
Experimental and Molecular Docking Studies of Cyclic Diphenyl Phosphonates as DNA Gyrase Inhibitors for Fluoroquinolone-Resistant Pathogens

 , , , , and
, , , , and
Abstract
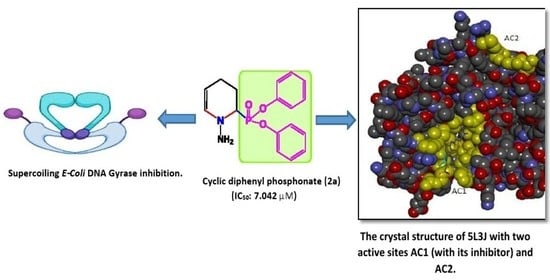
1. Introduction
2. Results and Discussion
2.1. Chemistry
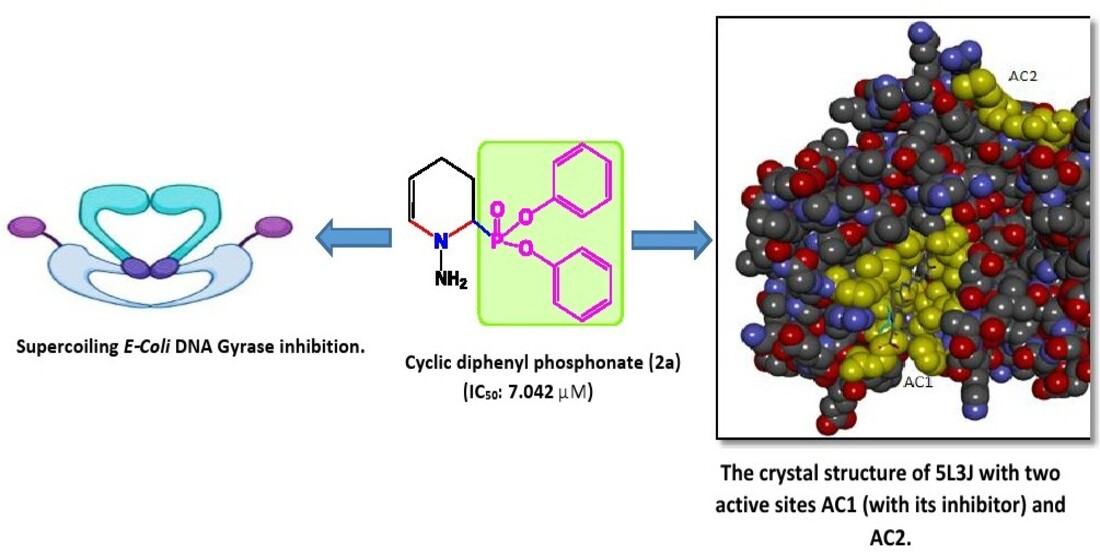
2.2. In Vitro Antibacterial Activity
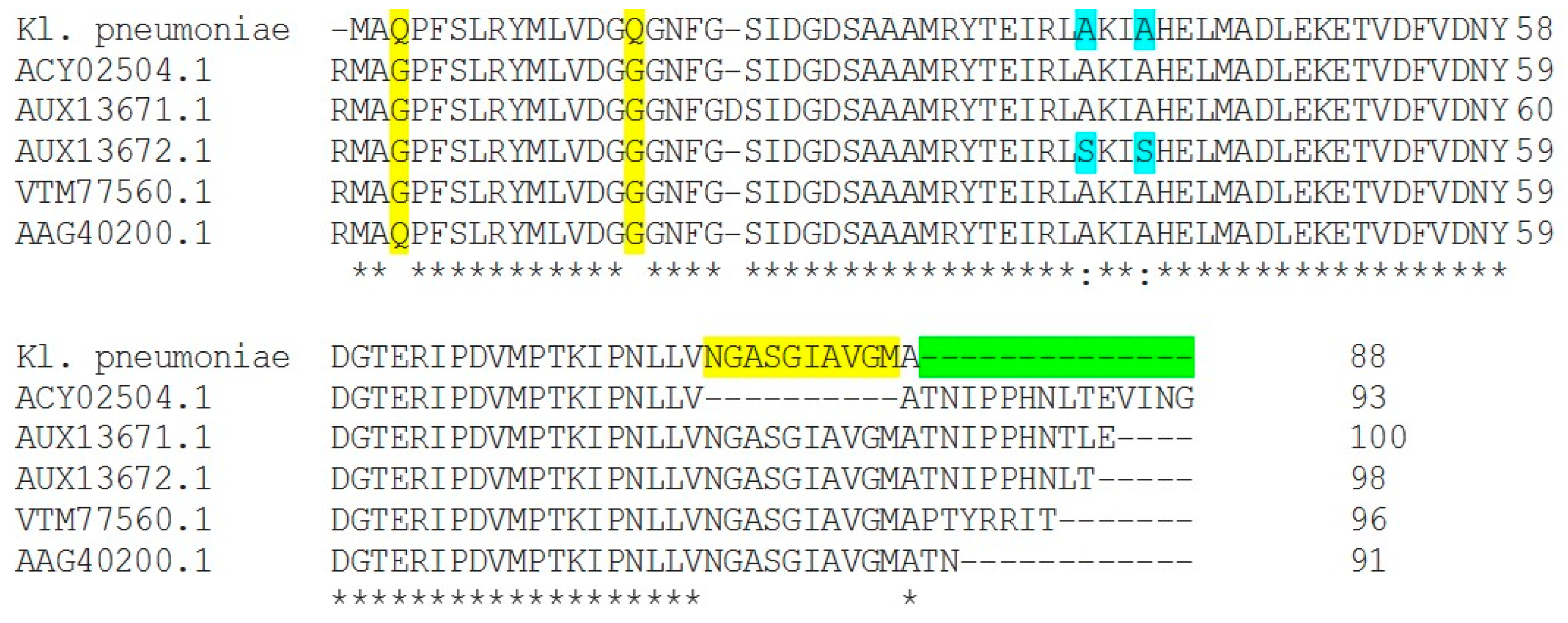
2.3. DNA Gyrase Mutation in Ciprofloxacin-Resistant Clinical Isolates E. coli (E17) and K. pneumonia (Kp8)
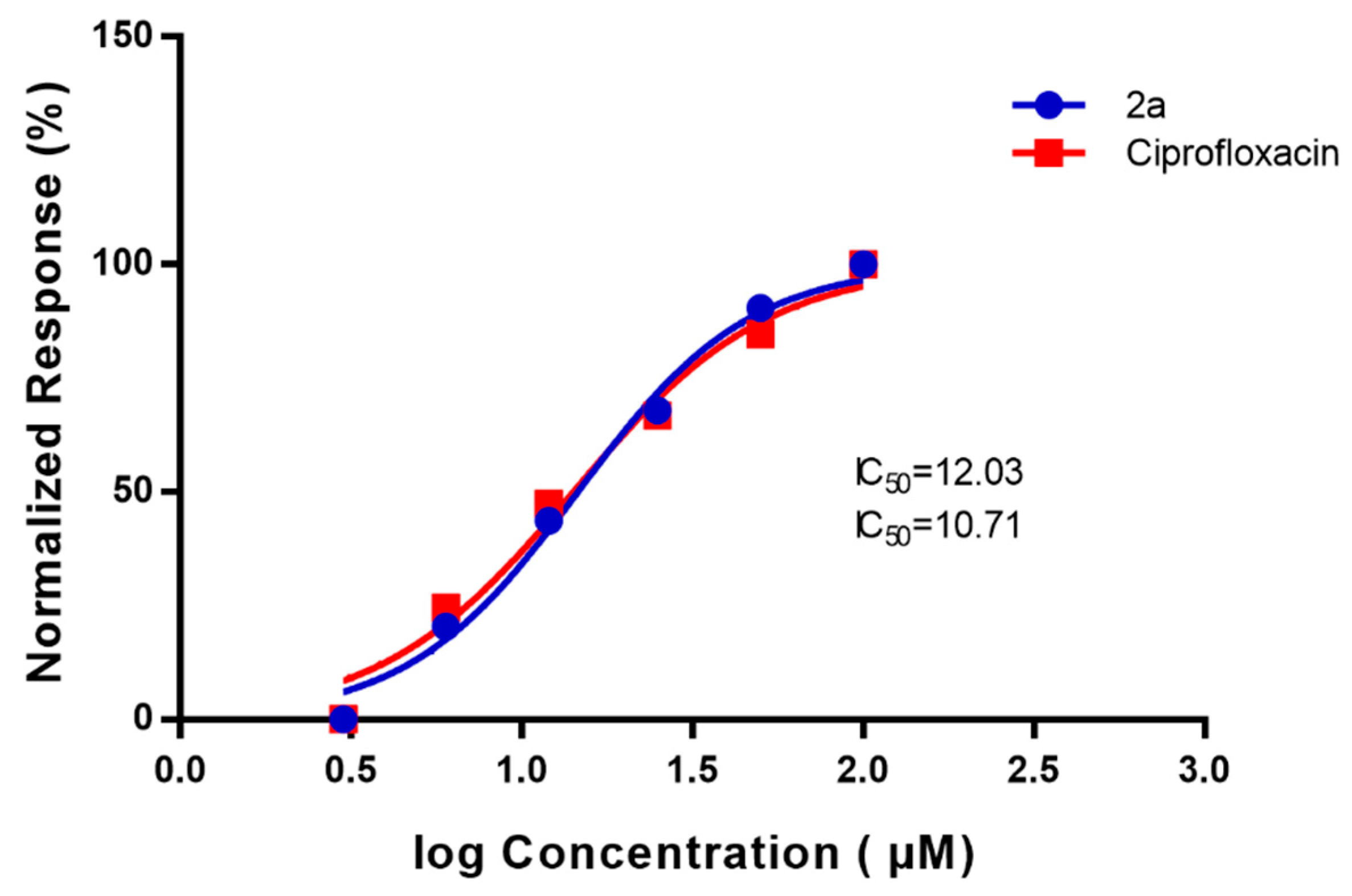
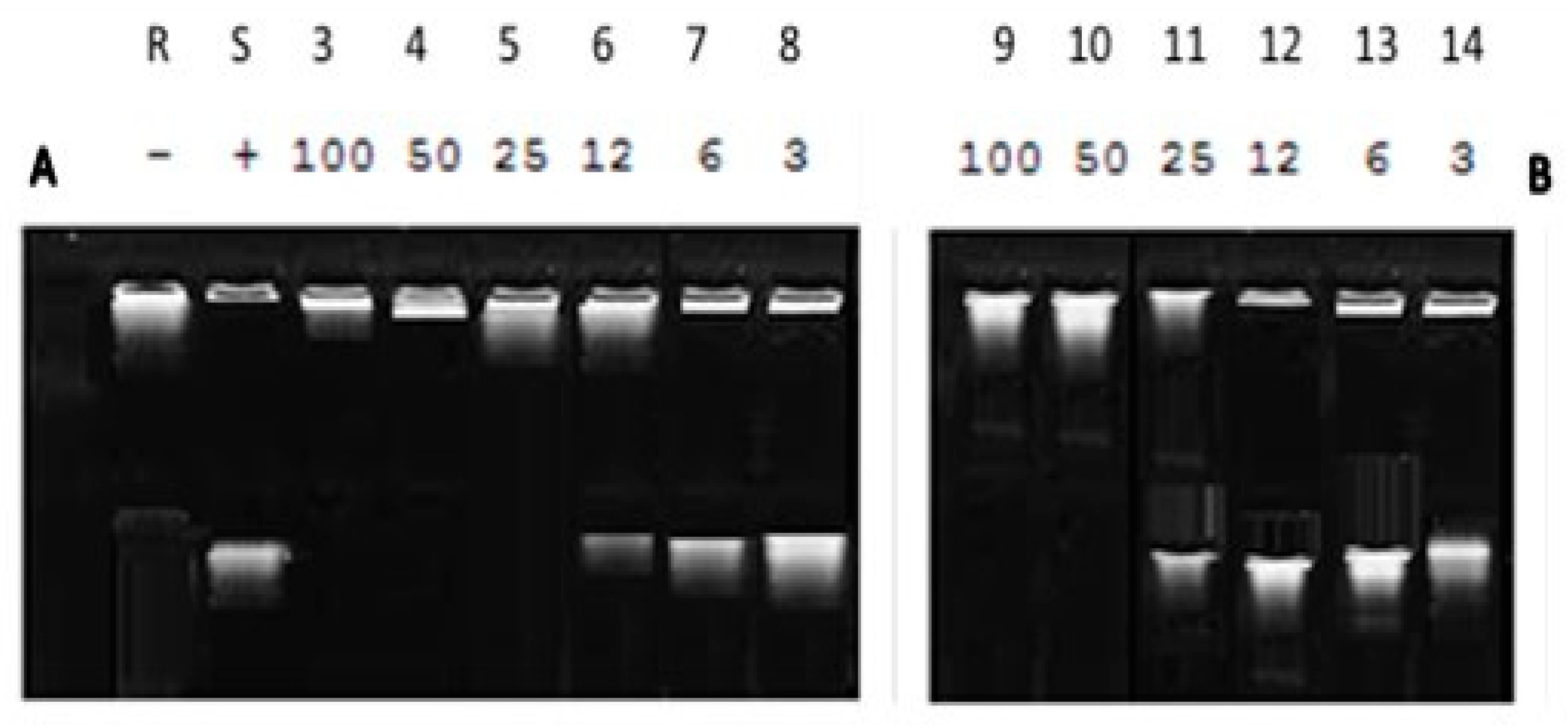
2.4. DNA Supercoiling Assay of Compound (2a) against DNA Gyrase Enzyme Activity
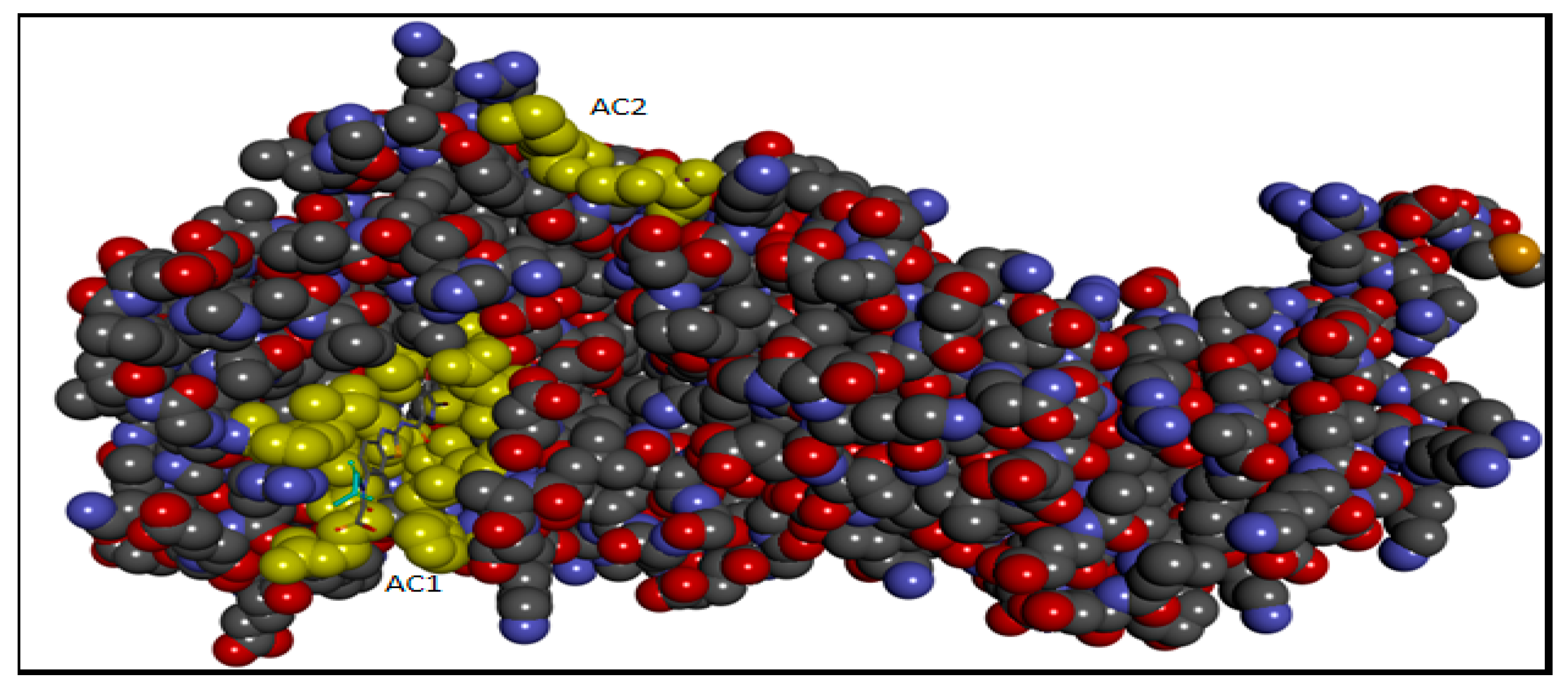
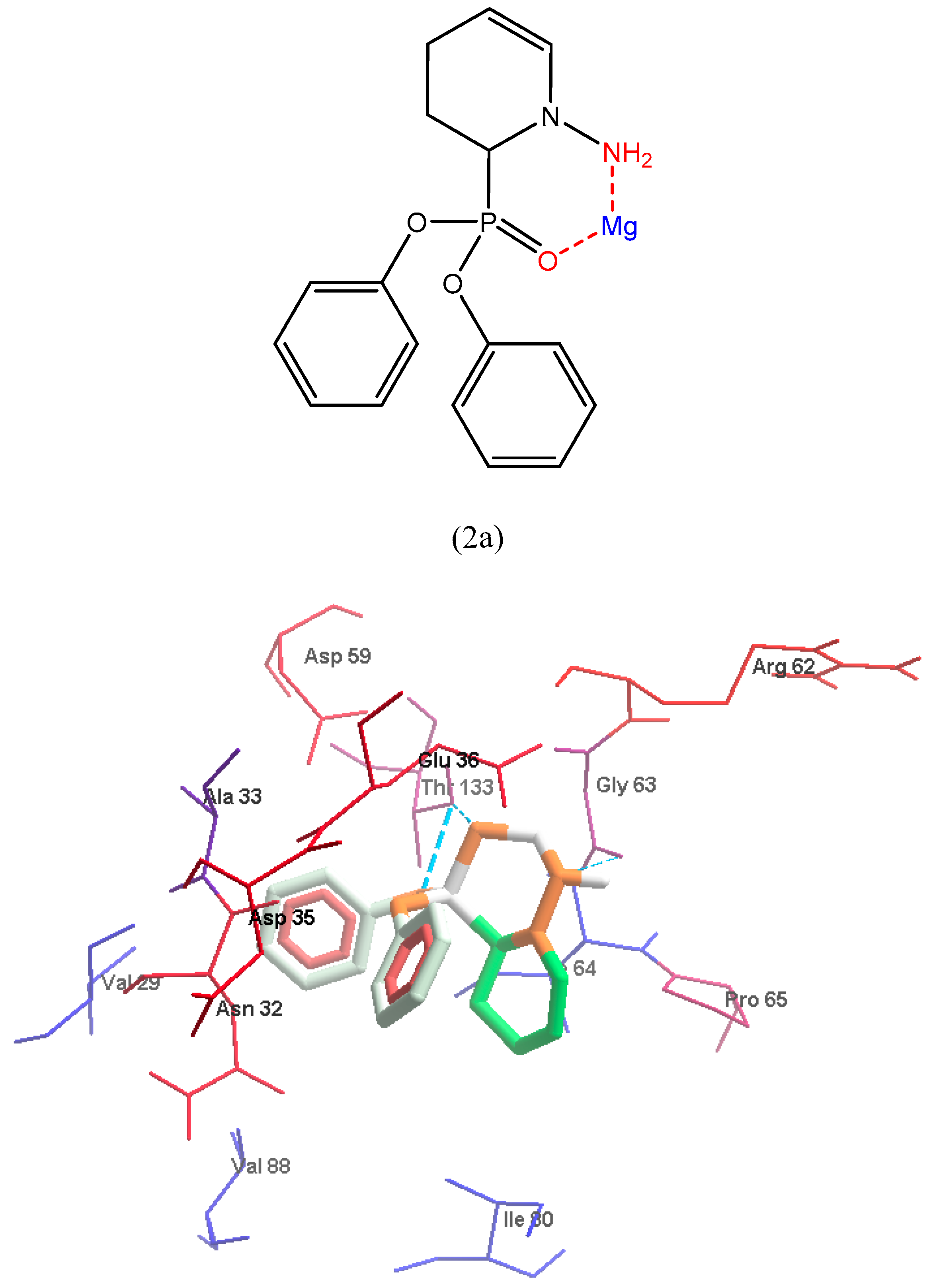
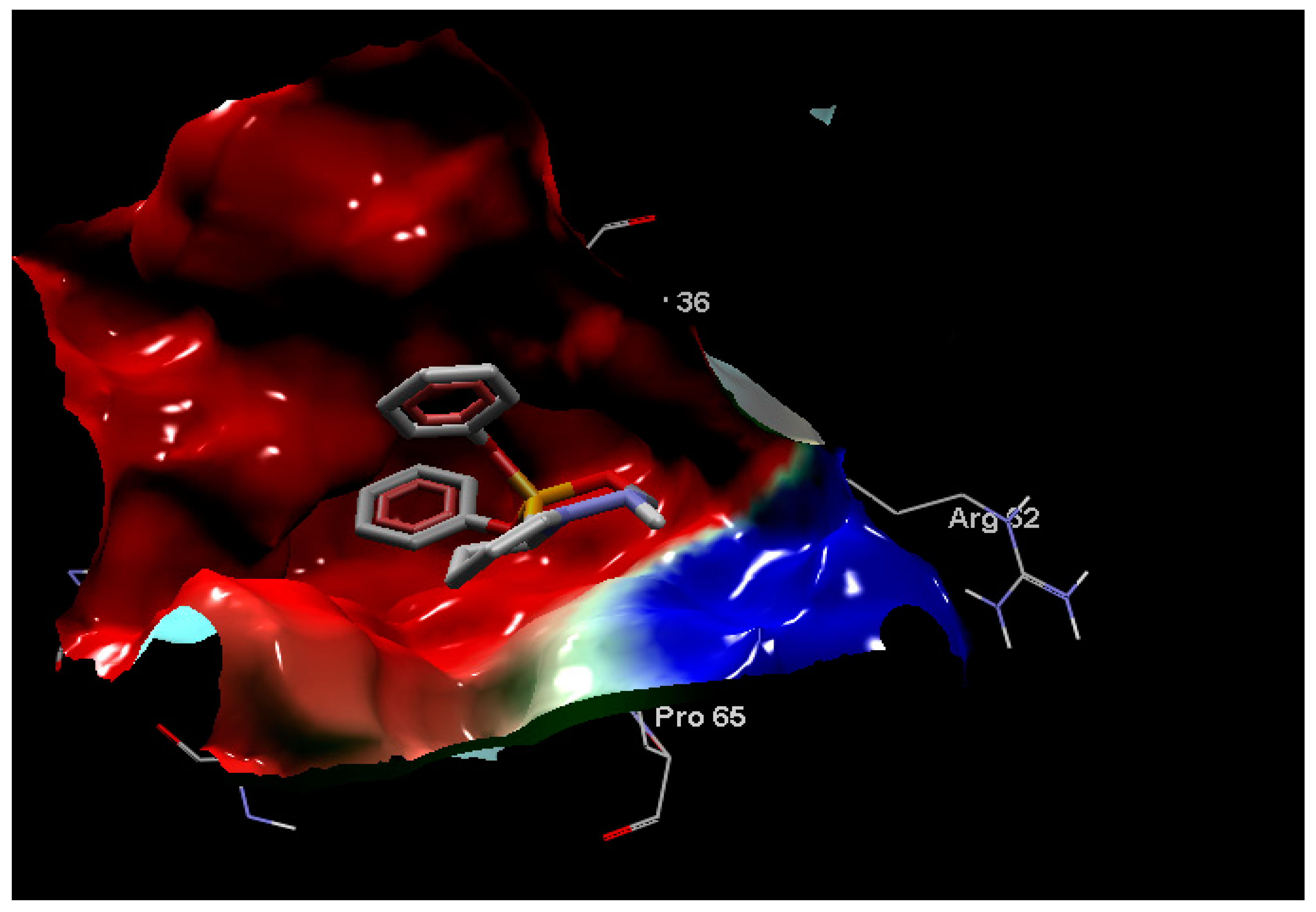
2.5. Molecular Docking
3. Materials and Methods
3.1. Microbiological Screening
3.1.1. Bacterial Strains
3.1.2. Screening of Synthesized Compounds against Tested Isolates Using Agar Well Diffusion Method
3.1.3. Quinolone Resistance Mechanism Detection (QRDR): Amplification and Sequencing of gyrA in QRDR DNA
3.1.4. Impact of Synthesized Compound (2a) against DNA Gyrase Activity Using DNA Supercoiling Assay
3.2. Molecular Docking
3.2.1. Preparation of Macromolecules and Ligands
3.2.2. Active Site Detection
3.2.3. Molecular Docking Implementation Process
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). Fact Sheet. 2018. Available online: https://www.who.int/news-room/fact-sheets/detail/antibiotic-resistance (accessed on 20 August 2020).
- De Kraker, M.E.A.; Stewardson, A.J.; Harbarth, S. Will 10 Million People Die a Year due to Antimicrobial Resistance by 2050? PLoS Med. 2016, 13, e1002184. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC). Antibiotic Resistance Threats in the United States; U.S. Department of Health and Human Services, Centers for Disease Control and Prevention (CDC): Atlanta, GA, USA, 2019. [CrossRef]
- Mohamed, S.H.; Khalil, M.S.; Azmy, M. In Vitro Efficiency of Ampicillin, Thymol and Their Combinations against Virulence Strains of Klebsiella pneumoniae. Int. J. Pharm. Sci. 2019, 11, 315–321. [Google Scholar]
- Norouzi, A.; Azizi, O.; Nave, H.H.; Shakibaie, S.; Shakibaie, M.R. Analysis of Amino Acid Substitution Mutations of gyrA and parC Genes in Clonal Lineage of Klebsiella pneumoniae Conferring High-level Quinolone Resistance. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 2, 109–117. [Google Scholar]
- Rushdy, A.A.; Mabrouk, M.I.; Abu-Sef, F.A.; Kheiralla, Z.H.; Abdel, S.M.; Saleh, N.M. Contribution of different mechanisms to the resistance to fluoroquinolones in clinical isolates of Salmonella enterica. Braz. J. Infect. Dis. 2013, 17, 431–437. [Google Scholar] [CrossRef]
- Mohamed, S.H.; Mohamed, M.S.M.; Khalil, M.; Azmy, M.; Mabrouk, M. Combination of essential oil and ciprofloxacin to inhibit/eradicate biofilms in multidrug-resistant Klebsiella pneumoniae. J. Appl. Microbiol. 2018, 125, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Boshta, N.M.; Elgamal, E.A.; El-Sayed, I.E.T. Bioactive amide and α-aminophosphonate inhibitors for methicillin-resistant Staphylococcus aureus (MRSA). Monatsh. Chem. 2018, 149, 2349–2358. [Google Scholar] [CrossRef]
- Elsherbiny, D.A.; Abdelgawad, A.M.; Elnaggar, M.; El-Sherbiny, R.A.; El-Rafie, M.H.; El Sayed, I.E.T. Synthesis, antimicrobial activity, and sustainable release of novel α-aminophosphonate derivatives loaded carrageenan cryogel. Int. J. Biol. Macromol. 2020, 163, 96–107. [Google Scholar] [CrossRef]
- El Gokha, A.A.; Ahmed, A.A.S.; Abdelwahed, N.A.M.; El Sayed, I.E.T. Synthesis and antimicrobial activity of novel mono- and bis-α-aminophosphonate derivatives. Int. J. Pharm. Sci. Rev. Res. 2016, 36, 35–39. [Google Scholar]
- Hamed, M.A.; El Gokha, A.A.; Ahmed, A.F.A.; Elsayed, M.S.A.E.; Tarabee, R.; El Megeed, A.S.; El Sayed, I.E.T. Synthesis and Antimicrobial Activity of Novel α-Aminophosphonates Bearing Pyrazoloquinoxaline Moiety. Int. J. Pharm. Sci. Rev. Res. 2015, 34, 205–213. [Google Scholar]
- Ouf, N.H.; Hamed, M.A.; El Sayed, I.E.T.; Sakeran, M.I. Anti-cancer, Anti-inflammatory, Cytotoxic and Biochemical Activities of a Novel Phosphonotripeptide Synthesized from Formyl Pyrazolofuran using TUBU as Condensing Agent. J. Adv. Chem. 2014, 6, 1093–1102. [Google Scholar] [CrossRef]
- Ahmed, A.A.S.; Awad, H.M.; El Sayed, I.E.T.; El Gokha, A.A. Synthesis and antiproliferative activity of new hybrids bearing neocryptolepine, acridine and α-aminophosphonate scaffolds. J. Iran. Chem. Soc. 2020, 17, 1211–1221. [Google Scholar] [CrossRef]
- Azzam, M.A.; El-Boraey, H.A.L.; El Sayed, I.E.T. Transition metal complexes of α-aminophosphonates part II: Synthesis, spectroscopic characterization, and in vitro anticancer activity of copper (II) complexes of α-aminophosphonates. Phosphorus Sulfur Silicon Relat. Elem. 2020, 195, 339–347. [Google Scholar] [CrossRef]
- El-Boraey, H.A.L.; El Gokha, A.A.; El Sayed, I.E.T.; Azzam, M.A. Transition metal complexes of α-aminophosphonates Part I: Synthesis, spectroscopic characterization, and in vitro anticancer activity of copper (II) complexes of α-aminophosphonates. Med. Chem. Res. 2015, 24, 2142–2153. [Google Scholar] [CrossRef]
- Joossens, J.; van der Veken, P.; Surpateanu, G.; Lambeir, A.M.; El Sayed, I.E.T.; Ali, O.M.; Augustyns, K.; Haemers, A. Diphenyl phosphonate inhibitors for the urokinase-type plasminogen activator: Optimization of the P4 position. J. Med. Chem. 2006, 49, 5785–5793. [Google Scholar] [CrossRef]
- Van der Veken, P.; El Sayed, I.E.T.; Joossens, J.; Stevens, C.V.; Augustyns, K.; Haemers, A. The Lewis acid catalyzed synthesis of N-protected diphenyl 1-aminoalkylphosphonates. Synthesis 2005, 634–638. [Google Scholar] [CrossRef]
- Galhoum, A.; Eisa, W.H.; El Sayed, I.E.T.; Tolba, A.A.; Shalaby, Z.M.; Mohamady, S.I.; Muhammad, S.S.; Hussien, S.S.; Akashi, T.; Guibal, E. A new route for manufacturing poly (aminophosphonic)-functionalized poly (glycidyl methacrylate)-magnetic nanocomposite-Application to uranium sorption from ore leachate. Environ. Pollut. 2020, 264, 114797–114812. [Google Scholar] [CrossRef]
- Hamed, M.A.; Elkhabiry, S.; Kafafy, H.; El Gokha, A.A.; El Sayed, I.E.T. Synthesis and Characterization of Novel Azo Disperse Dyes Containing α-amino Phosphonate and Their Dyeing Performance on Polyester Fabric. Egypt. J. Chem. 2017, 60, 89–95. [Google Scholar] [CrossRef][Green Version]
- Imam, E.A.; El Sayed, I.E.T.; Mahfouz, M.A.A.; Tolba, T.; Akashi, A.; Galhoum, A.; Gubial, E. Synthesis of α-aminophosphonate functionalized chitosan sorbents: Effect of methyl vs phenyl group on uranium sorption. Chem. Eng. J. 2018, 352, 1022–1034. [Google Scholar] [CrossRef]
- Mahmoud, M.E.; Adel, S.E.; El Sayed, I.E.T. Development of titanium oxide-bound-α-aminophosphonate nanocomposite for adsorptive removal of lead and copper from aqueous solution. Water Resour. Ind. 2020, 23, 100126–100138. [Google Scholar] [CrossRef]
- Amira, A.; Aouf, Z.; Ktir, H.; Chemam, Y.; Ghodbane, R.; Zerrouki, R.; Aouf, N.-E. Recent Advances in the Synthesis of α-Aminophosphonates: A Review. Chem. Select. 2021, 6, 6137–6149. [Google Scholar] [CrossRef]
- Kouznetsov, V.; Gómez, C.M.M.; Valencia Peña, J.L.; Vargas-Méndez, L.Y. Natural and synthetic quinoline molecules against tropical parasitic pathologies: An analysis of activity and structural evolution for developing new quinoline-based antiprotozoal agents. In Discovery and Development of Therapeutics from Natural Products Against Neglected Tropical Diseases; Elsevier: Amsterdam, The Netherlands, 2019; pp. 87–164. [Google Scholar]
- Kukowska, M. Amino acid or peptide conjugates of acridine/acridone and quinoline/quinolone-containing drugs. A critical examination of their clinical effectiveness within a twenty-year timeframe in antitumor chemotherapy and treatment of infectious diseases. Eur. J. Pharm. Sci. 2017, 15, 587–615. [Google Scholar] [CrossRef] [PubMed]
- Manjunath, G.; Mahesh, M.; Bheemaraju, G.; Ramana, P.V. Synthesis of New Pyrazole Derivatives Containing Quinoline Moiety via Chalcones: A Novel Class of Potential Antibacterial and Antifungal Agents. Chem. Sci. Trans. 2016, 5, 61–74. [Google Scholar]
- El Sayed, I.E.T.; Fathy, G.; Ahmed, A.A.S. Synthesis and Antibacterial Activity of Novel Cyclic α-Aminophsophonates. Biomed. J. Sci. Tech. Res. 2019, 23, 17609–17614. [Google Scholar]
- Mohamed, S.; Elshahed, M.; Saied, Y. Evaluation of Honey as an antibacterial agent against drug-resistant uropathogenic E. coli strains. Research, J. Pharm. Tech. 2020, 13, 3720–3724. [Google Scholar] [CrossRef]
- Mohamed, S.H.; Elshahed, M.M.S.; Saied, Y.M.; Mohamed, M.S.M.; Osman, G.H. Detection of heavy metal tolerance among different MLSB resistance phenotypes of methicillin-resistant S. aureus (MRSA). J. Pure. Appl. Microbiol. 2020, 5, 1905–1916. [Google Scholar] [CrossRef]
- Mohamed, S.H.; Khalil, M.S.; Mohamed, M.S.M.; Mabrouk, M.I. Prevalence of antibiotic resistance and biofilm formation in Klebsiella pneumoniae carrying fimbrial genes in Egypt. Res. J. Pharm. Technol. 2020, 13, 3051–3058. [Google Scholar] [CrossRef]
- Li, Z.; Deguchi, T.; Yasuda, M.; Kawamura, T.; Kanematsu, E.; Nishino, Y.; Ishihara, S.; Kawada, Y. Alteration in the GyrA Subunit of DNA Gyrase and the ParC Subunit of DNA Topoisomerase IV in Quinolone-Resistant Clinical Isolates of Staphylococcus epidermidis. Antimicrob. Agents Chemother. 1998, 42, 3293–3295. [Google Scholar] [CrossRef] [PubMed]
- Sekyere, J.O.; Amoako, D.G. Genomic and phenotypic characterisation of fluoroquinolone resistance mechanisms in Enterobacteriaceae in Durban South Africa. PLoS ONE 2017, 12, e0178888. [Google Scholar] [CrossRef]
- Ruiz, J. Mechanisms of resistance to quinolones: Target alterations, decreased accumulation and DNA gyrase protection. J. Antimicrob. Chemother. 2003, 51, 1109–1117. [Google Scholar] [CrossRef]
- Dasgupta, N.; Paul, D.; Chanda, D.D.; Chetri, S.; Chakravarty, A.; Bhattacharjee, A. Observation of a new pattern of mutations in gyrA and parC within Escherichia coli exhibiting fluroquinolone resistance. Indian J. Med. Microbiol. 2018, 36, 131–135. [Google Scholar] [CrossRef]
- Conrad, S.; Oethinger, M.; Kaifel, K.; Klotz, G.; Marre, R.; Kern, W.V. gyrA Mutations in high-level fluoroquinolone-resistant clinical isolates of Escherichia coli. J. Antimicrob. Chemother. 1996, 38, 443–455. [Google Scholar] [CrossRef]
- Fu, Y.; Guo, L.; Xu, Y.; Zhang, W.; Gu, J.; Xu, J.; Chen, X.; Zhao, Y.; Ma, J.; Liu, X.; et al. Alteration of GyrA Amino Acid Required for Ciprofloxacin Resistance in Klebsiella pneumoniae Isolates in China. Antimicrob. Agents Chemother. 2008, 52, 2980–2983. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hooper, D.; Jacoby, G. Mechanisms of drug resistance: Quinolone resistance. Ann. N. Y. Acad. Sci. 2017, 1354, 12–31. [Google Scholar] [CrossRef]
- Chatterji, M. Effect of different classes of inhibitors on DNA gyrase from Mycobacterium Smegmatis. J. Antimicrob. Chemother. 2001, 48, 479–485. [Google Scholar] [CrossRef]
- Vila, J.; Ruiz, J.; Goñi, P.; De Anta, T.J. Quinolone-resistance mutations in the topoisomerase IV parC gene of Acinetobacter baumannii. J. Antimicrob. Chemother. 1997, 39, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Doddaga, S.; Muttana, V.B.R.; Donka, R.; Meriga, B.; Chamarthi, N. Design, Synthesis and Antimicrobial Activity of α-Aminophosphonates of Quinoline and their Molecular Docking Studies Against DNA Gyrase A. Lett. Drug Des. Discov. 2013, 10, 967–976. [Google Scholar]
- Otter, R.; Cozzarelli, N.R. Eschenichia coli DNA gyrase. Method Enzymol. 1983, 100, 171–180. [Google Scholar]
- Heddle, J.G.; Blance, S.J.; Zamble, D.B.; Hollfelder, F.; Miller, D.A.; Wentzell, L.M.; Walsh, C.T.; Maxwell, A. The antibiotic microcin B17 is a DNA gyrase poison: Characterisation of the mode of inhibition. J. Mol. Biol. 2001, 307, 1223–1234. [Google Scholar] [CrossRef]
- Oyamada, Y.; Yamagishi, J.I.; Kihara, T.; Yoshida, H.; Wachi, M.; Ito, H. Mechanism of inhibition of DNA gyrase by ES-1273, a novel DNA gyrase inhibitor. Microbiol. Immunol. 2007, 51, 977–984. [Google Scholar] [CrossRef]
- Pierrat, O.A.; Maxwell, A. The Action of the Bacterial Toxin Microcin B17. Biochimie 2003, 278, 35016–35023. [Google Scholar] [CrossRef]
- Zamble, D.B.; Miller, D.A.; Heddle, J.G.; Maxwell, A.; Walsh, C.T.; Hollfelder, F. In vitro characterization of DNA gyrase inhibition by microcin B17 analogs with altered bisheterocyclic sites. Proc. Natl. Acad. Sci. USA 2001, 98, 7712–7717. [Google Scholar] [CrossRef]
- Bernard, P.; Kezdy, K.E.; van Melderen, L.; Steyaert, J.; Wyns, L.; Pato, M.L.; Higgins, N.P.; Couturier, M. The F Plasmid CcdB Protein Induces Efficient ATP-dependent DNA Cleavage by Gyrase. J. Mol. Biol. 1993, 234, 534–541. [Google Scholar] [CrossRef]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef]
- Zlatkov, A.B.; Peikov, P.T.; Momekov, G.C.; Pencheva, I.; Tsvetkova, B. Synthesis, stability and computational study of some ester derivatives of theophylline-7-acetic acid with antiproliferative activity. Der Pharma Chem. 2010, 2, 197–210. [Google Scholar]
- Vinoda, B.; Bodke, Y.; Vinuth, M.; Aruna, S.M.; Venkatesh, T.; Sandeep, T. One Pot Synthesis, Antimicrobial and In Silico Molecular Docking Study of 1,3-Benzoxazole-5-Sulfonamide Derivatives. Curr. Org. Chemie 2016, 5, 1000163. [Google Scholar] [CrossRef]
- Wayne, P.A. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard—Tenth Edition; CLSI document M07-A10; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2015. [Google Scholar]
- Liao, C.; Hsueh, P.; Jacoby, G.A.; Hooper, D.C. Risk factors and clinical characteristics of patients with qnr -positive Klebsiella pneumoniae bacteraemia. J. Antimicrob. Chemother. 2013, 68, 2907–2914. [Google Scholar] [CrossRef][Green Version]
- Fàbrega, A.; du Merle, L.; Le Bouguénec, C.; de Anta, M.J.; Vila, J. Repression of Invasion Genes and Decreased Invasion in a High-Level Fluoroquinolone-Resistant Salmonella Typhimurium Mutant. PLoS ONE 2009, 4, e8029. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, A.; Burton, N.P.; Hagan, N.O. High-throughput assays for DNA gyrase and other topoisomerases. Nucleic Acids Res. Spec. Publ. 2006, 34, e104. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T. E UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeerschd, T.; Zurek, E.; Hutchison, G.R. R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Gjorgjieva, M.; Tomasič, T.; Barancokova, M.; Katsamakas, S.; Ilas, J.; Tammela, P.; Masič, L.P.; Kikelj, D. Discovery of Benzothiazole Scaffold-Based DNA Gyrase B Inhibitors. J. Med. Chem. 2016, 59, 8941–8954. [Google Scholar] [CrossRef]
- Alland, C.; Moreews, F.; Boens, D.; Carpentier, M.; Chiusa, S.; Lonquety, M.; Renault, N.; Wong, Y.; Cantalloube, H.; Chomilier, J.; et al. RPBS: A web resource for structural bioinformatics. Nucleic Acids Res. 2005, 33, W44–W49. [Google Scholar] [CrossRef]
- Néron, B.; Ménager, H.; Maufrais, C.; Joly, N.; Maupetit, J.; Letort, S.; Carrere, S.; Tuffery, P.; Letondal, C. Mobyle: A new full web bioinformatics framework. Bioinformatics 2009, 25, 3005–3011. [Google Scholar] [CrossRef]
- Forli, S.; Olson, A.J. A force field with discrete displaceable waters and desolvations entropy for hydrated ligand docking. J. Med. Chem. 2012, 55, 623–638. [Google Scholar] [CrossRef] [PubMed]
- Moemen, Y.S.; El-Nahas, A.M.; Helmy, A.; Hassan, E.; Abdel-Azeim, S.; El-Bialy, S.A.A. Docking and 3D-QSAR Studies on Some HCV NS5b Inhibitors. J. Drug Des. Med. Chem. 2017, 3, 49–59. [Google Scholar]
- Adeniji, S.E.; Arthur, D.E.; Abdullahi, M.; Haruna, A. Quantitative structure–activity relationship model, molecular docking simulation and computational design of some novel compounds against DNA gyrase receptor. Chem. Afr. 2020, 3, 391–408. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
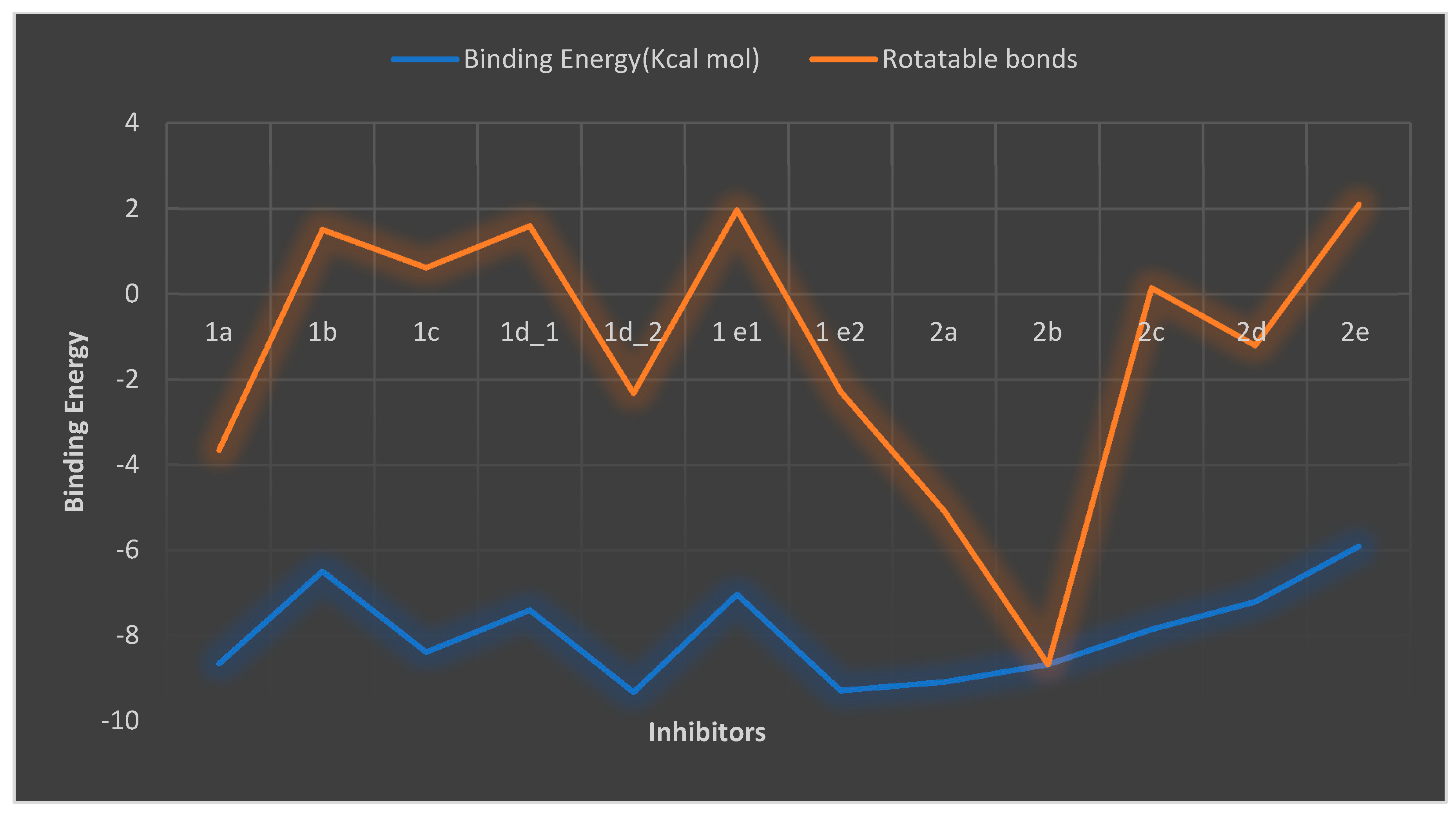
| Compound | Binding Energy (Kcal mol) | Rotatable Bonds |
|---|---|---|
| 1a | −8.65 | 5 |
| 1b | −6.49 | 8 |
| 1c | −8.38 | 9 |
| 1d1 | −7.4 | 9 |
| 1d2 | −9.32 | 7 |
| 1e1 | −7.03 | 9 |
| 1e2 | −9.28 | 7 |
| 2a | −9.08 | 4 |
| 2b | −8.67 | 0 |
| 2c | −7.85 | 8 |
| 2d | −7.2 | 6 |
| 2e | −5.9 | 8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saleh, N.M.; Moemen, Y.S.; Mohamed, S.H.; Fathy, G.; Ahmed, A.A.S.; Al-Ghamdi, A.A.; Ullah, S.; El Sayed, I.E.-T. Experimental and Molecular Docking Studies of Cyclic Diphenyl Phosphonates as DNA Gyrase Inhibitors for Fluoroquinolone-Resistant Pathogens. Antibiotics 2022, 11, 53. https://doi.org/10.3390/antibiotics11010053
Saleh NM, Moemen YS, Mohamed SH, Fathy G, Ahmed AAS, Al-Ghamdi AA, Ullah S, El Sayed IE-T. Experimental and Molecular Docking Studies of Cyclic Diphenyl Phosphonates as DNA Gyrase Inhibitors for Fluoroquinolone-Resistant Pathogens. Antibiotics. 2022; 11(1):53. https://doi.org/10.3390/antibiotics11010053
Chicago/Turabian StyleSaleh, Neveen M., Yasmine S. Moemen, Sara H. Mohamed, Ghady Fathy, Abdullah A. S. Ahmed, Ahmed A. Al-Ghamdi, Sami Ullah, and Ibrahim El-Tantawy El Sayed. 2022. "Experimental and Molecular Docking Studies of Cyclic Diphenyl Phosphonates as DNA Gyrase Inhibitors for Fluoroquinolone-Resistant Pathogens" Antibiotics 11, no. 1: 53. https://doi.org/10.3390/antibiotics11010053
APA StyleSaleh, N. M., Moemen, Y. S., Mohamed, S. H., Fathy, G., Ahmed, A. A. S., Al-Ghamdi, A. A., Ullah, S., & El Sayed, I. E.-T. (2022). Experimental and Molecular Docking Studies of Cyclic Diphenyl Phosphonates as DNA Gyrase Inhibitors for Fluoroquinolone-Resistant Pathogens. Antibiotics, 11(1), 53. https://doi.org/10.3390/antibiotics11010053