
In Vitro and In Silico Approaches for the Evaluation of Antimicrobial Activity, Time-Kill Kinetics, and Anti-Biofilm Potential of Thymoquinone (2-Methyl-5-propan-2-ylcyclohexa-2,5-diene-1,4-dione) against Selected Human Pathogens

 ,
,  , ,
, ,
Abstract
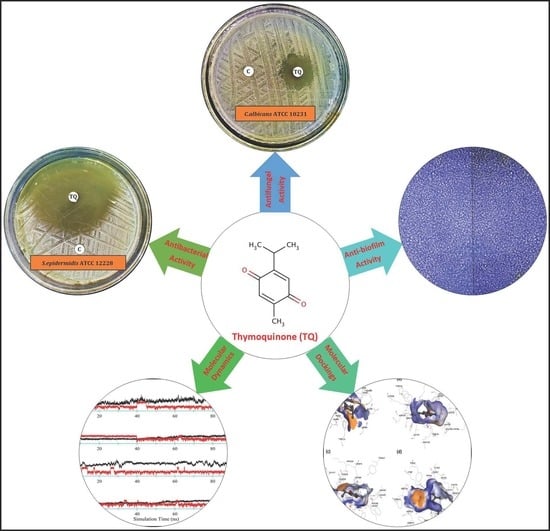
1. Introduction
2. Results
2.1. Antimicrobial Susceptibility Screening
2.2. Preliminary Antimicrobial Activity
2.3. Minimum Inhibitory Concentration (MIC) and Minimum Biocidal Concentration (MBC)
2.4. Time-Kill Kinetics Assay
2.5. Minimum Biofilm Inhibitory Concentration (MBIC) and Minimum Biofilm Eradication
Concentration (MBEC)
2.6. Statistical Analysis
2.7. In Silico Molecular Docking of TQ with Antimicrobial Enzymes
2.7.1. MD Simulations of TQ-Enzyme Complexes
Protein and TQ Stability
TQ-Induced Protein Flexibility
Distance Fluctuation between TQ and Enzymes and TQ Dynamics
Global Conformational Changes in Enzymes
Enzyme-TQ Binding Energy
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Test Organisms
4.3. Preliminary Antimicrobial Activity
4.4. MIC and MBC
4.5. Time-Kill Kinetics Assay
4.6. MBIC and MBEC Assay of TQ
4.7. Statistical Analysis
4.8. In Silico Molecular Dockings of TQ
4.8.1. Identification of Target Proteins
4.9. MD Simulations Protocol
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Qureshi, K.A.; Bholay, A.D.; Rai, P.K.; Mohammed, H.A.; Khan, R.A.; Azam, F.; Jaremko, M.; Emwas, A.-H.; Stefanowicz, P.; Waliczek, M.; et al. Isolation, characterization, anti-MRSA evaluation, and in-silico multi-target anti-microbial validations of actinomycin X2 and actinomycin D produced by novel Streptomyces smyrnaeus UKAQ_23. Sci. Rep. 2021, 11, 14539. [Google Scholar] [CrossRef] [PubMed]
- Alanazi, A.S.; Qureshi, K.A.; Elhassan, G.O.; El-Agam, E.I. Isolation, Purification and Characterization of Antimicrobial Agent Antagonistic to Escherichia coli ATCC 10536 Produced by Bacillus pumilus SAFR-032 Isolated from the Soil of Unaizah, Al Qassim Province of Saudi Arabia. Pak. J. Biol. Sci. 2016, 19, 191–201. [Google Scholar] [CrossRef]
- Qureshi, K.A.; Seroor, M.; AI-Masabi, A.; Saykhan, M.A.; Mutairi, Y.A.; Elhassan, G.O.; Khan, R.A. Bio-characterizations of some marine bacterial strains isolated from mangrove sediment samples of four major cities of Saudi Arabia. J. Environ. Biol. 2020, 41, 1003–1012. [Google Scholar] [CrossRef]
- Mohammed, H.A.; Al-Omar, M.S.; Khan, R.A.; Mohammed, S.A.A.; Qureshi, K.A.; Abbas, M.M.; Al Rugaie, O.; Abd-Elmoniem, E.; Ahmad, A.M.; Kandil, Y.I. Chemical Profile, Antioxidant, Antimicrobial, and Anticancer Activities of the Water-Ethanol Extract of Pulicaria undulata Growing in the Oasis of Central Saudi Arabian Desert. Plants 2021, 10, 1811. [Google Scholar] [CrossRef]
- Mohammed, H.A.; Ali, H.M.; Qureshi, K.A.; Alsharidah, M.; Kandil, Y.I.; Said, R.; Mohammed, S.A.A.; Al-Omar, M.S.; Al Rugaie, O.; Abdellatif, A.A.H.; et al. Comparative Phytochemical Profile and Biological Activity of Four Major Medicinal Halophytes from Qassim Flora. Plants 2021, 10, 2208. [Google Scholar] [CrossRef]
- Maiti, P.K.; Das, S.; Sahoo, P.; Mandal, S. Streptomyces sp SM01 isolated from Indian soil produces a novel antibiotic picolinamycin effective against multi drug resistant bacterial strains. Sci. Rep. 2020, 10, 10092. [Google Scholar] [CrossRef]
- Forouzanfar, F.; Bazzaz, B.S.F.; Hosseinzadeh, H. Black cumin (Nigella sativa) and its constituent (thymoquinone): A review on antimicrobial effects. Iran. J. Basic Med. Sci. 2014, 17, 929–938. [Google Scholar] [CrossRef]
- Halawani, E. Antibacterial Activity of Thymoquinone and Thymohydroquinone of Nigella sativa L. and Their Interaction with Some Antibiotics. Adv. Biol. Res. 2009, 3, 148–152. [Google Scholar]
- Darakhshan, S.; Pour, A.B.; Colagar, A.H.; Sisakhtnezhad, S. Thymoquinone and its therapeutic potentials. Pharmacol. Res. 2015, 95, 138–158. [Google Scholar] [CrossRef] [PubMed]
- Goyal, S.N.; Prajapati, C.P.; Gore, P.R.; Patil, C.R.; Mahajan, U.B.; Sharma, C.; Talla, S.P.; Ojha, S.K. Therapeutic Potential and Pharmaceutical Development of Thymoquinone: A Multitargeted Molecule of Natural Origin. Front. Pharmacol. 2017, 8, 656. [Google Scholar] [CrossRef]
- Woo, C.C.; Kumar, A.P.; Sethi, G.; Tan, K.H.B. Thymoquinone: Potential cure for inflammatory disorders and cancer. Biochem. Pharmacol. 2012, 83, 443–451. [Google Scholar] [CrossRef]
- Dera, A.A.; Ahmad, I.; Rajagopalan, P.; Al Shahrani, M.; Saif, A.; Alshahrani, M.Y.; Alraey, Y.; Alamri, A.M.; Alasmari, S.; Makkawi, M.; et al. Synergistic efficacies of thymoquinone and standard antibiotics against multi-drug resistant isolates. Saudi Med. J. 2021, 42, 196–204. [Google Scholar] [CrossRef]
- Liu, X.; Ouyang, S.; Yu, B.; Liu, Y.; Huang, K.; Gong, J.; Zheng, S.; Li, Z.; Li, H.; Jiang, H. PharmMapper server: A web server for potential drug target identification using pharmacophore mapping approach. Nucleic Acids Res. 2010, 38, W609–W614. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Pan, C.; Gong, J.; Liu, X.; Li, H. Enhancing the Enrichment of Pharmacophore-Based Target Prediction for the Polypharmacological Profiles of Drugs. J. Chem. Inf. Model. 2016, 56, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Singh, J.; Narasimhan, B.; Shah, S.A.A.; Lim, S.M.; Ramasamy, K.; Mani, V. Reverse pharmacophore mapping and molecular docking studies for discovery of GTPase HRas as promising drug target for bis-pyrimidine derivatives. Chem. Central J. 2018, 12, 106. [Google Scholar] [CrossRef]
- Alves, M.J.; Froufe, H.J.C.; Costa, A.F.T.; Santos, A.F.; Oliveira, L.G.; Osório, S.R.M.; Abreu, R.M.V.; Pintado, M.; Ferreira, I.C.F.R. Docking Studies in Target Proteins Involved in Antibacterial Action Mechanisms: Extending the Knowledge on Standard Antibiotics to Antimicrobial Mushroom Compounds. Molecules 2014, 19, 1672. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Kawai, F.; Obayashi, E.; Akashi, S.; Roper, D.I.; Tame, J.R.H.; Park, S.-Y. Crystal Structures of Penicillin-Binding Protein 3 (PBP3) from Methicillin-Resistant Staphylococcus aureus in the Apo and Cefotaxime-Bound Forms. J. Mol. Biol. 2012, 423, 351–364. [Google Scholar] [CrossRef]
- Chang, Y.-M.; Jeng, W.-Y.; Ko, T.-P.; Yeh, Y.-J.; Chen, C.K.-M.; Wang, A.H.-J. Structural study of TcaR and its complexes with multiple antibiotics from Staphylococcus epidermidis. Proc. Natl. Acad. Sci. USA 2010, 107, 8617–8622. [Google Scholar] [CrossRef] [PubMed]
- Aljohny, B.O.; Rauf, A.; Anwar, Y.; Naz, S.; Wadood, A. Antibacterial, Antifungal, Antioxidant, and Docking Studies of Potential Dinaphthodiospyrols from Diospyros lotus Linn Roots. ACS Omega 2021, 6, 5878–5885. [Google Scholar] [CrossRef] [PubMed]
- Schirner, K.; Marles-Wright, J.; Lewis, R.J.; Errington, J. Distinct and essential morphogenic functions for wall- and lipo-teichoic acids in Bacillus subtilis. EMBO J. 2009, 28, 830–842. [Google Scholar] [CrossRef]
- Gwenin, V.V.; Poornima, P.; Halliwell, J.; Ball, P.; Robinson, G.; Gwenin, C.D. Identification of novel nitroreductases from Bacillus cereus and their interaction with the CB1954 prodrug. Biochem. Pharmacol. 2015, 98, 392–402. [Google Scholar] [CrossRef]
- Chen, P.R.; Bae, T.; Williams, W.A.; Duguid, E.M.; Rice, P.A.; Schneewind, O.; He, C. An oxidation-sensing mechanism is used by the global regulator MgrA in Staphylococcus aureus. Nat. Chem. Biol. 2006, 2, 591–595. [Google Scholar] [CrossRef]
- Silvian, L.F.; Wang, J.; Steitz, T.A. Insights into Editing from an Ile-tRNA Synthetase Structure with tRNAIle and Mupirocin. Struct. Insights Gene Expr. Protein Synth. 2020, 415–418. [Google Scholar] [CrossRef]
- Nakamura, A.; Yao, M.; Chimnaronk, S.; Sakai, N.; Tanaka, I. Ammonia Channel Couples Glutaminase with Transamidase Reactions in GatCAB. Science 2006, 312, 1954–1958. [Google Scholar] [CrossRef]
- Tarbouriech, N.; Charnock, S.J.; Davies, G.J. Three-dimensional structures of the Mn and Mg dTDP complexes of the family GT-2 glycosyltransferase SpsA: A comparison with related NDP-sugar glycosyltransferases. J. Mol. Biol. 2001, 314, 655–661. [Google Scholar] [CrossRef]
- Lovering, A.L.; Lin, L.Y.-C.; Sewell, E.W.; Spreter, T.; Brown, E.D.; Strynadka, N.C.J. Structure of the bacterial teichoic acid polymerase TagF provides insights into membrane association and catalysis. Nat. Struct. Mol. Biol. 2010, 17, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Murray, D.S.; Schumacher, M.A.; Brennan, R.G. Crystal Structures of QacR-Diamidine Complexes Reveal Additional Multidrug-binding Modes and a Novel Mechanism of Drug Charge Neutralization. J. Biol. Chem. 2004, 279, 14365–14371. [Google Scholar] [CrossRef]
- Sundaramoorthy, R.; Iulek, J.; Barrett, M.P.; Bidet, O.; Ruda, G.F.; Gilbert, I.H.; Hunter, W.N. Crystal structures of a bacterial 6-phosphogluconate dehydrogenase reveal aspects of specificity, mechanism and mode of inhibition by analogues of high-energy reaction intermediates. FEBS J. 2007, 274, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Hargrove, T.Y.; Garvey, E.P.; Hoekstra, W.J.; Yates, C.M.; Wawrzak, Z.; Rachakonda, G.; Villalta, F.; Lepesheva, G.I. Crystal Structure of the New Investigational Drug Candidate VT-1598 in Complex with Aspergillus fumigatus Sterol 14α-Demethylase Provides Insights into Its Broad-Spectrum Antifungal Activity. Antimicrob. Agents Chemother. 2017, 61, e00570-17. [Google Scholar] [CrossRef]
- Raimi, O.G.; Hurtado-Guerrero, R.; Borodkin, V.; Ferenbach, A.; Urbaniak, M.D.; Ferguson, M.A.J.; van Aalten, D.M.F. A mechanism-inspired UDP-N-acetylglucosamine pyrophosphorylase inhibitor. RSC Chem. Biol. 2020, 1, 13–25. [Google Scholar] [CrossRef]
- Sansen, S.; De Ranter, C.J.; Gebruers, K.; Brijs, K.; Courtin, C.M.; Delcour, J.A.; Rabijns, A. Structural Basis for Inhibition of Aspergillus niger Xylanase by Triticum aestivum Xylanase Inhibitor-I. J. Biol. Chem. 2004, 279, 36022–36028. [Google Scholar] [CrossRef]
- Hast, M.A.; Beese, L.S. Structure of Protein Geranylgeranyltransferase-I from the Human Pathogen Candida albicans Complexed with a Lipid Substrate. J. Biol. Chem. 2008, 283, 31933–31940. [Google Scholar] [CrossRef]
- Hargrove, T.Y.; Friggeri, L.; Wawrzak, Z.; Qi, A.; Hoekstra, W.J.; Schotzinger, R.J.; York, J.D.; Guengerich, F.P.; Lepesheva, G.I. Structural analyses of Candida albicans sterol 14α-demethylase complexed with azole drugs address the molecular basis of azole-mediated inhibition of fungal sterol biosynthesis. J. Biol. Chem. 2017, 292, 6728–6743. [Google Scholar] [CrossRef]
- Aleshin, A.E.; Stoffer, B.; Firsov, L.M.; Svensson, B.; Honzatko, R.B. Crystallographic Complexes of Glucoamylase with Maltooligosaccharide Analogs: Relationship of Stereochemical Distortions at the Nonreducing End to the Catalytic Mechanism. Biochemistry 1996, 35, 8319–8328. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, K.L.; Klimacek, M.; Nidetzky, B.; Wilson, D.K. Structure of xylose reductase bound to NAD+ and the basis for single and dual co-substrate specificity in family 2 aldo-keto reductases. Biochem. J. 2003, 373, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Chaieb, K.; Kouidhi, B.; Jrah, H.; Mahdouani, K.; Bakhrouf, A. Antibacterial activity of Thymoquinone, an active principle of Nigella sativa and its potency to prevent bacterial biofilm formation. BMC Complement. Altern. Med. 2011, 11, 29. [Google Scholar] [CrossRef]
- Balouiri, M.; Sadiki, M.; Ibnsouda, S.K. Methods for in vitro evaluating antimicrobial activity: A review. J. Pharm. Anal. 2016, 6, 71–79. [Google Scholar] [CrossRef] [PubMed]
- European Committee on Antimicrobial Susceptibility Testing (EUCAST). Antimicrobial Susceptibility Testing EUCAST Disk Diffusion Method; Version 8.0; European Society of Clinical Microbiology and Infectious Diseases: Basel, Switzerland, 2020; pp. 1–21. [Google Scholar]
- Hudzicki, J. Kirby-Bauer Disk Diffusion Susceptibility Test Protocol. Am. Soc. Microbiol. 2009, pp. 1–13. Available online: https://asm.org/Protocols/Kirby-Bauer-Disk-Diffusion-Susceptibility-Test-Pro (accessed on 12 September 2021).
- Elshikh, M.; Ahmed, S.; Funston, S.; Dunlop, P.; McGaw, M.; Marchant, R.; Banat, I.M. Resazurin-based 96-well plate microdilution method for the determination of minimum inhibitory concentration of biosurfactants. Biotechnol. Lett. 2016, 38, 1015–1019. [Google Scholar] [CrossRef]
- Bhargava, B.; Tandon, N. Standard Operating Procedures for Fungal Identification and Detection of Antifungal Resistance; Indian Council of Medical Research (ICMR): Karnataka, India, 2015; Volume 7. [Google Scholar]
- Messick, C.R.; Rodvold, K.A.; Pendland, S.L. Modified time-kill assay against multidrug-resistant Enterococcus faecium with novel antimicrobial combinations. J. Antimicrob. Chemother. 1999, 44, 831–834. [Google Scholar] [CrossRef]
- Peck, K.R.; Kim, M.J.; Choi, J.Y.; Kim, H.S.; Kang, C.-I.; Cho, Y.K.; Park, D.W.; Lee, H.J.; Lee, M.S.; Ko, K.S. In vitro time-kill studies of antimicrobial agents against blood isolates of imipenem-resistant Acinetobacter baumannii, including colistin- or tigecycline-resistant isolates. J. Med. Microbiol. 2012, 61, 353–360. [Google Scholar] [CrossRef]
- Appiah, T.; Boakye, Y.D.; Agyare, C. Antimicrobial Activities and Time-Kill Kinetics of Extracts of Selected Ghanaian Mushrooms. Evid.-Based Complementary Altern. Med. 2017, 2017, 4534350. [Google Scholar] [CrossRef] [PubMed]
- Tutar, U.; Çelik, C.; Karaman, I.; Ataş, M.; Hepokur, C. Anti-biofilm and antimicrobial activity of Mentha pulegium L essential oil against multidrug-resistant Acinetobacter baumannii. Trop. J. Pharm. Res. 2016, 15, 1039–1046. [Google Scholar] [CrossRef]
- Thymoquinone|C10H12O2-PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/10281 (accessed on 26 November 2021).
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity—A rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nat. Cell Biol. 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Singh, U.C.; Kollman, P.A. A combinedab initio quantum mechanical and molecular mechanical method for carrying out simulations on complex molecular systems: Applications to the CH3Cl + Cl-exchange reaction and gas phase protonation of polyethers. J. Comput. Chem. 1986, 7, 718–730. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Software news and updates AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Martínez-Rosell, G.; Giorgino, T.; De Fabritiis, G. PlayMolecule ProteinPrepare: A Web Application for Protein Preparation for Molecular Dynamics Simulations. J. Chem. Inf. Model. 2017, 57, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Qureshi, K.A.; Al Nasr, I.; Koko, W.S.; Khan, T.A.; Fatmi, M.Q.; Imtiaz, M.; Khan, R.A.; Mohammed, H.A.; Jaremko, M.; Emwas, A.-H.; et al. In Vitro and In Silico Approaches for the Antileishmanial Activity Evaluations of Actinomycins Isolated from Novel Streptomyces smyrnaeus Strain UKAQ_23. Antibiotics 2021, 10, 887. [Google Scholar] [CrossRef] [PubMed]
- Bekker, H.; Berendsen, H.; Dijkstra, E.; Achterop, S.; Van Drunen, R.; Van der Spoel, D.; Sijbers, A.; Keegstra, H.; Reitsma, B.; Renardus, M. Gromacs: A parallel computer for molecular dynamics simulations. Phys. Comput. 1993, 92, 252–256. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2016, 14, 71–73. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microorganism | Zone of Inhibition Diameter (mm) | |
|---|---|---|
| TQ (200 µg/disc) | Control (20 µL/disc) | |
| S. aureus ATCC 29213 | 19.2 ± 0.2 | 6.0 ± 0.0 |
| S. saprophyticus ATCC 43867 | 29.4 ± 0.4 | 6.5 ± 0.1 |
| S. epidermidis ATCC 12228 | 50.3 ± 0.3 | 7.0 ± 0.2 |
| MRSA-1 * | 23.2 ± 0.2 | 6.0 ± 0.0 |
| MRSA-5 * | 23.5 ± 0.3 | 7.0 ± 0.1 |
| MRSA-7 * | 16.2 ± 0.2 | 6.0 ± 0.0 |
| B. cereus ATCC 10876 | 41.9 ± 0.1 | 6.5 ± 0.2 |
| S. pyogenes-A ATCC 19615 | 6.0 ± 0.0 | 6.0 ± 0.0 |
| S. pneumoniae ATCC 49619 | 6.0 ± 0.0 | 6.0 ± 0.0 |
| E. faecalis ATCC 29212 | 6.0 ± 0.0 | 6.5 ± 0.2 |
| E. coli ATCC 25922 | 6.0 ± 0.0 | 6.0 ± 0.0 |
| K. pneumoniae ATCC 27736 | 6.0 ± 0.0 | 6.5 ± 0.1 |
| P. aerugenosa ATCC 9027 | 6.0 ± 0.0 | 6.0 ± 0.0 |
| S. typhimurium ATCC 13311 | 6.0 ± 0.0 | 6.0 ± 0.0 |
| S. flexneri ATCC 12022 | 6.0 ± 0.0 | 6.0 ± 0.0 |
| P. vulgaris ATCC 6380 | 8.1 ± 0.2 | 6.5 ± 0.1 |
| P. mirabilis ATCC 29906 | 6.0 ± 0.0 | 6.5 ± 0.2 |
| C. albicans ATCC 10231 | 21.1 ± 0.1 | 6.0 ± 0.0 |
| A. niger ATCC 6275 | 8.7 ± 0.3 | 6.0 ± 0.0 |
| Microorganism | MIC (µg/mL) | MBC (µg/mL) |
|---|---|---|
| S. aureus ATCC 29213 | 50 | 50 |
| S. saprophyticus ATCC 43867 | 25 | 50 |
| S. epidermidis ATCC 12228 | 50 | 50 |
| MRSA-1 | 50 | 100 |
| MRSA-5 | 25 | 50 |
| MRSA-7 | 50 | 100 |
| B. cereus ATCC 10876 | 25 | 25 |
| P. vulgaris ATCC 6380 | 25 | 50 |
| C. albicans ATCC 10231 | 12.5 | 25 |
| A. niger ATCC 6275 | 12.5 | 25 |
| Microorganisms | MBIC (µg/mL) | MBEC (µg/mL) |
|---|---|---|
| S. aureus ATCC 29213 | 50 | 50 |
| S. saprophyticus ATCC 43867 | 25 | 50 |
| S. epidermidis ATCC 12228 | 50 | 50 |
| MRSA-1 | 50 | 100 |
| MRSA-5 | 25 | 50 |
| MRSA-7 | 50 | 100 |
| B. cereus ATCC 10876 | 25 | 25 |
| P. vulgaris ATCC 6380 | 25 | 50 |
| C. albicans ATCC 10231 | NT | NT |
| A. niger ATCC 6275 | NT | NT |
| ANOVA | |||||
|---|---|---|---|---|---|
| Sum of Squares | df | Mean Square | F | Sig. | |
| Between Groups | 9549.679 | 18 | 530.538 | 16,524.945 | 0.000 |
| Within Groups | 1.220 | 38 | 0.032 | ||
| Total | 9550.899 | 56 | |||
| # | Target Proteins | PDB ID | Source Organism | Function | Reference | Binding Energy from Docking (kcal/mol) |
|---|---|---|---|---|---|---|
| Bacterial Protein Targets | ||||||
| 1 | Alanine racemase (Alr) | 2SFP | Geobacillus stearothermophilus | Cell wall synthesis | [16] | −6.5 |
| 2 | D-alanyl-D-alanine synthetase (Ddl) | 2ZDQ | Thermus thermophilus | Cell wall synthesis | [16] | −7.8 |
| 3 | Penicillin-binding protein 3 (PBP3) | 3VSL | Methicillin-Resistant Staphylococcus aureus | Cell wall synthesis | [17] | −5.2 |
| 4 | Transcriptional regulator (TcaR) | 3KP3 | Staphylococcus epidermidis RP62A | Protein biosynthesis | [18] | −6.2 |
| 5 | Penicillin-binding protein 1a (PBP1a) | 3UDI | Acinetobacter baumannii | Cell wall synthesis | [16] | −6.7 |
| 6 | Dihydrofolate reductase (DHFR) | 3SRW | Staphylococcus aureus | Metabolite synthesis | [16] | −6.3 |
| 7 | Dihydropteroate synthetase (DHPS) | 2VEG | Streptococcus pneumoniae | Metabolite synthesis | [16] | −5.2 |
| 8 | DNA gyrase subunit B | 3TTZ | Staphylococcus aureus | Nucleic acid synthesis | [16] | −5.5 |
| 9 | Topoisomerase IV (TopoIV) | 3RAE | Streptococcus pneumoniae | Nucleic acid synthesis | [16] | −5.5 |
| 10 | Sortase A | 2MLM | Staphylococcus aureus CA-347 | (1) Attach proteins to the cell wall and (2) join proteins together to construct pili. | [19] | −5.7 |
| 11 | Glycerol phosphate lipoteichoic acid synthase 2 | 2W8D | Bacillus subtilis | Teichoic acid synthesis | [20] | −6.7 |
| 12 | Nitroreductase family protein | 1YWQ | Bacillus cereus ATCC 14579 | Reduction of nitrogen-containing compounds | [21] | −4.7 |
| 13 | HTH-type transcriptional regulator mgrA | 2BV6 | Staphylococcus aureus | Protein biosynthesis | [22] | −5.6 |
| 14 | Isoleucyl-tRNA synthetase (IleRS) | 1QU3 | Staphylococcus aureus | Protein biosynthesis | [23] | −7.3 * |
| 15 | Glutamyl-tRNA(Gln) amidotransferase subunit A | 2G5H | Staphylococcus aureus | Protein biosynthesis | [24] | −6.1 |
| 16 | Spore Coat Polysaccharide Biosynthesis Protein SPSA | 1H7L | Bacillus subtilis | Spore coat biogenesis | [25] | −4.9 |
| 17 | Teichoic acid biosynthesis protein F | 3L7L | Staphylococcus epidermidis RP62A | Teichoic acid biosynthesis | [26] | −5.9 |
| 18 | Transcriptional regulator qacR | 1RKW | Staphylococcus aureus | Negative regulation of transcription | [27] | −7.2 |
| 19 | YcgJ protein | 2GLU | Bacillus subtilis | Methyltransferase activity | To be published | −5.7 |
| 20 | 6-Phosphogluconate Dehydrogenase (Decarboxylating) | 2IZ0 | Lactococcus lactis | Involved in the production of ribulose 5-phosphate, which is used in nucleotide synthesis | [28] | −5.6 |
| Fungal Protein Targets | ||||||
| 1 | Sterol 14-alpha demethylase (CYP51B) | 5FRB | Aspergillus fumigatus | Sterol biosynthesis | [29] | −6.8 |
| 2 | UDP-N-acetylglucosamine pyrophosphorylase | 6TN3 | Aspergillus fumigatus Af293 | Cell wall synthesis | [30] | −5 |
| 3 | Aspergillus niger xylanase-I | 1T6G | Aspergillus niger | Cell wall metabolism | [31] | −5.4 |
| 4 | Dihydrofolate reductase (DHFR) | 4HOF | Candida albicans | Metabolite synthesis | [19] | −5.9 |
| 5 | Aspartic protease | 3Q70 | Candida albicans | Virulence factor | [19] | −5.3 |
| 6 | N-myristoyltransferase | 1IYL | Candida albicans | Protein biosynthesis | [19] | −7.2 |
| 7 | Geranylgeranyltransferase type-1 subunit alpha | 3DRA | Candida albicans | Metabolite synthesis | [32] | −7.6 * |
| 8 | Sterol 14-alpha demethylase (CYP51) | 5TZ1 | Candida albicans | Sterol biosynthesis | [33] | −6.3 |
| 9 | Glucoamylase-471 | 1GAH | Aspergillus awamori | Involved in the hydrolysis of starch | [34] | −6.8 |
| 10 | NADPH-dependent D-xylose reductase | 1MI3 | Candida tenuis | Involved in hydrolase activity, hydrolyzing O-glycosyl compounds | [35] | −7.5 |
| Enzymes in Complex with TQ | Residues Involved in Hydrophobic Interactions | Residues Involved in H-Bonds | Residues Involved in Pi-Pi Stacking | Residues Involved in Pi-Sigma Interactions |
|---|---|---|---|---|
| D-alanyl-D-alanine synthetase | Glu57, Glu58, Trp61, Gln64, Glu90, Gln96 | Tyr123, Thr89 | Tyr93 | - |
| Transcriptional regulator qacR | Val 131, Ile163, Lys190, Ala191, Leu192, Val195, Tyr223 | - | Phe151 | Phe272 |
| N-myristoyltransferase | His227, Gln226 Tyr335, Leu355, Asn392, Cys393, Leu415, Leu450, Val 449 | - | Tyr354 | Leu394 |
| NADPH-dependent D-xylose reductase | Asp47, Lys81, His114, Gln191, Ser224, Ile272, Pro273, Lys274, Asn310 | Tyr52, Ser218 | Tyr217 | Trp24 |
| TQ in Complex with: | ∆EVDW (van der Waal’s Energy) | ∆Eelec (Coulombic Energy) | ∆GGB (Generalized-Born Polar Solvation Energy) | ∆ESURF (Non-Polar Solvation Energy) | ∆GMMGBSA (Protein–Ligand Binding Energy) |
|---|---|---|---|---|---|
| D-alanyl-D-alanine synthetase | −21.04 | −2.73 | 13.30 | −3.12 | −13.59 ± 2.65 |
| Transcriptional regulator qacR | −17.84 | −6.74 | 14.98 | −3.03 | −12.62 ± 4.09 |
| N-myristoyltransferase | −18.35 | −3.90 | 14.26 | −2.85 | −10.83 ± 3.33 |
| NADPH-dependent D-xylose reductase | −21.41 | −2.13 | 13.68 | −3.06 | −12.92 ± 2.16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qureshi, K.A.; Imtiaz, M.; Parvez, A.; Rai, P.K.; Jaremko, M.; Emwas, A.-H.; Bholay, A.D.; Fatmi, M.Q. In Vitro and In Silico Approaches for the Evaluation of Antimicrobial Activity, Time-Kill Kinetics, and Anti-Biofilm Potential of Thymoquinone (2-Methyl-5-propan-2-ylcyclohexa-2,5-diene-1,4-dione) against Selected Human Pathogens. Antibiotics 2022, 11, 79. https://doi.org/10.3390/antibiotics11010079
Qureshi KA, Imtiaz M, Parvez A, Rai PK, Jaremko M, Emwas A-H, Bholay AD, Fatmi MQ. In Vitro and In Silico Approaches for the Evaluation of Antimicrobial Activity, Time-Kill Kinetics, and Anti-Biofilm Potential of Thymoquinone (2-Methyl-5-propan-2-ylcyclohexa-2,5-diene-1,4-dione) against Selected Human Pathogens. Antibiotics. 2022; 11(1):79. https://doi.org/10.3390/antibiotics11010079
Chicago/Turabian StyleQureshi, Kamal A., Mahrukh Imtiaz, Adil Parvez, Pankaj K. Rai, Mariusz Jaremko, Abdul-Hamid Emwas, Avinash D. Bholay, and Muhammad Qaiser Fatmi. 2022. "In Vitro and In Silico Approaches for the Evaluation of Antimicrobial Activity, Time-Kill Kinetics, and Anti-Biofilm Potential of Thymoquinone (2-Methyl-5-propan-2-ylcyclohexa-2,5-diene-1,4-dione) against Selected Human Pathogens" Antibiotics 11, no. 1: 79. https://doi.org/10.3390/antibiotics11010079
APA StyleQureshi, K. A., Imtiaz, M., Parvez, A., Rai, P. K., Jaremko, M., Emwas, A.-H., Bholay, A. D., & Fatmi, M. Q. (2022). In Vitro and In Silico Approaches for the Evaluation of Antimicrobial Activity, Time-Kill Kinetics, and Anti-Biofilm Potential of Thymoquinone (2-Methyl-5-propan-2-ylcyclohexa-2,5-diene-1,4-dione) against Selected Human Pathogens. Antibiotics, 11(1), 79. https://doi.org/10.3390/antibiotics11010079