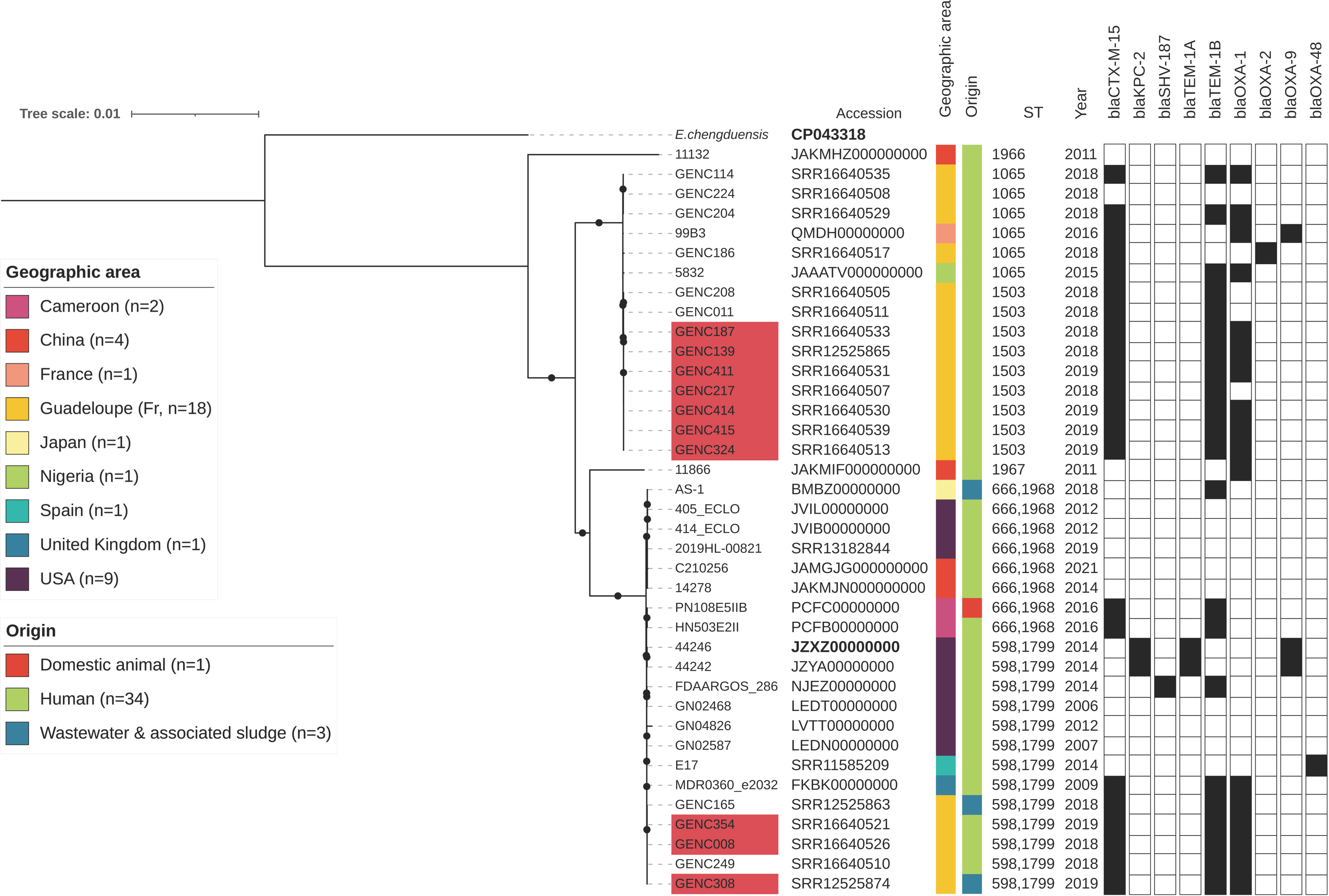
2.5. Literature Search for Similar IncHI2/ST1 Resistance Plasmids
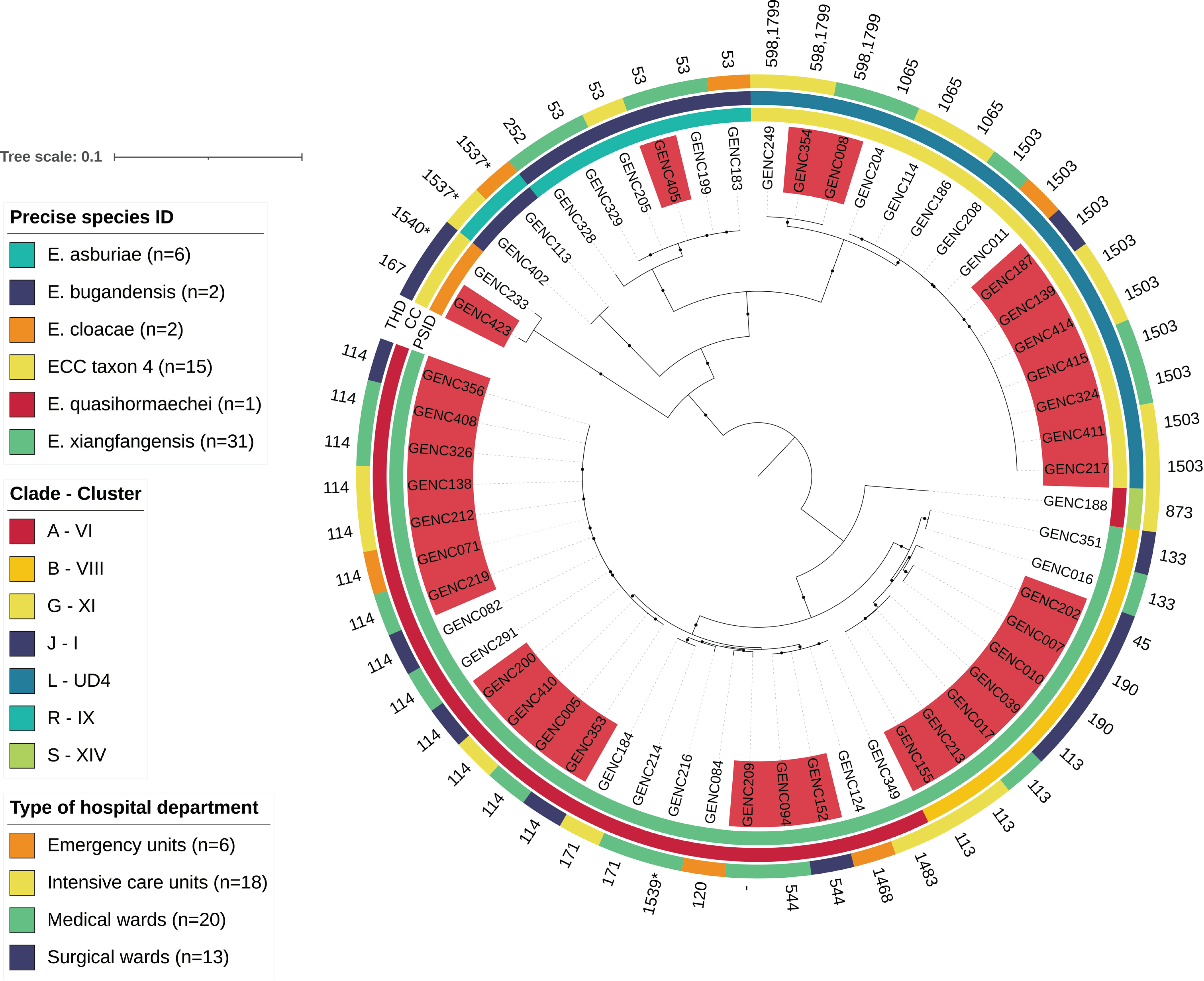
2.6. Phylogenetic Analysis of ECC Sutton’s Clade L in Guadeloupe and Global Comparison
3. Discussion
3.1. Nosocomial Incidence of ESBL-Producers in Guadeloupe
3.2. The Local hsp60 Cluster Distribution of Clinical ECC Does Not Mirror That in the National Population
3.3. Dissemination of Resistant Lineages through Hospital Wards
3.4. A Successful blaCTXM-15/IncHI2/ST1 Plasmid Is Circulating in ECC Nosocomial Populations
3.5. Limitations of the Study
4. Materials and Methods
4.1. Collection of Metadata on ESBL Enterobacteriaceae Associated Infections
4.2. Collection of Clinical ECC Strains and Associated Data
4.3. DNA Extraction and hsp60 Cluster Identification
4.4. Assembly and Core-Genome Phylogenetic Analyses of ESBL ECC
4.5. Sequence Type and Precise Clonal Relatedness among Major Enterobacter
4.6. Plasmid Profiles and Genetic Contents
4.7. Sequencing and Synteny of a Major Local Resistance Plasmid
4.8. Literature Search for Similar Plasmids
4.9. Global Phylogenetic Analysis of ECC Sutton’s Clade L
4.10. Data Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Davin-Regli, A.; Lavigne, J.P.; Pagès, J.M. Enterobacter spp.: Update on taxonomy, clinical aspects, and emerging antimicrobial resistance. Clin. Microbiol. Rev. 2019, 32, e00002-19. [Google Scholar] [CrossRef]
- Beyrouthy, R.; Barets, M.; Marion, E.; Dananché, C.; Dauwalder, O.; Robin, F.; Gauthier, L.; Jousset, A.; Dortet, L.; Guérin, F.; et al. Novel Enterobacter lineage as leading cause of nosocomial outbreak involving carbapenemase-producing strains. Emerg. Infect. Dis. 2018, 24, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Sutton, G.G.; Brinkac, L.M.; Clarke, T.H.; Fouts, D.E. Enterobacter hormaechei subsp. hoffmannii subsp. nov., Enterobacter hormaechei subsp. xiangfangensis comb. nov., Enterobacter roggenkampii sp. nov., and Enterobacter muelleri is a later heterotypic synonym of Enterobacter asburiae based on computational analysis of sequenced Enterobacter genomes. F1000Research 2018, 7, 521. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, H.; Roggenkamp, A. Population genetics of the nomenspecies Enterobacter cloacae. Appl. Environ. Microbiol. 2003, 69, 5306–5318. [Google Scholar] [CrossRef]
- Wu, W.; Feng, Y.; Zong, Z. Precise species identification for Enterobacter: A genome sequence-based study with reporting of two novel species, Enterobacter quasiroggenkampii sp. nov. and Enterobacter quasimori sp. nov. mSystems 2020, 5, e00527-20. [Google Scholar] [CrossRef]
- Feng, Y.; Hu, Y.; Zong, Z. Reexamining the association of AmpC variants with Enterobacter species in the context of updated taxonomy. Antimicrob. Agents Chemother. 2021, 65, e0159621. [Google Scholar] [CrossRef]
- Rice, L.B. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: No ESKAPE. J. Infect. Dis. 2008, 197, 1079–1081. [Google Scholar] [CrossRef]
- World Health Organization. WHO Publishes List of Bacteria for Which New Antibiotics Are Urgently Needed. 2017. Available online: https://www.who.int/news/item/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed on 2 February 2020).
- Rozwandowicz, M.; Brouwer, M.S.M.; Fischer, J.; Wagenaar, J.A.; Gonzalez-Zorn, B.; Guerra, B.; Mevius, D.J.; Hordijk, J. Plasmids carrying antimicrobial resistance genes in Enterobacteriaceae. J. Antimicrob. Chemother. 2018, 73, 1121–1137. [Google Scholar] [CrossRef] [PubMed]
- Carattoli, A.; Zankari, E.; García-Fernández, A.; Voldby Larsen, M.; Lund, O.; Villa, L.; Møller Aarestrup, F.; Hasman, H. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef]
- Krawczyk, P.S.; Lipinski, L.; Dziembowski, A. PlasFlow: Predicting plasmid sequences in metagenomic data using genome signatures. Nucleic Acids Res. 2018, 46, e35. [Google Scholar] [CrossRef]
- van der Graaf-van Bloois, L.; Wagenaar, J.A.; Zomer, A.L. RFPlasmid: Predicting plasmid sequences from short-read assembly data using machine learning. Microb. Genom. 2021, 7, 000683. [Google Scholar] [CrossRef]
- Robertson, J.; Nash, J.H.E. MOB-suite: Software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microb. Genom. 2018, 4, e000206. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Pankhurst, L.; Hubbard, A.; Votintseva, A.; Stoesser, N.; Sheppard, A.E.; Mathers, A.; Norris, R.; Navickaite, I.; Eaton, C.; et al. Resolving plasmid structures in Enterobacteriaceae using the MinION Nanopore sequencer: Assessment of MinION and MinION/Illumina hybrid data assembly approaches. Microb. Genom. 2017, 3, e000118. [Google Scholar] [CrossRef]
- Guyomard-Rabenirina, S.; Malespine, J.; Ducat, C.; Sadikalay, S.; Falord, M.; Harrois, D.; Richard, V.; Dozois, C.; The Laboratory Working Group; Breurec, S.; et al. Temporal trends and risks factors for antimicrobial resistant Enterobacteriaceae urinary isolates from outpatients in Guadeloupe. BMC Microbiol. 2016, 16, 121. [Google Scholar] [CrossRef]
- Heinz, E.; Brindle, R.; Morgan-McCalla, A.; Peters, K.; Thomson, N.R. Caribbean multi-centre study of Klebsiella pneumoniae: Whole-genome sequencing, antimicrobial resistance and virulence factors. Microb. Genom. 2019, 5, e000266. [Google Scholar] [CrossRef]
- Piednoir, P.; Clarac, U.; Rolle, A.; Bastian, S.; Gruel, G.; Martino, F.; Mehdaoui, H.; Valette, M.; Breurec, S.; Carles, M. Spontaneous community-acquired bacterial meningitis in adults admitted to the intensive care units in the Caribbean French West Indies: Unusual prevalence of Klebsiella pneumonia. Int. J. Infect. Dis. 2020, 100, 473–475. [Google Scholar] [CrossRef]
- Pot, M.; Reynaud, Y.; Couvin, D.; Ducat, C.; Ferdinand, S.; Gravey, F.; Gruel, G.; Guérin, F.; Malpote, E.; Breurec, S.; et al. Wide distribution and specific Resistance pattern to third-generation cephalosporins of Enterobacter cloacae complex members in humans and in the environment in Guadeloupe (French West Indies). Front. Microbiol. 2021, 12, 628058. [Google Scholar] [CrossRef]
- Pot, M.; Guyomard-Rabenirina, S.; Couvin, D.; Ducat, C.; Enouf, V.; Ferdinand, S.; Gruel, G.; Malpote, E.; Talarmin, A.; Breurec, S.; et al. Dissemination of extended-spectrum-β-Lactamase-producing Enterobacter cloacae complex from a hospital to the nearby environment in Guadeloupe (French West Indies): ST114 Lineage coding for a successful IncHI2/ST1 plasmid. Antimicrob. Agents Chemother. 2021, 65, e02146-20. [Google Scholar] [CrossRef]
- Kieffer, N.; Royer, G.; Decousser, J.W.; Bourrel, A.S.; Palmieri, M.; Ortiz de la Rosa, J.M.; Jacquier, H.; Denamur, E.; Nordmann, P.; Poirel, L. mcr-9, an inducible gene encoding an acquired phosphoethanolamine transferase in Escherichia coli, and its origin. Antimicrob. Agents Chemother. 2019, 63, e00965-19. [Google Scholar] [CrossRef]
- Redondo-Salvo, S.; Bartomeus-Peñalver, R.; Vielva, L.; Tagg, K.A.; Webb, H.E.; Fernández-López, R.; de la Cruz, F. COPLA, a taxonomic classifier of plasmids. BMC Bioinform. 2021, 22, 390. [Google Scholar] [CrossRef]
- Redondo-Salvo, S.; Fernández-López, R.; Ruiz, R.; Vielva, L.; de Toro, M.; Rocha, E.P.C.; Garcillán-Barcia, M.P.; de la Cruz, F. Pathways for horizontal gene transfer in bacteria revealed by a global map of their plasmids. Nat. Commun. 2020, 11, 3602. [Google Scholar] [CrossRef]
- Schmartz, G.P.; Hartung, A.; Hirsch, P.; Kern, F.; Fehlmann, T.; Müller, R.; Keller, A. PLSDB: Advancing a comprehensive database of bacterial plasmids. Nucleic Acids Res. 2022, 50, D273–D278. [Google Scholar] [CrossRef]
- NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2016, 44, D7–D19. [Google Scholar] [CrossRef] [PubMed]
- Pot, M.; Ducat, C.; Reynaud, Y.; Couvin, D.; Ferdinand, S.; Breurec, S.; Talarmin, A.; Guyomard-Rabenirina, S. Draft Genome Sequence of Enterobacter chengduensis ECC445, Isolated from Fresh Water in the West Indies. [Figure S1—Manuscript under Revision Process]. 2022. Available online: https://doi.org/10.6084/m9.figshare.21294879 (accessed on 7 October 2022).
- Wu, W.; Feng, Y.; Zong, Z. Characterization of a strain representing a new Enterobacter species, Enterobacter chengduensis sp. nov. Antonie Leeuwenhoek 2019, 112, 491–500. [Google Scholar] [CrossRef]
- Santé Publique France. Mission Spares. In Surveillance de L’antibiorésistance en Établissement de Santé—Données 2018, Partie 2: Résistance Bactérienne aux Antibiotiques; Santé Publique France: Saint-Maurice, France, 2020; pp. 1–54. [Google Scholar]
- Robin, F.; Beyrouthy, R.; Bonacorsi, S.; Aissa, N.; Bret, L.; Brieu, N.; Cattoir, V.; Chapuis, A.; Chardon, H.; Degand, N.; et al. Inventory of extended-spectrum-β-Lactamase-producing Enterobacteriaceae in France as assessed by a multicenter study. Antimicrob. Agents Chemother. 2017, 61, e01911-16. [Google Scholar] [CrossRef]
- Santé Publique France. GEODES. Available online: https://geodes.santepubliquefrance.fr (accessed on 2 January 2021).
- Girlich, D.; Ouzani, S.; Emeraud, C.; Gauthier, L.; Bonnin, R.A.; Le Sache, N.; Mokhtari, M.; Langlois, I.; Begasse, C.; Arangia, N.; et al. Uncovering the novel Enterobacter cloacae complex species responsible for septic shock deaths in newborns: A cohort study. Lancet Microbe. 2021, 5247, e536–e544. [Google Scholar] [CrossRef]
- Chang, C.Y.; Huang, P.H.; Lu, P.L. The resistance mechanisms and clinical impact of resistance to the third generation cephalosporins in species of Enterobacter cloacae complex in Taiwan. Antibiotics 2022, 11, 1153. [Google Scholar] [CrossRef]
- Garinet, S.; Fihman, V.; Jacquier, H.; Corvec, S.; Le Monnier, A.; Guillard, T.; Cattoir, V.; Zahar, J.R.; Woerther, P.L.; Carbonnelle, E.; et al. Elective distribution of resistance to beta-lactams among Enterobacter cloacae genetic clusters. J. Infect. 2018, 77, 178–182. [Google Scholar] [CrossRef]
- Kremer, A.; Hoffmann, H. Prevalences of the Enterobacter cloacae complex and its phylogenetic derivatives in the nosocomial environment. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 2951–2955. [Google Scholar] [CrossRef]
- Moradigaravand, D.; Reuter, S.; Martin, V.; Peacock, S.J.; Parkhill, J. The dissemination of multidrug-resistant Enterobacter cloacae throughout the UK and Ireland. Nat. Microbiol. 2016, 1, 16173. [Google Scholar] [CrossRef]
- Zhou, K.; Yu, W.; Cao, X.; Shen, P.; Lu, H.; Luo, Q.; Rossen, J.W.A.; Xiao, Y. Characterization of the population structure, drug resistance mechanisms and plasmids of the community-associated Enterobacter cloacae complex in China. J. Antimicrob. Chemother. 2018, 73, 66–76. [Google Scholar] [CrossRef]
- Morand, P.C.; Billoet, A.; Rottman, M.; Sivadon-Tardy, V.; Eyrolle, L.; Jeanne, L.; Tazi, A.; Anract, P.; Courpied, J.P.; Poyart, C.; et al. Specific distribution within the Enterobacter cloacae complex of strains isolated from infected orthopedic implants. J. Clin. Microbiol. 2009, 47, 2489–2495. [Google Scholar] [CrossRef]
- Mateos, M.; Hernández-García, M.; Del Campo, R.; Martínez-García, L.; Gijón, D.; Morosini, M.I.; Ruiz-Garbajosa, P.; Cantón, R. Emergence and persistence over time of carbapenemase-producing Enterobacter isolates in a Spanish university hospital in Madrid, Spain (2005–2018). Microb. Drug Resist. 2021, 27, 895–903. [Google Scholar] [CrossRef]
- Ji, Y.; Wang, P.; Xu, T.; Zhou, Y.; Chen, R.; Zhu, H.; Zhou, K. Development of a one-step multiplex PCR assay for differential detection of four species (Enterobacter cloacae, Enterobacter hormaechei, Enterobacter roggenkampii, and Enterobacter kobei) belonging to Enterobacter cloacae complex with clinical significance. Front. Cell. Infect. Microbiol. 2021, 11, 677089. [Google Scholar] [CrossRef]
- Hernandez-Alonso, E.; Barreault, S.; Augusto, L.A.; Jatteau, P.; Villet, M.; Tissieres, P.; Doucet-Populaire, F.; Bourgeois-Nicolaos, N.; SENSE Group. dnaJ: A new approach to identify species within the genus Enterobacter. Microbiol. Spectr. 2021, 9, e0124221. [Google Scholar] [CrossRef] [PubMed]
- Peirano, G.; Matsumura, Y.; Adams, M.D.; Bradford, P.; Motyl, M.; Chen, L.; Kreiswirth, B.N.; Pitout, J.D.D. Genomic epidemiology of global carbapenemase-producing Enterobacter spp., 2008–2014. Emerg. Infect. Dis. 2018, 24, 1010–1019. [Google Scholar] [CrossRef] [PubMed]
- Izdebski, R.; Baraniak, A.; Herda, M.; Fiett, J.; Bonten, M.J.; Carmeli, Y.; Goossens, H.; Hryniewicz, W.; Brun-Buisson, C.; Gniadkowski, M.; et al. MLST reveals potentially high-risk international clones of Enterobacter cloacae. J. Antimicrob. Chemother. 2015, 70, 48–56. [Google Scholar] [CrossRef]
- Gomez-Simmonds, A.; Annavajhala, M.K.; Wang, Z.; Macesic, N.; Hu, Y.; Giddins, M.J.; O’Malley, A.; Toussaint, N.C.; Whittier, S.; Torres, V.J.; et al. Genomic and geographic context for the evolution of high-risk carbapenem-resistant Enterobacter cloacae complex clones ST171 and ST78. mBio 2018, 9, e00542-18. [Google Scholar] [CrossRef]
- Harada, K.; Shimizu, T.; Mukai, Y.; Kuwajima, K.; Sato, T.; Kajino, A.; Usui, M.; Tamura, Y.; Kimura, Y.; Miyamoto, T.; et al. Phenotypic and molecular characterization of antimicrobial resistance in Enterobacter spp. isolates from companion animals in Japan. PLoS ONE 2017, 12, e0174178. [Google Scholar] [CrossRef]
- Haenni, M.; Saras, E.; Ponsin, C.; Dahmen, S.; Petitjean, M.; Hocquet, D.; Madec, J.Y. High prevalence of international ESBL CTX-M-15-producing Enterobacter cloacae ST114 clone in animals. J. Antimicrob. Chemother. 2016, 71, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Tetsuka, N.; Hirabayashi, A.; Matsumoto, A.; Oka, K.; Hara, Y.; Morioka, H.; Iguchi, M.; Tomita, Y.; Suzuki, M.; Shibayama, K.; et al. Molecular epidemiological analysis and risk factors for acquisition of carbapenemase-producing Enterobacter cloacae complex in a Japanese university hospital. Antimicrob. Resist. Infect. Control 2019, 8, 126. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.W.; Harris, P.N.A.; Forde, B.M.; Ben Zakour, N.L.; Catchpoole, E.; Stanton-Cook, M.; Phan, M.D.; Sidjabat, H.E.; Bergh, H.; Heney, C.; et al. Integrating multiple genomic technologies to investigate an outbreak of carbapenemase-producing Enterobacter hormaechei. Nat. Commun. 2020, 11, 466. [Google Scholar] [CrossRef] [PubMed]
- Kizny Gordon, A.; Phan, H.T.T.; Lipworth, S.I.; Cheong, E.; Gottlieb, T.; George, S.; Peto, T.E.A.; Mathers, A.J.; Walker, A.S.; Crook, D.W.; et al. Genomic dynamics of species and mobile genetic elements in a prolonged blaIMP-4-associated carbapenemase outbreak in an Australian hospital. J. Antimicrob. Chemother. 2020, 75, 873–882. [Google Scholar] [CrossRef]
- Zong, Z.; Feng, Y.; McNally, A. Carbapenem and colistin resistance in Enterobacter: Determinants and clones. Trends Microbiol. 2021, 29, 473–476. [Google Scholar] [CrossRef] [PubMed]
- Haenni, M.; Métayer, V.; Jarry, R.; Drapeau, A.; Puech, M.P.; Madec, J.Y.; Keck, N. Wide spread of blaCTX-M-9/mcr-9 IncHI2/ST1 plasmids and CTX-M-9-Producing Escherichia coli and Enterobacter cloacae in rescued wild animals. Front. Microbiol. 2020, 11, 601317. [Google Scholar] [CrossRef] [PubMed]
- Macesic, N.; Blakeway, L.V.; Stewart, J.D.; Hawkey, J.; Wyres, K.L.; Judd, L.M.; Wick, R.R.; Jenney, A.W.; Holt, K.E.; Peleg, A.Y. Silent spread of mobile colistin resistance gene mcr-9.1 on IncHI2 “superplasmids” in clinical carbapenem-resistant Enterobacterales. Clin. Microbiol. Infect. 2021, 27, 1856.e7–1856.e13. [Google Scholar] [CrossRef] [PubMed]
- Pati, N.B.; Doijad, S.P.; Schultze, T.; Mannala, G.K.; Yao, Y.; Jaiswal, S.; Ryan, D.; Suar, M.; Gwozdzinski, K.; Bunk, B.; et al. Enterobacter bugandensis: A novel enterobacterial species associated with severe clinical infection. Sci. Rep. 2018, 8, 5392. [Google Scholar] [CrossRef] [PubMed]
- Kubicek-Sutherland, J.Z.; Xie, G.; Shakya, M.; Dighe, P.K.; Jacobs, L.L.; Daligault, H.; Davenport, K.; Stromberg, L.R.; Stromberg, Z.R.; Cheng, Q.; et al. Comparative genomic and phenotypic characterization of invasive non-typhoidal Salmonella isolates from Siaya, Kenya. PLoS Negl. Trop. Dis. 2021, 15, e0008991. [Google Scholar] [CrossRef]
- Gibert, M.; Paytubi, S.; Madrid, C.; Balsalobre, C. Temperature dependent control of the R27 conjugative plasmid genes. Front. Mol. Biosci. 2020, 7, 124. [Google Scholar] [CrossRef]
- Forns, N.; Baños, R.C.; Balsalobre, C.; Juárez, A.; Madrid, C. Temperature-dependent conjugative transfer of R27: Role of chromosome- and plasmid-encoded Hha and H-NS proteins. J. Bacteriol. 2005, 187, 3950–3959. [Google Scholar] [CrossRef] [PubMed]
- Buelow, E.; Rico, A.; Gaschet, M.; Lourenço, J.; Kennedy, S.P.; Wiest, L.; Ploy, M.C.; Dagot, C. Hospital discharges in urban sanitation systems: Long-term monitoring of wastewater resistome and microbiota in relationship to their eco-exposome. Water Res. X 2020, 7, 100045. [Google Scholar] [CrossRef] [PubMed]
- Astolfi, M.C.T.; Carvalho, E.B.S.; de Barros, A.M.; Pinto, M.V.; de Lacerda, L.B.; Nogueira, V.B.; Lopes, E.F.; Astolfi-Filho, S. Draft genome sequence of the novel Enterobacter cloacae strain amazonensis, a highly heavy metal-resistant bacterium from a contaminated stream in Amazonas, Brazil. Genome Announc. 2018, 6, e00450-18. [Google Scholar] [CrossRef]
- Vornhagen, J.; Bassis, C.M.; Ramakrishnan, S.; Hein, R.; Mason, S.; Bergman, Y.; Sunshine, N.; Fan, Y.; Holmes, C.L.; Timp, W.; et al. A plasmid locus associated with Klebsiella clinical infections encodes a microbiome-dependent gut fitness factor. PLoS Pathog. 2021, 17, e1009537. [Google Scholar] [CrossRef]
- Paauw, A.; Caspers, M.P.M.; Leverstein-van Hall, M.A.; Schuren, F.H.J.; Montijn, R.C.; Verhoef, J.; Fluit, A.C. Identification of resistance and virulence factors in an epidemic Enterobacter hormaechei outbreak strain. Microbiology 2009, 155 Pt 5, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.H.; Kikuchi, T.; Tokunaga, T.; Iyoda, S.; Iguchi, A. Diversity of the tellurite resistance gene operon in Escherichia coli. Front. Microbiol. 2021, 12, 681175. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, C.; Perry, N.T.; Godbole, G.; Gharbia, S. Evaluation of chromogenic selective agar (CHROMagar STEC) for the direct detection of Shiga toxin-producing Escherichia coli from faecal specimens. J. Med. Microbiol. 2020, 69, 487–491. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Prevention of Hospital-Acquired Infections: A Practical Guide, 2nd ed.; World Health Organization: Geneva, Switzerland, 2002; pp. 1–72. [Google Scholar]
- Patel, R. Matrix-assisted laser desorption ionization–time of flight mass spectrometry in clinical microbiology. Clin. Infect. Dis. 2013, 57, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Société Française de Microbiologie. CA-SFM/EUCAST: Comité de L’antibiogramme de la Société Française de Microbiologie—Recommandation 2018; Société Française de Microbiologie: Paris, France, 2018; pp. 1–132. [Google Scholar]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef]
- Couvin, D.; Dereeper, A.; Meyer, D.F.; Noroy, C.; Gaete, S.; Bhakkan, B.; Poullet, N.; Gaspard, S.; Bezault, E.; Marcelino, I.; et al. KaruBioNet: A network and discussion group for a better collaboration and structuring of bioinformatics in Guadeloupe (French West Indies). Bioinform. Adv. 2022, 2, vbac010. [Google Scholar] [CrossRef]
- Galaxy Karubionet. Available online: http://www.pasteur-guadeloupe.fr/karubionet.html (accessed on 9 November 2021).
- GitHub—hsp60ECCtool. Available online: https://github.com/karubiotools/hsp60ECCtool (accessed on 9 November 2021).
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods. 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Criscuolo, A.; Brisse, S. AlienTrimmer removes adapter oligonucleotides with high sensitivity in short-insert paired-end reads. Commentary on Turner (2014) Assessment of insert sizes and adapter content in FASTQ data from NexteraXT libraries. Front Genet. 2014, 5, 130. [Google Scholar] [CrossRef]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes de novo assembler. Curr. Protoc. Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Jain, C.; Rodriguez, R.L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 2022, 50, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed]
- GitHub—Mlst. Available online: https://github.com/tseemann/mlst (accessed on 24 February 2020).
- Jolley, K.A.; Bray, J.E.; Maiden, M.C.J. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 2018, 3, 124. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi-Akiyama, T.; Hayakawa, K.; Ohmagari, N.; Shimojima, M.; Kirikae, T. Multilocus sequence typing (MLST) for characterization of Enterobacter cloacae. PLoS ONE 2013, 8, e66358. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef]
- GitHub—Snippy. Available online: https://github.com/tseemann/snippy (accessed on 28 November 2017).
- Didelot, X.; Wilson, D.J. ClonalFrameML: Efficient inference of recombination in whole bacterial genomes. PLoS Comput. Biol. 2015, 11, e1004041. [Google Scholar] [CrossRef]
- GitHub—Abricate. Available online: https://github.com/tseemann/abricate (accessed on 19 April 2020).
- Florensa, A.F.; Kaas, R.S.; Clausen, P.T.L.C.; Aytan-Aktug, D.; Aarestrup, F.M. ResFinder—An open online resource for identification of antimicrobial resistance genes in next-generation sequencing data and prediction of phenotypes from genotypes. Microb. Genom. 2022, 8, 000748. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Matlock, W.; Chau, K.K.; AbuOun, M.; Stubberfield, E.; Barker, L.; Kavanagh, J.; Pickford, H.; Gilson, D.; Smith, R.P.; Gweon, H.S.; et al. Genomic network analysis of environmental and livestock F-type plasmid populations. ISME J. 2021, 15, 2322–2335. [Google Scholar] [CrossRef]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef]
- Alikhan, N.F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple prokaryote genome comparisons. BMC Genom. 2011, 12, 402. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef]
- Davis, J.J.; Wattam, A.R.; Aziz, R.K.; Brettin, T.; Butler, R.; Butler, R.M.; Chlenski, P.; Conrad, N.; Dickerman, A.; Dietrich, E.M.; et al. The PATRIC bioinformatics resource center: Expanding data and analysis capabilities. Nucleic Acids Res. 2020, 48, D606–D612. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pot, M.; Reynaud, Y.; Couvin, D.; Dereeper, A.; Ferdinand, S.; Bastian, S.; Foucan, T.; Pommier, J.-D.; Valette, M.; Talarmin, A.; et al. Emergence of a Novel Lineage and Wide Spread of a blaCTX-M-15/IncHI2/ST1 Plasmid among Nosocomial Enterobacter in Guadeloupe. Antibiotics 2022, 11, 1443. https://doi.org/10.3390/antibiotics11101443
Pot M, Reynaud Y, Couvin D, Dereeper A, Ferdinand S, Bastian S, Foucan T, Pommier J-D, Valette M, Talarmin A, et al. Emergence of a Novel Lineage and Wide Spread of a blaCTX-M-15/IncHI2/ST1 Plasmid among Nosocomial Enterobacter in Guadeloupe. Antibiotics. 2022; 11(10):1443. https://doi.org/10.3390/antibiotics11101443
Chicago/Turabian StylePot, Matthieu, Yann Reynaud, David Couvin, Alexis Dereeper, Séverine Ferdinand, Sylvaine Bastian, Tania Foucan, Jean-David Pommier, Marc Valette, Antoine Talarmin, and et al. 2022. "Emergence of a Novel Lineage and Wide Spread of a blaCTX-M-15/IncHI2/ST1 Plasmid among Nosocomial Enterobacter in Guadeloupe" Antibiotics 11, no. 10: 1443. https://doi.org/10.3390/antibiotics11101443
APA StylePot, M., Reynaud, Y., Couvin, D., Dereeper, A., Ferdinand, S., Bastian, S., Foucan, T., Pommier, J.-D., Valette, M., Talarmin, A., Guyomard-Rabenirina, S., & Breurec, S. (2022). Emergence of a Novel Lineage and Wide Spread of a blaCTX-M-15/IncHI2/ST1 Plasmid among Nosocomial Enterobacter in Guadeloupe. Antibiotics, 11(10), 1443. https://doi.org/10.3390/antibiotics11101443