

Antileishmanial Activity of 4,8-Dimethoxynaphthalenyl Chalcones on Leishmania amazonensis

 , , , , , ,
, , , , , ,  and
and 
Abstract
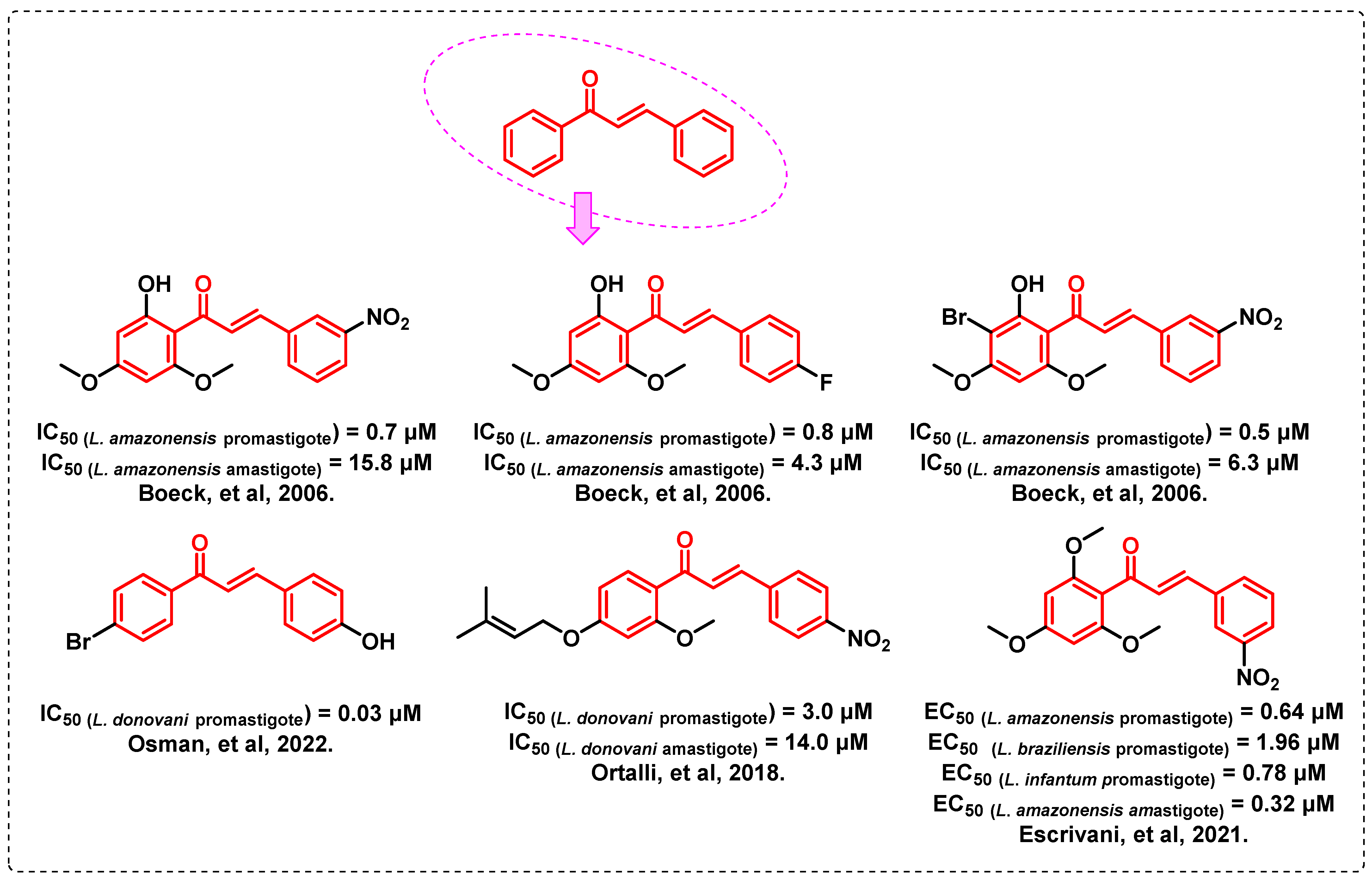
1. Introduction
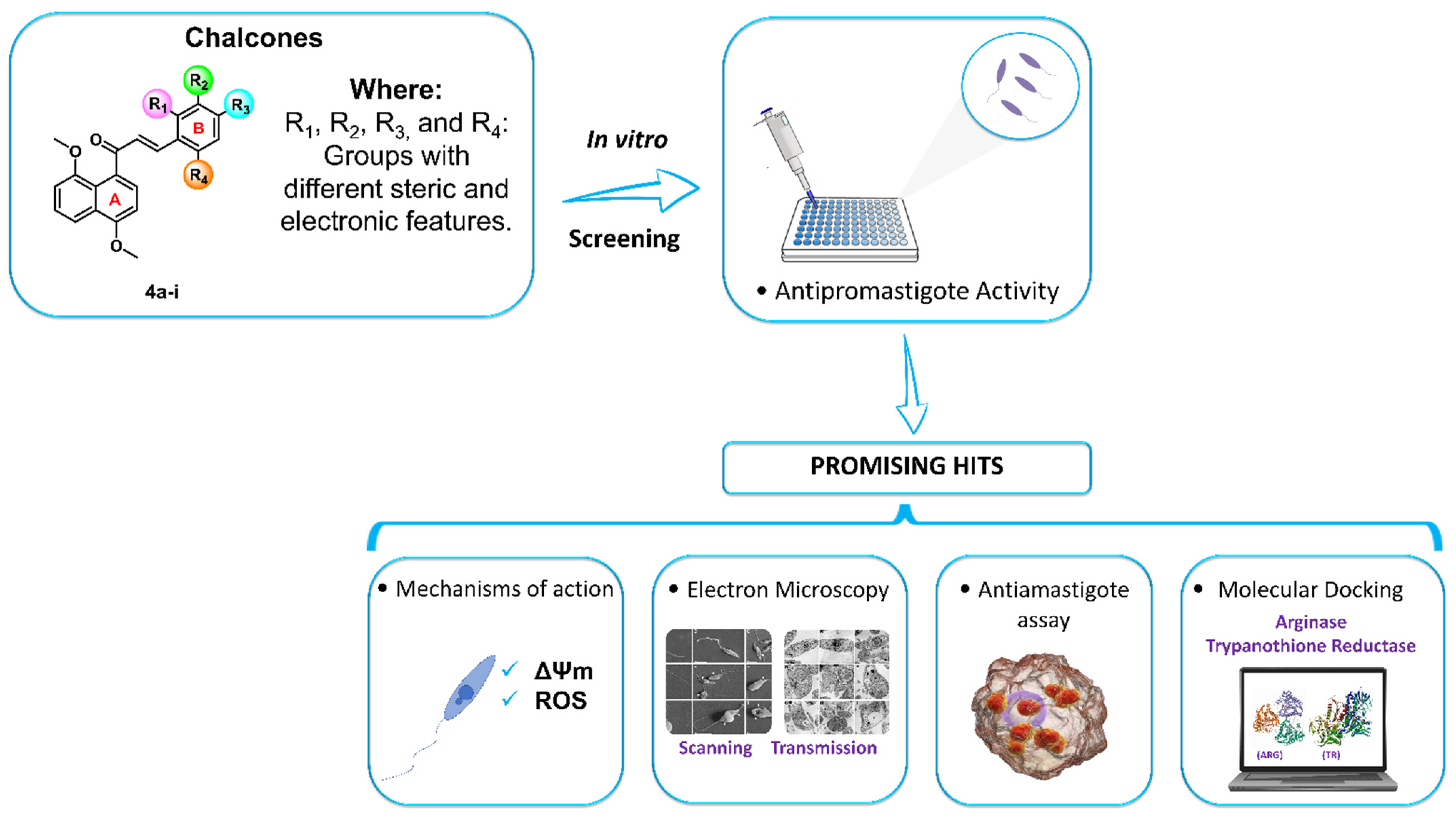
2. Results and Discussion
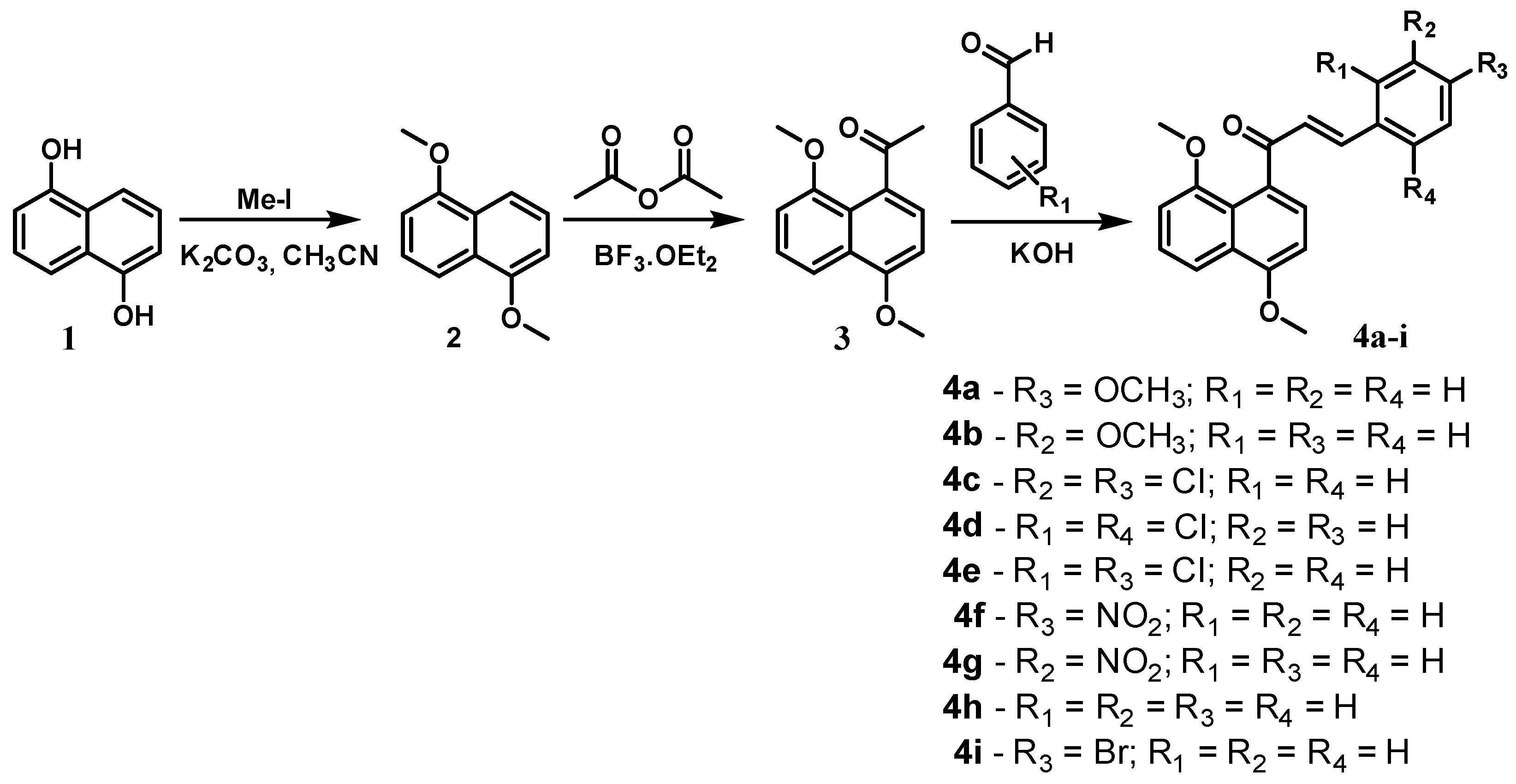
2.1. Chemistry
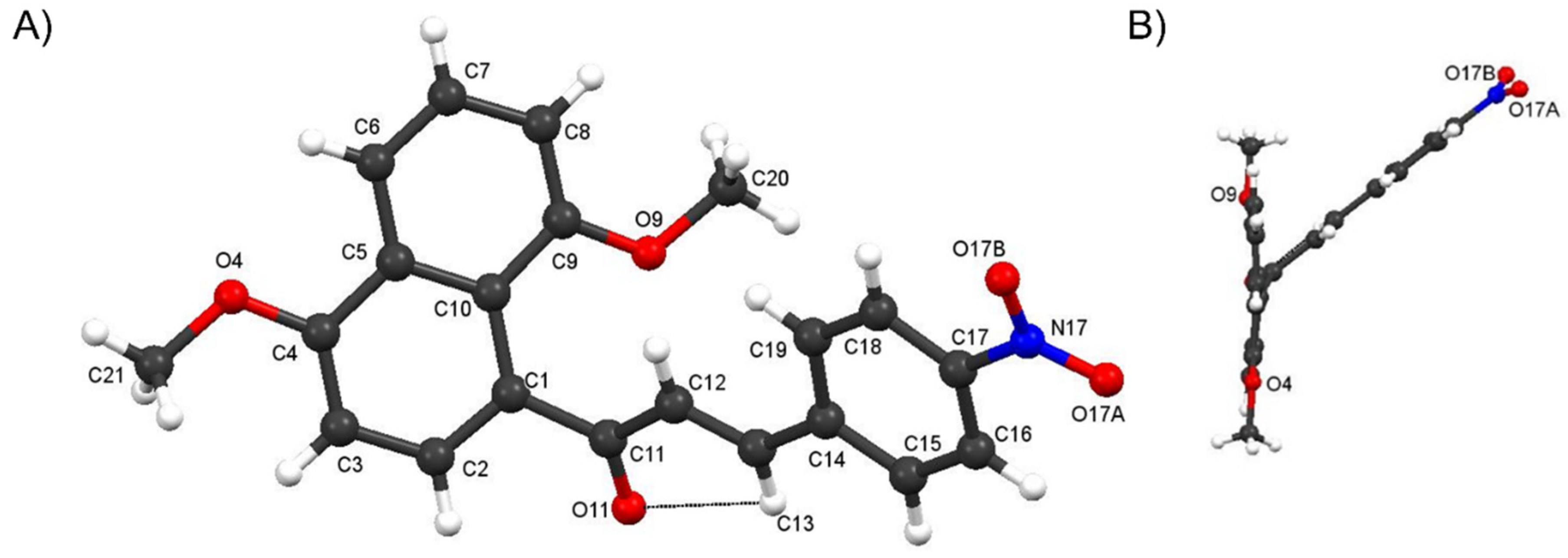
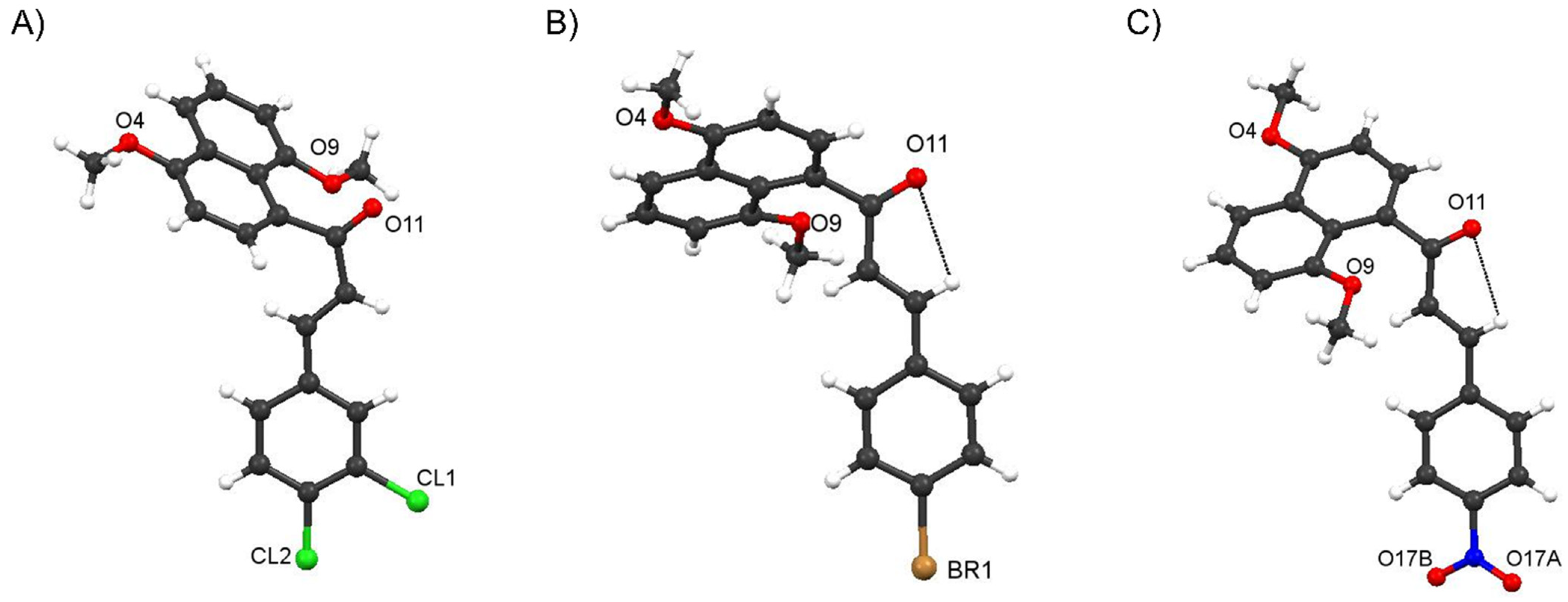
Crystal Structure of Compound 4f and comparisons with those of 4c and 4i
2.2. Biological Evaluation
2.2.1. Cytotoxicity and Antipromastigote Activity In Vitro of the 4,8-Dimethoxynaphthalenyl Chalcone Derivatives (4a–i)
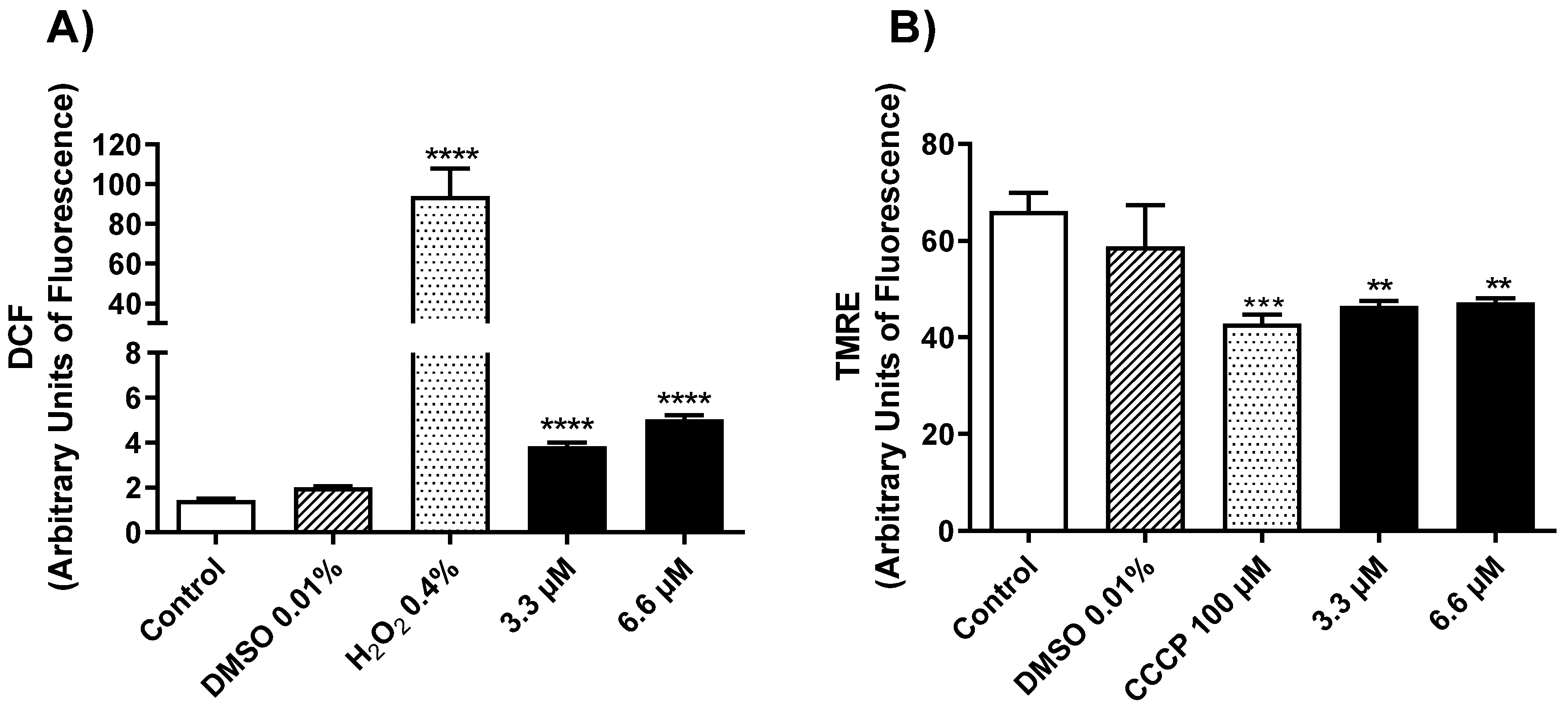
2.2.2. Mechanism of Action in Promastigotes of L. amazonensis
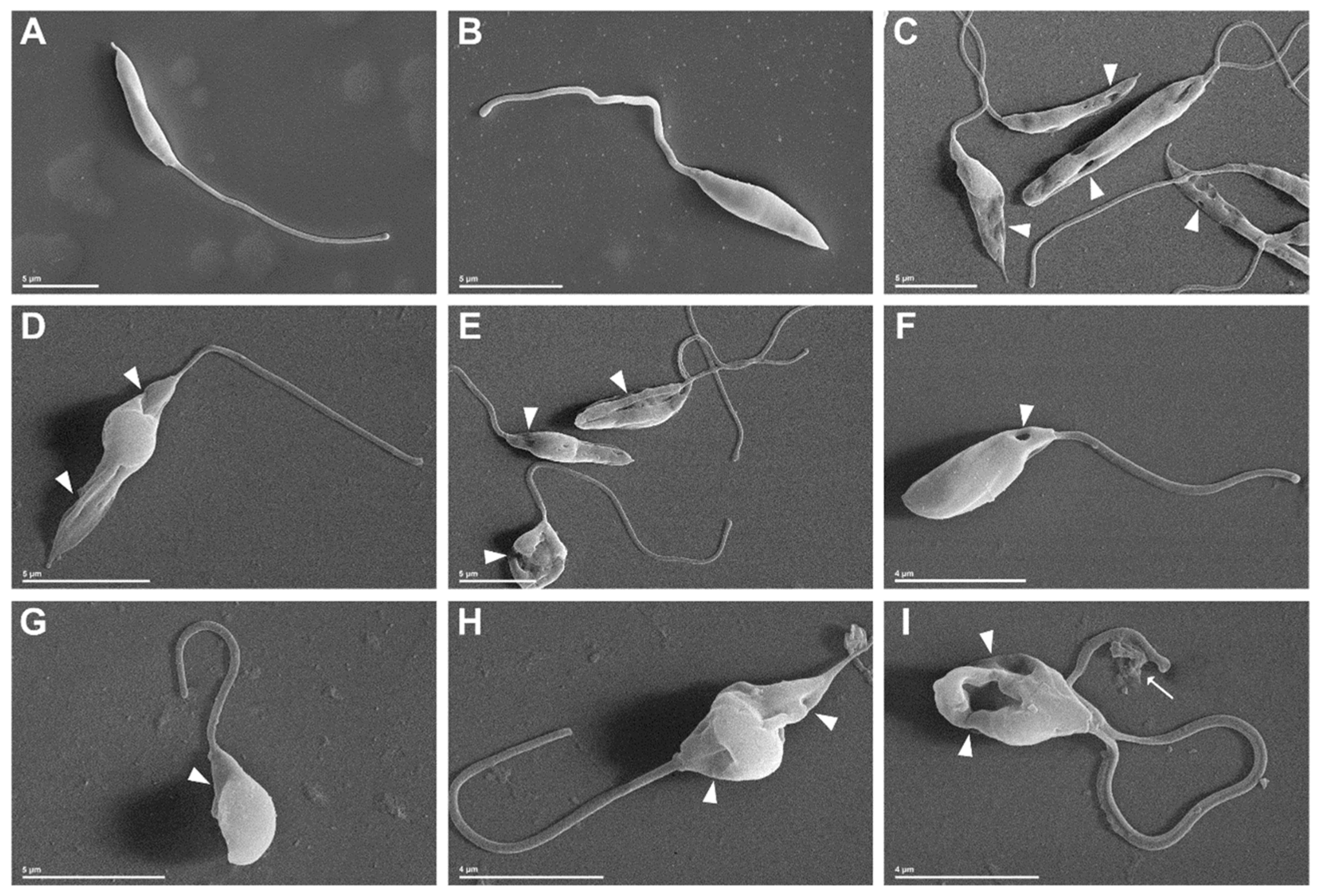
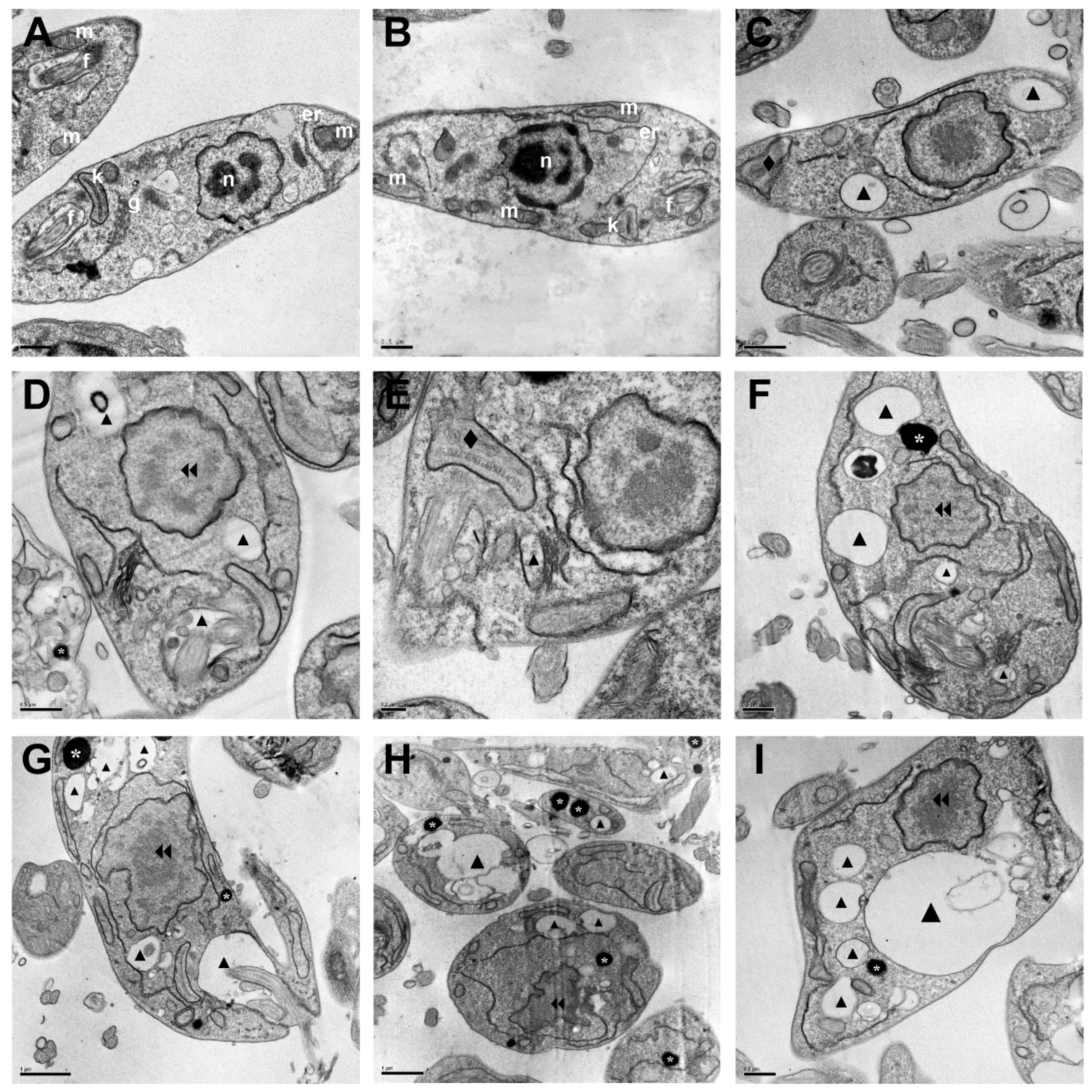
2.2.3. Morphological and Ultrastructural Changes in Promastigotes
2.2.4. Evaluation of Antileishmanial Activity on L. amazonensis-Intracellular Amastigotes Forms
2.2.5. In Silico Study to Predict Pharmacokinetic and Toxicity Parameters ADME Prediction
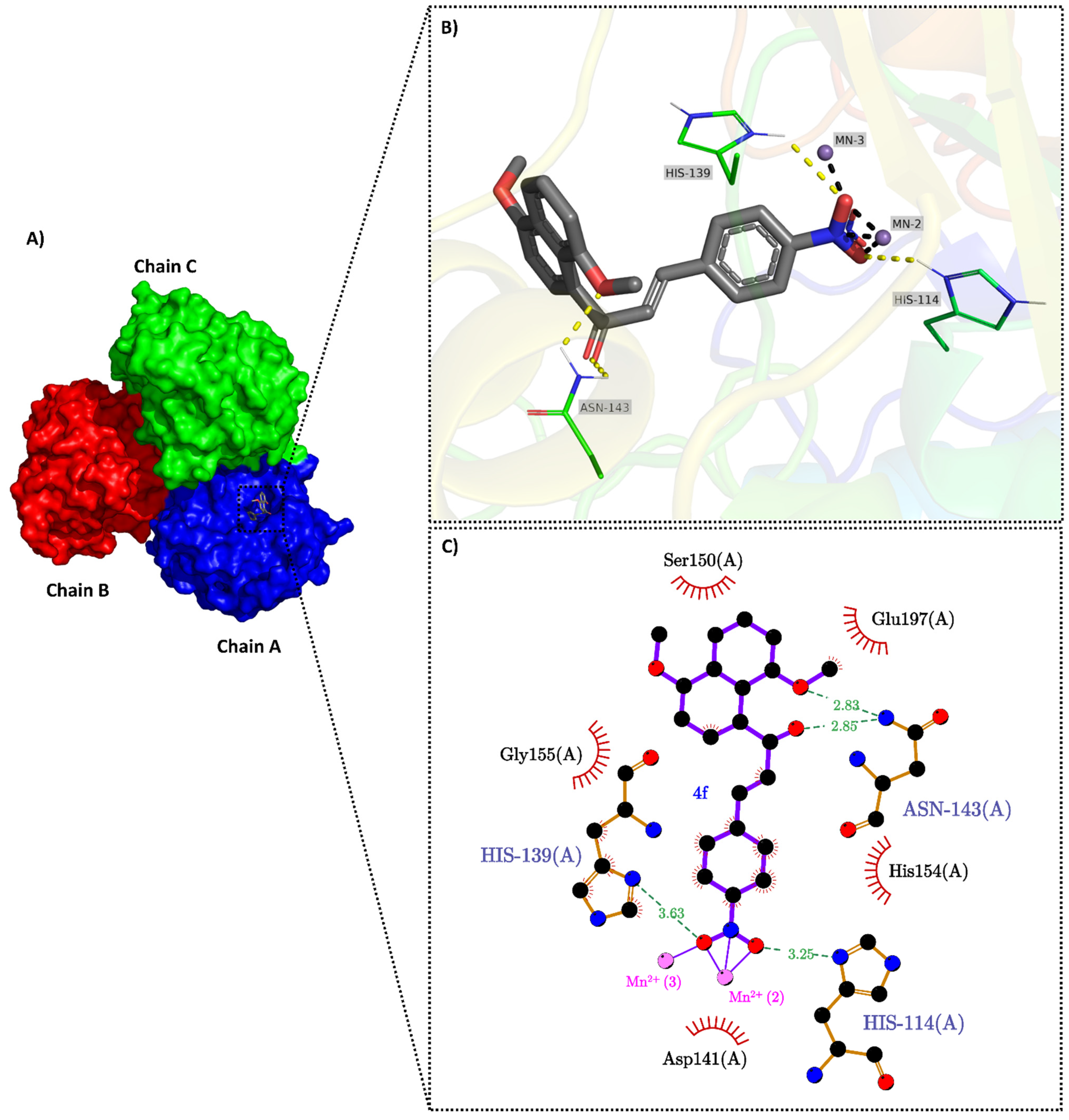
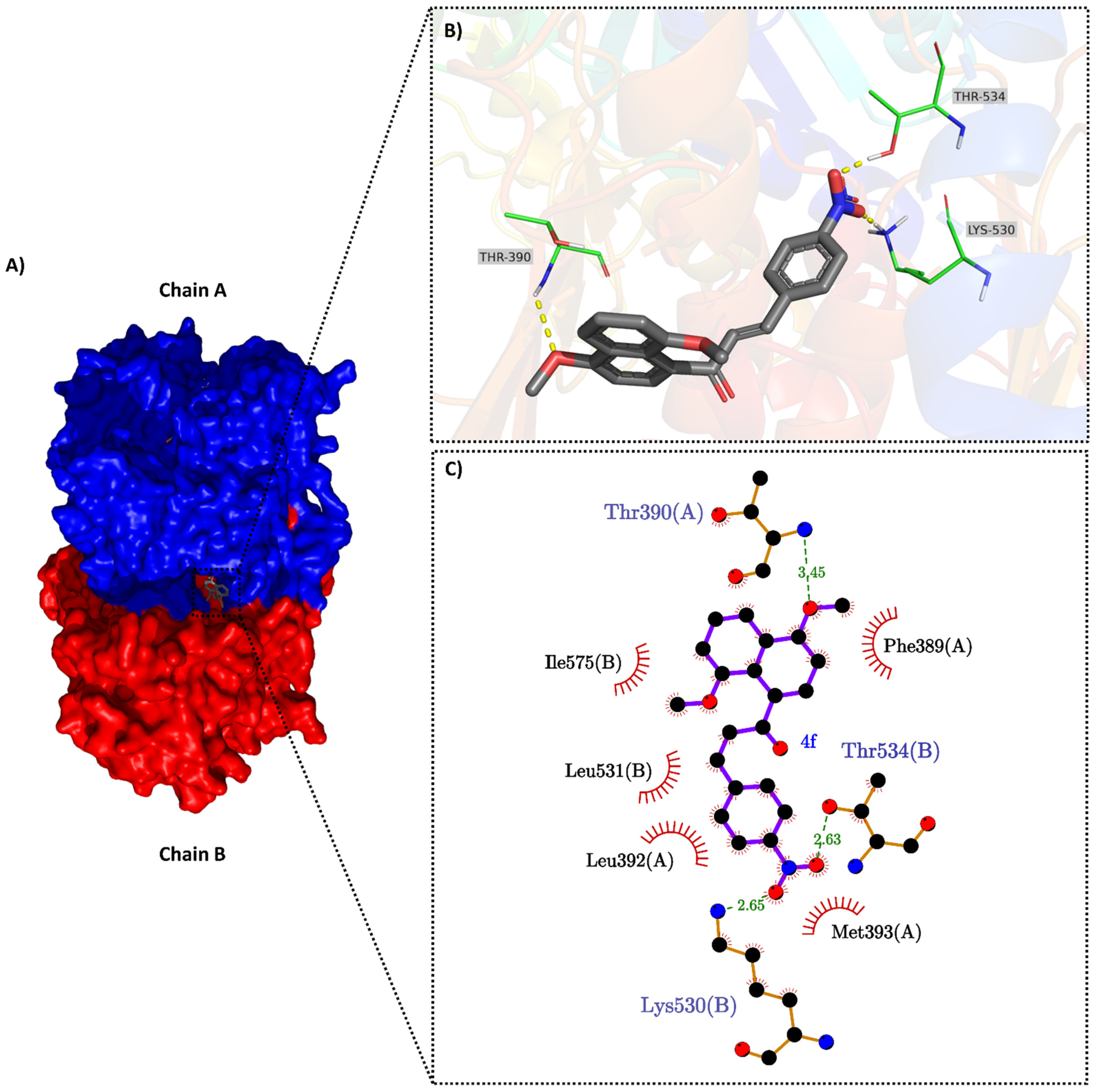
2.3. Molecular Docking
3. Materials and Methods
3.1. General Information
3.2. Chemistry
3.2.1. Synthesis of 1,5-Dimethoxynaphthalene (2)
3.2.2. Synthesis of 1-(1,5-Dimethoxynaphthalen-8-yl)ethenone (3)
3.2.3. General Procedure for the Synthesis of 4,8-Dimethoxynaphthalenyl Chalcone Derivatives (4a–i)
3.2.4. Experimental X-ray Structure Determination
3.3. Biological Assays
3.3.1. Leishmania (Leishmania) Amazonensis Maintenance
3.3.2. Effect of the Compounds on L. amazonensis Promastigotes
3.3.3. Evaluation of Cytotoxicity on Murine Macrophages
3.3.4. Selectivity Index (SI)
3.3.5. Determination of Mitochondrial Membrane Potential (ΔΨm)
3.3.6. Reactive Oxygen Species (ROS) Generation in L. amazonensis Promastigotes
3.3.7. Scanning Electron Microscopy (SEM) and Transmission Electron Microscopy (TEM)
3.3.8. Antiamastigote Assay
3.3.9. Statistical Analysis
3.4. In Silico ADME Predictions
3.5. Molecular Docking
3.5.1. Proteins and Ligands Preparation for Docking
3.5.2. Consensus Molecular Docking
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Akhoundi, M.; Kuhls, K.; Cannet, A.; Votýpka, J.; Marty, P.; Delaunay, P.; Sereno, D. A Historical Overview of the Classification, Evolution, and Dispersion of Leishmania Parasites and Sandflies. PLoS Negl. Trop. Dis. 2016, 10, e0004349. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Leishmaniasis. 2022. Available online: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (accessed on 8 March 2022).
- Sundar, S.; Chakravarty, J. An update on pharmacotherapy for leishmaniasis. Expert Opin. Pharmacother. 2015, 16, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, M.; Simoens, M.; Falchi, G.; Lavaggi, M.L.; Piro, O.E.; Castellano, E.E.; Vidal, A.; Azqueta, A.; Monge, A.; de Ceráin, A.L.; et al. Synthetic chalcones, flavanones, and flavones as antitumoral agents: Biological evaluation and structure–activity relationships. Bioorg. Med. Chem. 2007, 15, 3356–3367. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef] [PubMed]
- Sahu, N.K.; Balbhadra, S.S.; Choudhary, J.; Kohli, D.V. Exploring Pharmacological Significance of Chalcone Scaffold: A Review. Curr. Med. Chem. 2012, 19, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Boeck, P.; Bandeira Falcão, C.A.; Leal, P.C.; Yunes, R.A.; Filho, V.C.; Torres-Santos, E.C.; Rossi-Bergmann, B. Synthesis of chalcone analogues with increased antileishmanial activity. Bioorg. Med. Chem. 2006, 14, 1538–1545. [Google Scholar] [CrossRef] [PubMed]
- Osman, M.S.; Awad, T.A.; Shantier, S.W.; Garelnabi, E.A.; Osman, W.; Mothana, R.A.; Nasr, F.A.; Elhag, R.I. Identification of some chalcone analogues as potential antileishmanial agents: An integrated in vitro and in silico evaluation. Arab. J. Chem. 2022, 15, 103717. [Google Scholar] [CrossRef]
- Ortalli, M.; Ilari, A.; Colotti, G.; De Ionna, I.; Battista, T.; Bisi, A.; Gobbi, S.; Rampa, A.; Di Martino, R.M.C.; Gentilomi, G.A.; et al. Identification of chalcone-based antileishmanial agents targeting trypanothione reductase. Eur. J. Med. Chem. 2018, 152, 527–541. [Google Scholar] [CrossRef] [PubMed]
- Escrivani, D.O.; Charlton, R.L.; Caruso, M.B.; Burle-Caldas, G.A.; Borsodi, M.P.G.; Zingali, R.B.; Arruda-Costa, N.; Palmeira-Mello, M.V.; de Jesus, J.B.; Souza, A.M.T.; et al. Chalcones identify cTXNPx as a potential antileishmanial drug target. PLoS Negl. Trop. Dis. 2021, 15, e0009951. [Google Scholar] [CrossRef]
- Aoki, J.I.; Laranjeira-Silva, M.F.; Muxel, S.M.; Floeter-Winter, L.M. The impact of arginase activity on virulence factors of Leishmania amazonensis. Curr. Opin. Microbiol. 2019, 52, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Battista, T.; Colotti, G.; Ilari, A.; Fiorillo, A. Targeting Trypanothione Reductase, a Key Enzyme in the Redox Trypanosomatid Metabolism, to Develop New Drugs against Leishmaniasis and Trypanosomiases. Molecules 2020, 25, 1924. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, M.G.; Gonçalves, R.S.B.; Lima, C.H.d.S.; Maia, F.L.d.A.; Machado, S.d.P.; Oliveira, L.d.N.; da Silva, T.U.; Wardell, J.L.; Wardell, S.M.S.V. Crystal structures, DFT calculations and Hirshfeld surface analysis of two (E)-3-(aryl)-1-(naphthalen-1-yl)prop-2-en-1-one chalcone derivatives, potential Mycobacterium tuberculosis Enoyl ACP reductase (InhA) inhibitors and optical materials: Conformation. J. Mol. Struct. 2021, 1246, 131091. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Fujita, T. The ortho effect in quantitative structure—Activity correlations. Anal. Chim. Acta 1981, 133, 667–676. [Google Scholar] [CrossRef]
- Katsuno, K.; Burrows, J.N.; Duncan, K.; van Huijsduijnen, R.H.; Kaneko, T.; Kita, K.; Mowbray, C.E.; Schmatz, D.; Warner, P.; Slingsby, B.T. Hit and lead criteria in drug discovery for infectious diseases of the developing world. Nat. Rev. Drug Discov. 2015, 14, 751–758. [Google Scholar] [CrossRef]
- Fang, F.C. Antimicrobial reactive oxygen and nitrogen species: Concepts and controversies. Nat. Rev. Microbiol. 2004, 2, 820–832. [Google Scholar] [CrossRef]
- Smirlis, D.; Duszenko, M.; Ruiz, A.; Scoulica, E.; Bastien, P.; Fasel, N.; Soteriadou, K. Targeting essential pathways in trypanosomatids gives insights into protozoan mechanisms of cell death. Parasit. Vectors 2010, 3, 107. [Google Scholar] [CrossRef]
- de Mello, T.F.P.; Bitencourt, H.R.; Pedroso, R.B.; Aristides, S.M.A.; Lonardoni, M.V.C.; Silveira, T.G.V. Leishmanicidal activity of synthetic chalcones in Leishmania (Viannia) braziliensis. Exp. Parasitol. 2014, 136, 27–34. [Google Scholar] [CrossRef]
- Aponte, J.C.; Castillo, D.; Estevez, Y.; Gonzalez, G.; Arevalo, J.; Hammond, G.B.; Sauvain, M. In vitro and in vivo anti-Leishmania activity of polysubstituted synthetic chalcones. Bioorg. Med. Chem. Lett. 2010, 20, 100–103. [Google Scholar] [CrossRef]
- Gupta, S.; Shivahare, R.; Korthikunta, V.; Singh, R.; Gupta, S.; Tadigoppula, N. Synthesis and biological evaluation of chalcones as potential antileishmanial agents. Eur. J. Med. Chem. 2014, 81, 359–366. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Delaney, J.S. ESOL: Estimating Aqueous Solubility Directly from Molecular Structure. J. Chem. Inf. Comput. Sci. 2004, 44, 1000–1005. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, S.; Chhibber, T.; Lahooti, B.; Verma, A.; Borse, V.; Jayant, R.D. In-vitro blood-brain barrier models for drug screening and permeation studies: An overview. Drug Des. Devel. Ther. 2019, 13, 3591–3605. [Google Scholar] [CrossRef] [PubMed]
- Vilar, S.; Sobarzo-Sánchez, E.; Uriarte, E. In Silico Prediction of P-glycoprotein Binding: Insights from Molecular Docking Studies. Curr. Med. Chem. 2019, 26, 1746–1760. [Google Scholar] [CrossRef]
- Wasukan, N.; Kuno, M.; Maniratanachote, R. Molecular Docking as a Promising Predictive Model for Silver Nanoparticle-Mediated Inhibition of Cytochrome P450 Enzymes. J. Chem. Inf. Model. 2019, 59, 5126–5134. [Google Scholar] [CrossRef]
- Desta, Z.; Zhao, X.; Shin, J.-G.; Flockhart, D.A. Clinical Significance of the Cytochrome P450 2C19 Genetic Polymorphism. Clin. Pharmacokinet. 2002, 41, 913–958. [Google Scholar] [CrossRef]
- Zhou, S.-F. Drugs Behave as Substrates, Inhibitors and Inducers of Human Cytochrome P450 3A4. Curr. Drug Metab. 2008, 9, 310–322. [Google Scholar] [CrossRef]
- Miners, J.O.; Birkett, D.J. Cytochrome P4502C9: An enzyme of major importance in human drug metabolism. Br. J. Clin. Pharmacol. 1998, 45, 525–538. [Google Scholar] [CrossRef]
- Ortiz de Montellano, P.R. Hydrocarbon Hydroxylation by Cytochrome P450 Enzymes. Chem. Rev. 2010, 110, 932–948. [Google Scholar] [CrossRef]
- D’Antonio, E.L.; Ullman, B.; Roberts, S.C.; Dixit, U.G.; Wilson, M.E.; Hai, Y.; Christianson, D.W. Crystal structure of arginase from Leishmania mexicana and implications for the inhibition of polyamine biosynthesis in parasitic infections. Arch. Biochem. Biophys. 2013, 535, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.; Smith, K.; Fairlamb, A.H.; Hunter, W.N. Substrate interactions between trypanothione reductase and N1-glutathionylspermidine disulphide at 0.28-nm resolution. Eur. J. Biochem. 1993, 213, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Feher, M. Consensus scoring for protein–ligand interactions. Drug Discov. Today 2006, 11, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Reczkowski, R.S.; Ash, D.E. EPR evidence for binuclear manganese(II) centers in rat liver arginase. J. Am. Chem. Soc. 1992, 114, 10992–10994. [Google Scholar] [CrossRef]
- Méndez-Cuesta, C.A.; Méndez-Lucio, O.; Castillo, R. Homology modeling, docking and molecular dynamics of the Leishmania mexicana arginase: A description of the catalytic site useful for drug design. J. Mol. Graph. Model. 2012, 38, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, R.B.; Toque, H.A.; Narayanan, S.P.; Caldwell, R.W. Arginase: An old enzyme with new tricks. Trends Pharmacol. Sci. 2015, 36, 395–405. [Google Scholar] [CrossRef]
- Baiocco, P.; Colotti, G.; Franceschini, S.; Ilari, A. Molecular Basis of Antimony Treatment in Leishmaniasis. J. Med. Chem. 2009, 52, 2603–2612. [Google Scholar] [CrossRef]
- O’Sullivan, M.C.; Durham, T.B.; Valdes, H.E.; Dauer, K.L.; Karney, N.J.; Forrestel, A.C.; Bacchi, C.J.; Baker, J.F. Dibenzosuberyl substituted polyamines and analogs of clomipramine as effective inhibitors of trypanothione reductase; molecular docking, and assessment of trypanocidal activities. Bioorg. Med. Chem. 2015, 23, 996–1010. [Google Scholar] [CrossRef]
- Salmon-Chemin, L.; Buisine, E.; Yardley, V.; Kohler, S.; Debreu, M.-A.; Landry, V.; Sergheraert, C.; Croft, S.L.; Krauth-Siegel, R.L.; Davioud-Charvet, E. 2- and 3-Substituted 1,4-Naphthoquinone Derivatives as Subversive Substrates of Trypanothione Reductase and Lipoamide Dehydrogenase from Trypanosomacruzi: Synthesis and Correlation between Redox Cycling Activities and in Vitro Cytotoxicity. J. Med. Chem. 2001, 44, 548–565. [Google Scholar] [CrossRef]
- Armarego, W.L.F. Purification of Laboratory Chemicals, 8th ed.; Armarego, W.L.F., Ed.; Butterworth-Heinemann: Oxford, UK, 2017; ISBN 978-0-12-805457-4. [Google Scholar]
- Buu-Hoi, N.P.; Lavit, D. Compounds with potential activity against lethal radiations. v. 1 methyl homologs of 1,5-dihydroxynaphthalene. J. Org. Chem. 1955, 20, 1191–1196. [Google Scholar] [CrossRef]
- CrysAlisPRO Software System; Version 1.171.40.39; Rigaku Oxford Diffraction, Rigaku Corporation: Oxford, UK, 2017.
- Farrugia, L.J. ORTEP -3 for Windows—A version of ORTEP -III with a Graphical User Interface (GUI). J. Appl. Crystallogr. 1997, 30, 565. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef] [PubMed]
- Rudik, A.; Dmitriev, A.; Lagunin, A.; Filimonov, D.; Poroikov, V. SOMP: Web server for in silico prediction of sites of metabolism for drug-like compounds. Bioinformatics 2015, 31, 2046–2048. [Google Scholar] [CrossRef] [PubMed]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Cassarino, T.G.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef] [PubMed]
- Ilari, A.; Genovese, I.; Fiorillo, F.; Battista, T.; De Ionna, I.; Fiorillo, A.; Colotti, G. Toward a Drug Against All Kinetoplastids: From LeishBox to Specific and Potent Trypanothione Reductase Inhibitors. Mol. Pharm. 2018, 15, 3069–3078. [Google Scholar] [CrossRef]
- Camargo, P.G.; Bortoleti, B.T.d.S.; Fabris, M.; Gonçalves, M.D.; Tomiotto-Pellissier, F.; Costa, I.N.; Conchon-Costa, I.; Lima, C.H.d.S.; Pavanelli, W.R.; Bispo, M.d.L.F.; et al. Thiohydantoins as anti-leishmanial agents: N vitro biological evaluation and multi-target investigation by molecular docking studies. J. Biomol. Struct. Dyn. 2022, 40, 3213–3222. [Google Scholar] [CrossRef]
- Evans, D.A. History of the Harvard ChemDraw Project. Angew. Chemie Int. Ed. 2014, 53, 11140–11145. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J. MOPAC2016. Stewart Computational Chemistry. 2016. Available online: http://openmopac.net (accessed on 1 September 2022).
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| # | R1 | R2 | R3 | R4 | IC50 (μM) a | CC50 (μM) b | SI c |
| 4a | H | H | OCH3 | H | 264.1 ± 0.12 | 414.8 ± 0.04 | 1.7 |
| 4b | H | OCH3 | H | H | 67.1 ± 0.16 | 246.7 ± 0.03 | 3.7 |
| 4c | H | Cl | Cl | H | 13.4 ± 0.08 | 194.3 ± 0.06 | 14.5 |
| 4d | Cl | H | H | Cl | 94.1 ± 0.06 | 126.8 ± 0.09 | 3.4 |
| 4e | Cl | H | Cl | H | 37.7 ± 0.06 | 561.1 ± 0.03 | 5.9 |
| 4f | H | H | NO2 | H | 3.3 ± 0.34 | 372.9 ± 0.04 | 112.6 |
| 4g | H | NO2 | H | H | 14.5 ± 0.09 | 366.0 ± 0.13 | 23.2 |
| 4h | H | H | H | H | 26.1 ± 0.09 | 872.3 ± 0.1338 | 33.44 |
| 4i | H | H | Br | H | 24.3 ± 0.05 | 434.0 ± 0.05 | 17.8 |
| AmB d | ------ | ------ | ------ | ------ | 0.68 ± 0.21 | 49.72 ± 0.00 | 73.17 |
| # | 4f | ||
|---|---|---|---|
 | Physicochemical | MW (g∙mol−1) (≤500) * | 363.36 |
| cLog Po/w (≤5) * | 3.53 | ||
| HBA (≤10) * | 5 | ||
| HBD (≤5) * | 0 | ||
| HBA + HBD (≤12) * | 5 | ||
| RB (≤10) * | 6 | ||
| tPSA (≤140 Å2) * | 81.35 | ||
| N° Violations * | 0 | ||
| Log S (ESOL) * | −5.10 | ||
| Pharmacokinetic | Absorption GI * | High | |
| BBB permeability * | No | ||
| P-glycoprotein substrate * | No | ||
| CYP1A2 inhibitor ** | No | ||
| CYP1A2 substrate ** | Yes | ||
| CYP2C19 inhibitor ** | Yes | ||
| CYP2C19 substrate ** | No | ||
| CYP2C9 inhibitor ** | Yes | ||
| CYP2C9 substrate ** | Yes | ||
| CYP2D6 inhibitor ** | No | ||
| CYP2D6 substrate ** | Yes | ||
| CYP3A4 inhibitor | Yes | ||
| CYP3A4 substrate | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Santiago-Silva, K.M.; Bortoleti, B.T.d.S.; Oliveira, L.d.N.; Maia, F.L.d.A.; Castro, J.C.; Costa, I.C.; Lazarin, D.B.; Wardell, J.L.; Wardell, S.M.S.V.; Albuquerque, M.G.; et al. Antileishmanial Activity of 4,8-Dimethoxynaphthalenyl Chalcones on Leishmania amazonensis. Antibiotics 2022, 11, 1402. https://doi.org/10.3390/antibiotics11101402
de Santiago-Silva KM, Bortoleti BTdS, Oliveira LdN, Maia FLdA, Castro JC, Costa IC, Lazarin DB, Wardell JL, Wardell SMSV, Albuquerque MG, et al. Antileishmanial Activity of 4,8-Dimethoxynaphthalenyl Chalcones on Leishmania amazonensis. Antibiotics. 2022; 11(10):1402. https://doi.org/10.3390/antibiotics11101402
Chicago/Turabian Stylede Santiago-Silva, Kaio Maciel, Bruna Taciane da Silva Bortoleti, Laudicéa do Nascimento Oliveira, Fernanda Lima de Azevedo Maia, Joyce Cristina Castro, Ivete Conchon Costa, Danielle Bidóia Lazarin, James L. Wardell, Solange M. S. V. Wardell, Magaly Girão Albuquerque, and et al. 2022. "Antileishmanial Activity of 4,8-Dimethoxynaphthalenyl Chalcones on Leishmania amazonensis" Antibiotics 11, no. 10: 1402. https://doi.org/10.3390/antibiotics11101402
APA Stylede Santiago-Silva, K. M., Bortoleti, B. T. d. S., Oliveira, L. d. N., Maia, F. L. d. A., Castro, J. C., Costa, I. C., Lazarin, D. B., Wardell, J. L., Wardell, S. M. S. V., Albuquerque, M. G., Lima, C. H. d. S., Pavanelli, W. R., Bispo, M. d. L. F., & Gonçalves, R. S. B. (2022). Antileishmanial Activity of 4,8-Dimethoxynaphthalenyl Chalcones on Leishmania amazonensis. Antibiotics, 11(10), 1402. https://doi.org/10.3390/antibiotics11101402