1. Introduction
Clinical microbiology is increasingly confronted with the consequences of biofilm formation, causing problems of chronic infections and chronic dysbiosis. This is also the case for bacterial vaginosis (BV), which often develops into a chronic and refractory condition and which is now generally recognized to be largely due to biofilm formation with Gardnerella as one of the principal bacterial taxa. Since antibiotic treatment only causes transient relief in many BV cases, alternative therapies such as endolysin treatment are increasingly being considered and developed. However, the effect of any therapy on a biofilm is difficult to assess because bacteria have already decreased metabolic activity in biofilms, which makes it challenging to evaluate therapy success. One option is qPCR, which has the disadvantage that by amplifying DNA from dead cells that is still present in the sample, it easily provides a false negative result (positive = therapy success). The second option, culturing, may provide false positive results if alive but dormant biofilm cells cannot be cultured under the same conditions as metabolically active planktonic cells.
In this study, we assessed to what extent viability-qPCR was a valuable tool to quantify biofilm eradication from vaginal samples by means of endolysin specific for
Gardnerella. Viability-qPCR in principle is based on the ability of propidium monoazide (PMA) to block amplification of DNA outside of cells with intact cell membranes and of DNA inside of cells with damaged cell membranes, leaving only DNA from living cells amplifiable. In principle, PMA does not easily enter living cells, and therefore will bind only covalently with DNA from lysed cells, inhibiting amplification of DNA from lysed cells and enabling amplification of DNA from live cells only [
1,
2,
3,
4]. However, the method needs finetuning, since a PMA-treatment too weak will not inactivate all DNA from killed cells and PMA-treatment too harsh might also inactivate DNA inside living cells [
5].
Gardnerella can be isolated from the urogenital tract of women and men [
6,
7,
8] and can form biofilms on vaginal epithelial cells, observed as so-called clue cells in a condition known as BV [
8,
9], the most common reason of (malodorous) vaginal discharge. BV increases the risk of preterm delivery, human immunodeficiency virus infection and transmission as well as other sexually transmitted infections (STIs), intrauterine growth retardation, postpartum endometritis and pelvic inflammatory disease [
10,
11,
12]. When growing as biofilm,
Gardnerella changes gene expression, which may be one of the reasons of biofilm resistance towards antibiotics and the chronic nature of BV [
9,
13].
According to the Centers for Disease Control and Prevention, the antibiotics clindamycin or metronidazole are recommended to treat BV [
14,
15]. The level of their efficacy reaches only around 60%, with a high recurrence rate of 30–40% [
14,
16,
17,
18], which may also increase the dissemination of antibiotic resistance among non-
Gardnerella BV-related species. Due to the lack of suitable and effective ways of treatment, there is the continuing need to find effective alternatives for antibiotics, with endolysins as a recent development e.g., [
19,
20]. Endolysins are bacteriophage-encoded enzymes that specifically degrade bacterial peptidoglycans resulting in a rapid bacteriolysis [
19]. They are produced at the end of the phage replication cycle to lyse the bacterial host cell from within and enable phage progeny release [
21]. Due to the lack of an outer membrane in the cell wall of Gram-positive bacteria, endolysins can be easily applied externally, leading to bacterial cell lysis. Moreover, endolysins were shown to be effective against bacterial biofilms and were characterized as antimicrobials against which the level of generated resistance is very low [
20,
22,
23]. Together with their high killing specificity, leaving the commensal microflora unaffected, these characteristics make endolysins potential tools to fight antibiotic-resistant bacterial infections.
Here, we assessed the usefulness of PMA-based viability-qPCR to quantify killing by PM-477, a recently developed engineered endolysin, active against different
Gardnerella species [
20]. Therefore, we first optimized this approach for planktonic cells of
G. swidsinskii. Finally, we quantified the efficacy of PM-477 on (dormant)
Gardnerella cells in original biofilm as present in vaginal samples from women with BV.
2. Materials and Methods
2.1. Gardnerella Strains and Culture Conditions
G. vaginalis ATCC 14018T, G. leopoldii UGent 06.41T, G. piotii UGent 18.01T and G. swidsinskii GS 10234T were grown on chocolate (Choc) agar plates (Becton Dickinson, Franklin Lakes, NJ, USA) at 37 °C under anaerobic conditions (10% CO2, 10% H2, 80%N2, Concept 400, Anaerobic Workstation). Liquid cultures were prepared in New York City Broth III (10 mM HEPES (Sigma Aldrich, Burlington, MA, USA), 15 g/L Proteose Peptone No. 3 (Becton Dickinson), 3.8 g/L yeast extract (Thermo Fisher Scientific, Waltham, MA, USA), 86 mM sodium chloride (Carl Roth, Karlsruhe, Germany), 28 mM α-D-glucose (Sigma–Aldrich)), at pH 5 (NYB5), supplemented with 10% horse serum (HS, Thermo Fisher Scientific) (NYB5 + HS).
2.2. Preparation of PM-477 Phage Endolysin
Engineered phage endolysin PM-477 was described previously [
20]. PM-477 was expressed and purified as presented previously [
20]. Briefly, PM-477 expression in
E. coli BL21 (DE3) was performed in Terrific Broth Medium, with induction with 1.5% α-lactose monohydrate (Carl Roth) at 25 °C for 24 h, with shaking at 250 rpm. Protein purification was performed by affinity chromatography on a nickel–nitrilotriacetic acid (Ni–NTA) HISTrap column. Elution with 50 mM MES (Carl Roth) pH 7, 150 mM NaCl (Carl Roth), 250 mM imidazole (Carl Roth) was followed by size exclusion chromatography and dialysis against MES buffer (50 mM MES (2-morpholinoethanesulfonic acid) pH 5.5, 100 mM NaCl, 8 mM MgSO
4 (Sigma–Aldrich)). Protein concentration was determined at OD 260/280 nm or by using the PierceTM BCA (bicinchoninic acid) protein assay kit (Thermo Fisher Scientific). Purified PM-477 aliquots (0.5 mg/mL) of 450 µL were stored at −80 °C till the moment of use. The His tag used for purification was not cleaved off PM-477 for the scope of this study.
2.3. Inactivation of Extracted Gardnerella DNA Using PMAxx
Extracted G. swidsinskii GS 10234T DNA at log8.00 genomes/mL was treated with PMAxx (PMA molecule optimized by Biotium, Fremont, CA, USA) according to the manufacturer’s protocol: 25 µM final concentration of PMAxx, 10 min incubation at room temperature, followed by 15 min light exposure in the PMA-Lite equipment (PMA-Lite™ LED Photolysis Device, for photoactivation of PMAxx™, PMA or EMA (Biotium), the PMA-Lite equipment illuminates each tube with 3 LEDs, one at the bottom and two at the side, of each 600–800 milliCandela (0.6–0.8 lumen) at a wavelength of 465–475 nm). Finally, DNA PMAxx-treated and non-treated were used for qPCR specific for G. swidsinskii.
2.4. Viability-qPCR on Live and Heat Killed Gardnerella Cells
Cells of G. swidsinski GS 10234T, freshly cultured on Choc agar plates, were used to prepare a bacterial suspension at OD600 = 0.1 in NYB5 + HS, corresponding to log8.3 CFU/mL. Suspension was divided into “live” and “dead” samples. “Dead” samples were heat killed by incubation at 95 °C for 10 min. Aliquots were centrifuged for 10 min at 4000× g in a benchtop centrifuge (Biofuge Pico, Heraeus, Sysmex, Hoeilaart, Belgium) and the supernatant was removed. Standard PMAxx treatment conditions was performed (25 µM final concentration of PMAxx, 10 min incubation at room temperature, followed by 15 min light exposure in the PMA-Lite equipment) and cells were pelleted (10 min 5000× g). Finally, DNA was extracted and used for qPCR specific for G. swidsinskii.
2.5. Optimization of Viability-qPCR
Viability-qPCR was optimized using
G. swidsinski GS 10234
T. Cells of
G. swidsinski GS 10234
T, freshly cultured on Choc agar plates, were used to prepare a bacterial suspension at OD
600 = 0.1 in NYB5 + HS, corresponding to log8.3 CFU/mL. Then
G. swidsinkii cells were treated with 0.05 mg/mL PM-477 overnight at 37 °C in anaerobic conditions (Concept 400, Anaerobic Workstation, Ruskinn, Bridgend, United Kingdom, 75% humidity). As a control, treatment with MES buffer was performed. After overnight treatment, aliquots were centrifuged for 10 min at 4000×
g in a benchtop centrifuge (Biofuge Pico, Heraeus) and the supernatant was removed. Different PMAxx treatment conditions and addition of the apolar solvent dimethylsulfoxide (DMSO 2%-which might increase permeability of the cell membranes of killed cells and is used as the solvent for the alternative dye, ethidium monoazide bromide (EMA) [
1]) were tested on pelleted cells, as indicated in
Table 1. During PMAxx treatment, the samples were protected from light. After PMAxx treatment, vials were exposed to high intensity light for 15 min in the (PMA-Lite equipment and pelleted (10 min 5000×
g). Finally, DNA was extracted, and the DNA extracts were used for qPCR specific for
G. swidsinskii.As an additional control to assess the bactericidal effect of PM-477, each endolysin-treated sample was diluted in 10-fold dilution series, plated on Choc agar plates and cultured. After 48 h of incubation at 37 °C in anaerobic conditions, colonies were counted and CFU/mL was calculated.
2.6. Optimized Viability-qPCR Protocol (PMAxx Treatment Followed by qPCR) for Differentiation between Live and Dead Cells
Two aliquots of vaginal samples were incubated with MES and two with PM-477 endolysin, and all four were treated with 50 µM PMAxx for 15 min on ice protected from light followed by 15 min light exposure in the PMA-Lite equipment. This procedure was repeated three times. Subsequently, the aliquots were centrifuged for 10 min at 5000× g in a benchtop centrifuge and supernatant was removed. Pellets were used for DNA extraction and qPCRs, as described below.
2.7. Influence of Dead Cells on the Quantification of Live Cells
Cells of G. swidsinskii GS 10234T, freshly cultured on Choc agar plates, were used to prepare a bacterial suspension at OD600 = 1 and OD600 = 0.1 in NYB5 + HS, corresponding to log9.3 and log8.3 CFU/mL respectively. Part of the cell suspensions was heat killed by incubation at 95 °C for 10 min. Ten-fold dilution series of living cells were prepared in heat killed suspensions to obtain variable titers of living cells from log9.3 to log3.3 in a constant titer of log9 killed cells and from log8.3 to log3.3 of living cells in a constant titer of log8 killed cells. Aliquots were centrifuged for 10 min at 4000× g in a benchtop centrifuge (Biofuge Pico, Heraeus) and the supernatant was removed. Optimized PMAxx treatment protocol was performed (50 µM final concentration of PMAxx, 15 min incubation at room temperature, followed by 15 min light exposure in the PMA-Lite equipment) and cells were pelleted (10 min 5000× g). Finally, DNA was extracted, and the DNA extracts were used for qPCR specific for G. swidsinskii.
2.8. Validation of Viability-qPCR on Planktonic Cells of Different Gardnerella Species
Freshly cultured cells of Gv17 (G. piotii UGent 18.01T), Gv10 (G. leopoldii UGent 06.41T), Gv9 (G. vaginalis ATCC 14018T) and Gv23 (G. swidsinskii GS 10234T) were used to prepare aliquots at OD600 = 0.1 in NYB5 + HS. Then Gardnerella cells were treated with 0.05 mg/mL PM-477 overnight at 37 °C in anaerobic conditions at 75% humidity (Concept 400, Anaerobic Workstation). As a control, treatment with MES buffer was performed. After overnight treatment, aliquots were centrifuged for 10 min at 4000× g in a benchtop centrifuge and supernatant was removed. PMAxx treatment was performed on the pelleted cells according to the PMAxx treatment protocol that had been optimized (as described above), and followed by DNA extraction and species-specific qPCRs.
2.9. Vaginal Swabs Collection
Vaginal samples were collected from randomly selected women who were previously diagnosed with BV. Fresh vaginal smears were collected with ESwab™ (COPAN, Murrieta, CA, USA) and resuspended in liquid Amies. The vials were frozen in −80 °C and stored until further use.
2.10. Differentiation between Live and Dead Gardnerella after PM-477 Treatment in Vaginal Samples from Women with Bacterial Vaginosis
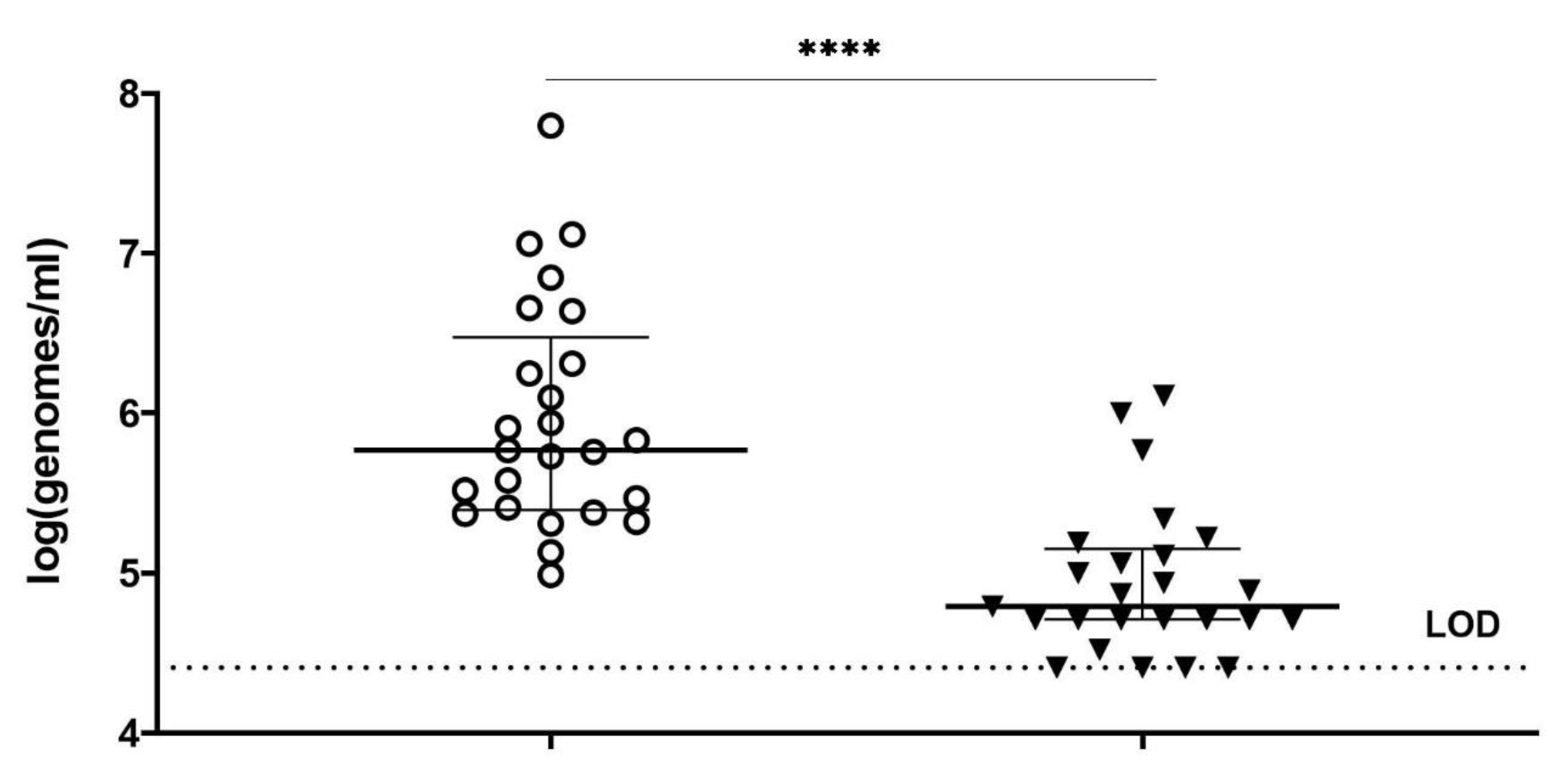
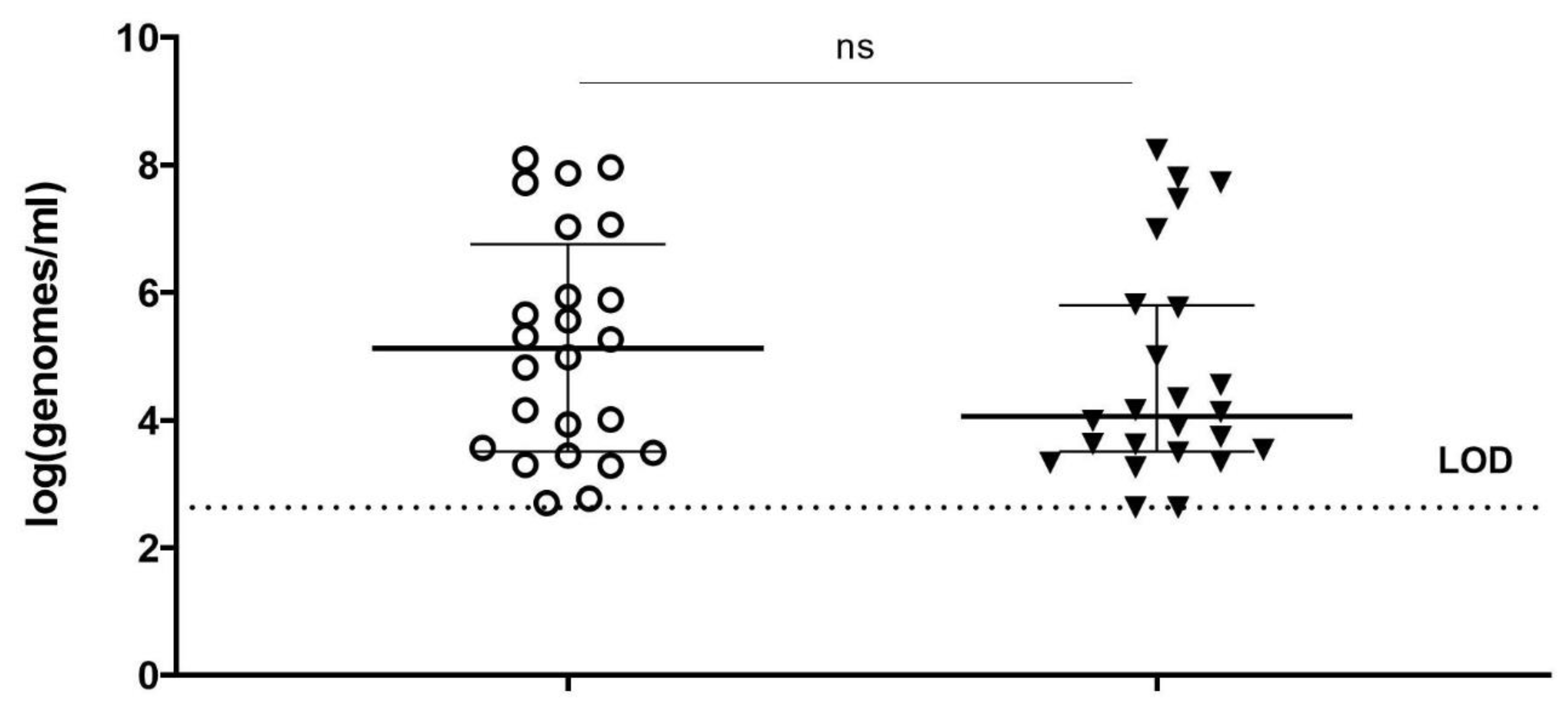
Each vaginal sample was vortexed and divided into five equal aliquots of 45 µL. Respectively three aliquots were treated with MES buffer (50 mM MES, 100 mM NaCl, 8 mM MgSO4, pH 5.5) and two were treated with PM-477 (final conc. 0.1 mg/mL in MES) overnight at 37 °C in anaerobic conditions at 75% humidity (Concept 400, Anaerobic Workstation). After overnight treatment, the aliquots were centrifuged for 10 min at 4000× g in a benchtop centrifuge and the supernatant was removed. PMAxx treatment was performed on the pelleted cells of 2 MES treated and 2 PM-477-treated samples according to the optimized conditions (as described above). One aliquot was treated with MES buffer only and remained without PMAxx incubation. Thereafter, DNA extraction and species-specific qPCRs were carried out.
2.11. DNA Extraction of Gardnerella Planktonic Cells and Vaginal Samples after PM-477 Treatment Followed by PMAxx Treatment
Samples, after PM-477 treatment followed by PMAxx treatment, were centrifuged for 10 min at 5000× g in a benchtop microcentrifuge (Biofuge Pico, Heraeus), and the supernatant was removed. Pellets were protease digested by adding 400 µL of protease buffer (9.5 mL of a 20 mM Tris-HCl pH 8.0, 0.5 mL of 10% SDS) containing 0.6 mg/mL Proteinase K (Merck, Darmstadt, Germany) per sample. Thereafter, samples were pre-lysed with Nuclisens EasyMag Lysis Buffer (bioMerieux, Marcy-l’Étoile, France) and frozen at −80 °C. Finally, DNA was extracted with the High Pure PCR Template Preparation Kit (Roche, Basel, Switzerland) and stored at −20 °C until further use. On the DNA extracts, Gardnerella species-specific or Lactobacillus genus-specific qPCRs were performed.
2.12. Design of Gardnerella Species-Specific Primers
After a literature search, six different household genes (i.e.,
Xfp,
clpC,
dnaJ,
priA,
rpoB and
dnaG) were considered for the design of
Gardnerella species-specific primers. The annotation tool, DFAST, was used to find the sequences of these genes. The sequences from the different
Gardnerella species were aligned using Clustal Omega, with most variation found for
dnaG. Therefore,
Gardnerella species-specific primers (
Table 2) targeting the DnaG region were designed using SnapGene and ordered at Eurogentec.
2.13. Quantification of G. leopoldii, G. piotii, G. swidsinskii, G. vaginalis and the Genus Lactobacillus by Means of qPCR
Gardnerella species-specific qPCRs were performed using SYBR Green qPCR Master Mix (Roche) and species-specific primers (500 nM final concentration,) with the following cyclic conditions: 95 °C denaturation for 5 min, followed by 40 amplification cycles of 15 s denaturation at 95 °C, 30 s annealing at 56 °C and 30 s extension at 72 °C, followed by melting analysis between 55 and 95 °C with a ramp rate of 2.5 °C/s.
Lactobacillus genus-specific qPCR were performed using LightCycler
® 480 High Resolution Melting Master (Roche) and genus specific primers (300 nM final concentration,
Table 2) and with the following cyclic conditions: initial denaturation by heating to 95 °C during 10 min, followed by 45 cycles of 15 sec at 95 °C, 40 sec at 58 °C and 30 sec at 72 °C, followed by melting analysis between 55 and 95 °C with a ramp rate of 2.5 °C/s.
In each qPCR assay, a 10-fold standard dilution series of genomic DNA was run in duplicate, to calculate the concentrations of the unknown samples. All concentrations were expressed in genome equivalents per ml (genomes/mL).
The obtained Cq-values (quantification cycle) of these qPCR assays were expressed as bacterial concentrations (genomes/mL) by calculations of the software based on a standard curve (genomes/mL = 10(Cq–intercept)/slope)).
Values of efficiency, slope and intercept were as follows: 1.864, −3.698, 49.20 for G. leopoldii 1.785, −3.973, 52.57 for G. piotii, 1.822, −3.839, 51.97 for G. swidsinskii, 1.910, −3.559, 47.38 for G. vaginalis and 1.903, −3579, 46.76 for Lactobacillus, respectively.
2.14. Statistical Analysis
One-way ANOVA followed by Tukey’s multiple comparisons test (GraphPad Prism 9, San Diego, CA, USA) were run to test if there was a statistically significant difference in log(genomes/mL) reduction between samples treated and not treated with PM-477 followed by PMAxx as well as to test if PM-477 and PMaxx treatment had a significant effect in log(genomes/mL) reduction of different Gardnerella strains. Normality and lognormality (Anderson–Darling test, D’Agostino & Pearson test, Shapiro–Wilk test and Kolmogorov–Smirnov test) test followed by Wilcoxon test were run to compare if there was a statistically significant difference in log(genomes/mL) between vaginal samples treated and not treated with PM-477. Spearman’s rank correlation coefficient was calculated to assess the correlation between initial load of Gardnerella and reduction caused by PM-477 treatment. p-values of < 0.05 were considered statistically significant, and this was indicated with asterisks in the graphs.
4. Discussion
The goals of this study were: (1) to determine the optimal conditions of viability-qPCR to specifically differentiate between live and dead Gardnerella/Lactobacillus cells in vitro and in vaginal samples of women with bacterial vaginosis (BV), and (2) to quantify the effect of Gardnerella-specific endolysin PM-477 in vaginal samples of women with BV.
The in vitro efficacy of PM-477 (tested in this study) to lyse
Gardnerella cells was recently shown [
20] on a broad panel of
Gardnerella strains belonging to four
Gardnerella species (
G. leopoldii,
G. piotii,
G. vaginalis and
G. swidsinskii), known to be important in vaginal microbiology [
25]. At the same time, PM-477 is inactive against
Lactobacillus species [
20] typically present in the vagina [
26]. Quantification of the number of viable cells of vaginal bacterial taxa such as
Gardnerella in vaginal samples is cumbersome because of (i) the polymicrobial vaginal environment, (ii) the difficulty to selectively culture
Gardnerella strains and (iii) the likeliness that culture is false-negative due to dormancy of the cells in vaginal biofilms [
8,
9]. As such, culture could lead to false-positive results when trying to assess the in vitro antimicrobial effectiveness of agents such as PM-477, an engineered endolysin specifically active against
Gardnerella species [
20]. However, DNA and RNA qPCR methods are also problematic when it comes to quantifying the number of viable cells. DNA qPCR may lead to false-negative results, since also DNA from cells that were efficiently killed will be amplified. RNA qPCR might lead to false-positive results since living cells in biofilms may reduce metabolism and consequently also may reduce DNA transcription into RNA. Since differentiation between viable versus killed cells is crucial to quantify the bactericidal efficacy of potential antimicrobials, we decided to optimize a viability-qPCR method, which consisted of a combination of propidium monoazide (PMAxx, molecule optimized by Biotium) prior treatment, DNA extraction and
Gardnerella species-specific qPCRs.
Viability-qPCR assesses cell viability on the basis of the impermeability of the cell envelope of living cells for propidium monoazide [
1,
27,
28]. Propidium monoazide can bind to unprotected DNA molecules (from nonviable cells with impaired membranes and with free nucleic acids from lysed cell) by means of photoactivation, such that these nucleic acids can no longer serve as template for the polymerase chain reaction. Viable cells, on the other hand, prevent the contact of PMA with their genomic DNA, which can further serve as the matrix for amplification. In principle, only DNA from viable cells is amplified and quantified.
In practice, at least some DNA from dead cells escapes PMA binding, for different reasons, such as adsorption onto debris of degraded cells or due to insufficient cell lysis of killed cells, and this results in overestimation of the number of living cells by means of viability-qPCR. Conversely, some DNA from viable cells may become covalently bound to PMA, for different reasons, such as penetration of (excess) PMA into intact cells or killing and lysis of live cells by the treatment with PMA. This might result in an underestimation of the number of living cells by means of viability-qPCR. The importance of the contribution of these factors can differ between target organisms, bactericidal/bacteriolytic agents, sample types as well as DNA binding agents [
29,
30].
Therefore, the reliability of this approach relies on an appropriate trade-off between sufficiently harsh conditions to ensure that all DNA from dead cells is bound to the PMA dye, but at the same time, conditions that avoid damage and subsequent PMA-labelling of DNA from cells that were alive prior to the PMA treatment. Moreover, the reliability of viability-qPCR also depends on the correlation between the death of a bacterial cell and the increase of cell membrane impermeability. It is conceivable that no longer viable cells can still have intact membranes. Cell death without cell lysis or cell death not resulting in sufficient cell permeability of the cytoplasmic membrane will cause overestimation of the number of living cells. Therefore, the effect of bacteriostatic and bactericidal agents cannot be quantified by means of viability-qPCR. In contrast, killing by bacteriolytic agents, such as endolysins, should be quantifiable in a reliable manner.
Because of all the above-mentioned possible constraints and biases of viability-qPCR, we compared different parameters of the application of PMA to inhibit as completely as possible the amplification of DNA from dead cells without jeopardizing the integrity of the membranes of non-killed cells. This was carried out to quantify the ex vivo efficacy of PM-477, a genetically-engineered endolysin recently described [
20] and aimed to be a therapy for BV, that may be more efficient than current antibiotic-based treatments.
First, we opted to use PMA instead of EMA, because PMA has been shown to be more efficiently excluded from cells with intact cell membranes [
1], possibly due to its higher charge relative to EMA [
1]. PMA is related to propidium iodide, used for life/dead staining, but with an azide group instead of an amino group on the phenanthridine ring, which enables photo-induced cross-linking to dsDNA such as removing the chemically modified DNA as a template for qPCR.
Dye permeability of lipid bilayers depends to a large extent on temperature, whereby elevated temperatures during sample treatment have been shown to increase dye uptake. One study found that for
Salmonella typhimurium and
Listeria monocytogenes, PMA-based inhibition of amplification of DNA from dead cells was improved at temperatures up to 40 °C, while signals from live cells were not (
Salmonella) or only slightly affected (
Listeria) [
4]. Therefore, we compared PMAxx incubation on ice, at RT, 37 °C and 42 °C. We observed significantly better PMAxx penetration into the dead cells for incubation on ice, compared to incubation at RT, 37 and 42 °C.
Pan and Breidt [
2] reported no penetration of PMA to live
L. monocytogenes cells. For single PMAxx treatment, we found a log0.60 (±0.04) signal decrease of viable
G. swidsinskii and for triple treatment, we observed a log0.6–log1.65 decrease of signal for four
Gardnerella species, which were statistically significant (
p value < 0.05).
Kralik et al. [
31] compared single, double and triple PMA treatment (incubation during 5 min at RT) of around log8 CFU/mL cultured cells of a
Mycobacterium avium isolate and found that in case of double PMA treatment, the signal decrease of dead cells was about 1.5 cycles higher than in case of single treatment, thus increasing sensitivity. However, triple PMA treatment resulted in signal decrease for both live and dead cells. Although
Gardnerella has a thin Gram-positive cell wall [
32], whereas
M. avium is characterized by a thick cell wall, whereby the presence of mycolic acids may further decrease PMA penetration [
33]. To understand how many cycles of PMA treatment may be optimal for
Gardnerella, we also compared single, double and triple PMA treatments. In our study, double and triple treatment of live
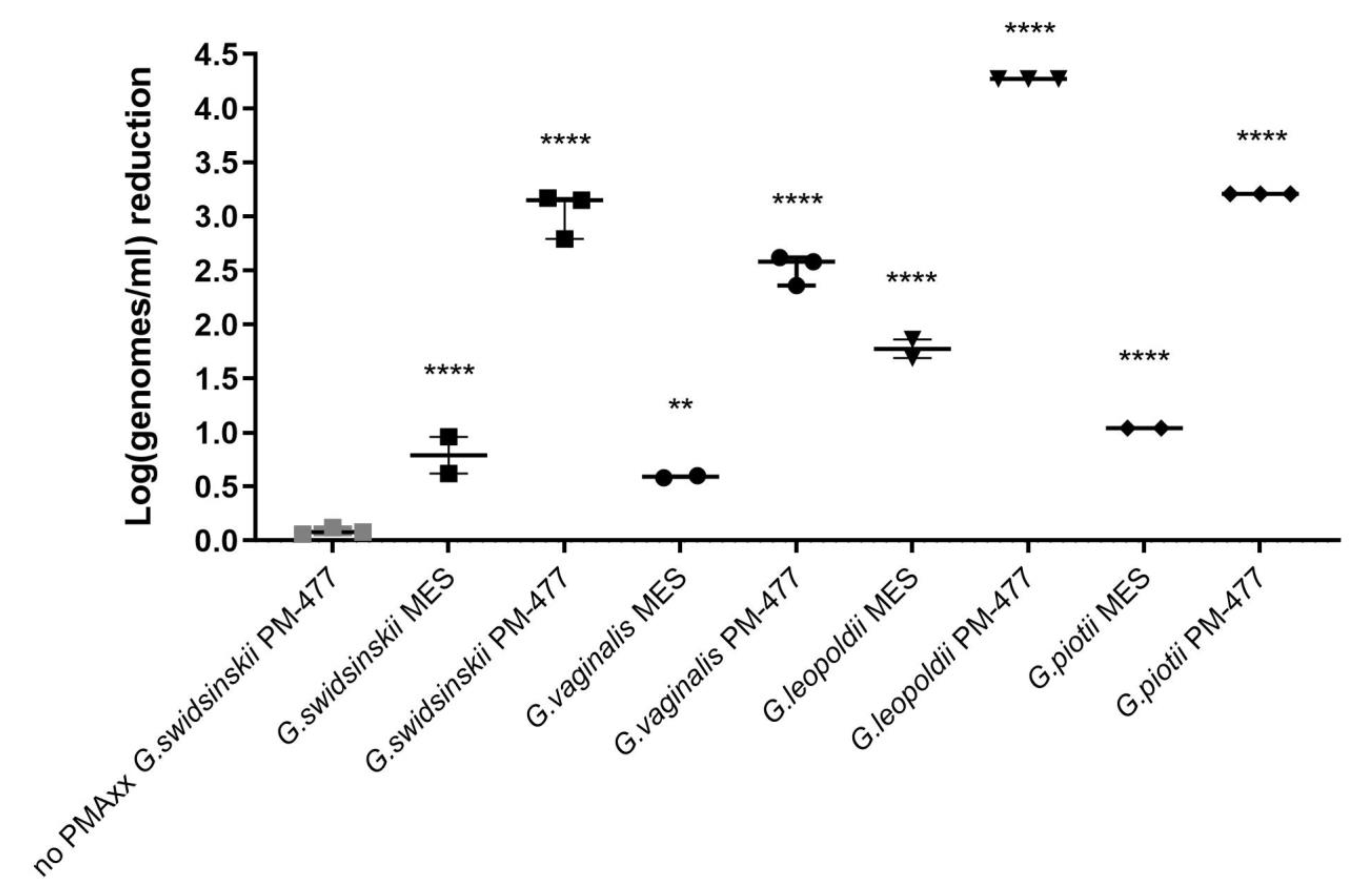
Gardnerella cells with 0.05 mM PMAxx resulted in the same level of signal decrease (log0.78 ± 0.02 and log0.75 ± 0.03, respectively). Signal reduction for PM-477 killed cells was log0.5 higher in case of three times PMAxx treatment compared to treatment performed two times (log1.7 ± 0.05 and log2.2 ± 0.06, respectively), which was statistically significant.
In the study of Pan and Breidt [
2], double 0.05 mM PMA treatment of approx. 2.4 log7
L. monocytogenes cells at room temperature was considered as optimal, causing 2.8 cycles reduction compared to single PMA treatment. No further reduction was obtained with 3 × PMA treatment. The same authors reported that neither increased dye concentrations (0.10 and 0.20 mM) nor increased incubation temperatures (23 °C and 40 °C) showed significant additional reduction. In the present study, the repetitive treatment was also the most efficient, with the highest log reduction observed for three times PMA. Moreover, in agreement with Pan and Breidt [
2], higher temperatures did not result in statistically significant stronger reduction than on ice incubation.
Viability-qPCR has been widely used, although the above literature review suggests that it is difficult to standardize and that treatment conditions (concentration of the dye, incubation time and temperature, number of treatments) should be adapted to each organism (and each sample type) studied. The efficacy of PMA-covalent binding as a means to inhibit PCR-based amplification of target DNA can be further jeopardized in case bacterial cells grow within clusters or biofilms. Pisz et al. [
34] suggested that the composition of the biofilms, particularly their extracellular polymeric substances, may interfere with either the DNA binding or the photoactivation of EMA. This interference may be similar to the way biofilm matrix polymers protect matrix-bound extracellular DNA from environmental nucleases [
35]. Furthermore, the extracellular biofilm DNA may reduce the concentrations of unbound PMA by binding it.
Another problem may be caused by the fact that the dye could adsorb (differently) onto different compounds present in the sample [
36]. EMA/PMA efficacy can be complicated by the composition of natural samples, as was found even for samples such as environmental water [
37], and which can be expected to be the case for clinical samples. Moreover, even samples of the same type, but from different individuals, may differ substantially with regard to their interaction with the DNA-binding compounds, as can be the case for vaginal swabs from women with BV. That might be one of the reasons why in our study, viability-PCR reached the detection limit for only half of the samples treated with PM-477. Viability-qPCR may be hampered as well by the presence of dead cells and by biofilms.
The presence of substantial amounts of dead cells also could interfere with DNA extraction or with amplification from viable cells. For example, the number of viable cells of
Escherichia coli was underestimated by viability-qPCR, when log8 dead and live cells per ml were present [
38]. We observed that when the ratio of dead to live cells exceeded 100:1, the number of live cells was overestimated. For
Listeria monocytogenes, Pan and Breidt [
2] reported that the ratio of dead cells to the live cells could be no greater than log5. Our results are consistent with those from Yang et al. [
3], who showed that a linear relationship between the log numbers and Cq value of live cells can be achieved only when viable cells comprise between 1 and 100% of the mixture of live and dead bacteria. When live bacteria were below 0.1% of the mixture, an increase in Cq value was not corresponding to the decrease in live cells. Similarly, Lovdal and coworkers [
39] discovered that for live cells being <1% of the live/dead mixture, the quantification by viability-qPCR was not reliable.
It has been suggested that the amplification of longer target regions might increase the chance that at least one dye molecule will bind to the targeted DNA region in damaged cells and as such decrease the probability that DNA from damaged cells is amplified [
40,
41]. In the present study, the region amplified was 95 bp, which might be prolonged to approximately 200 bp (which is the maximal target length for qPCR).
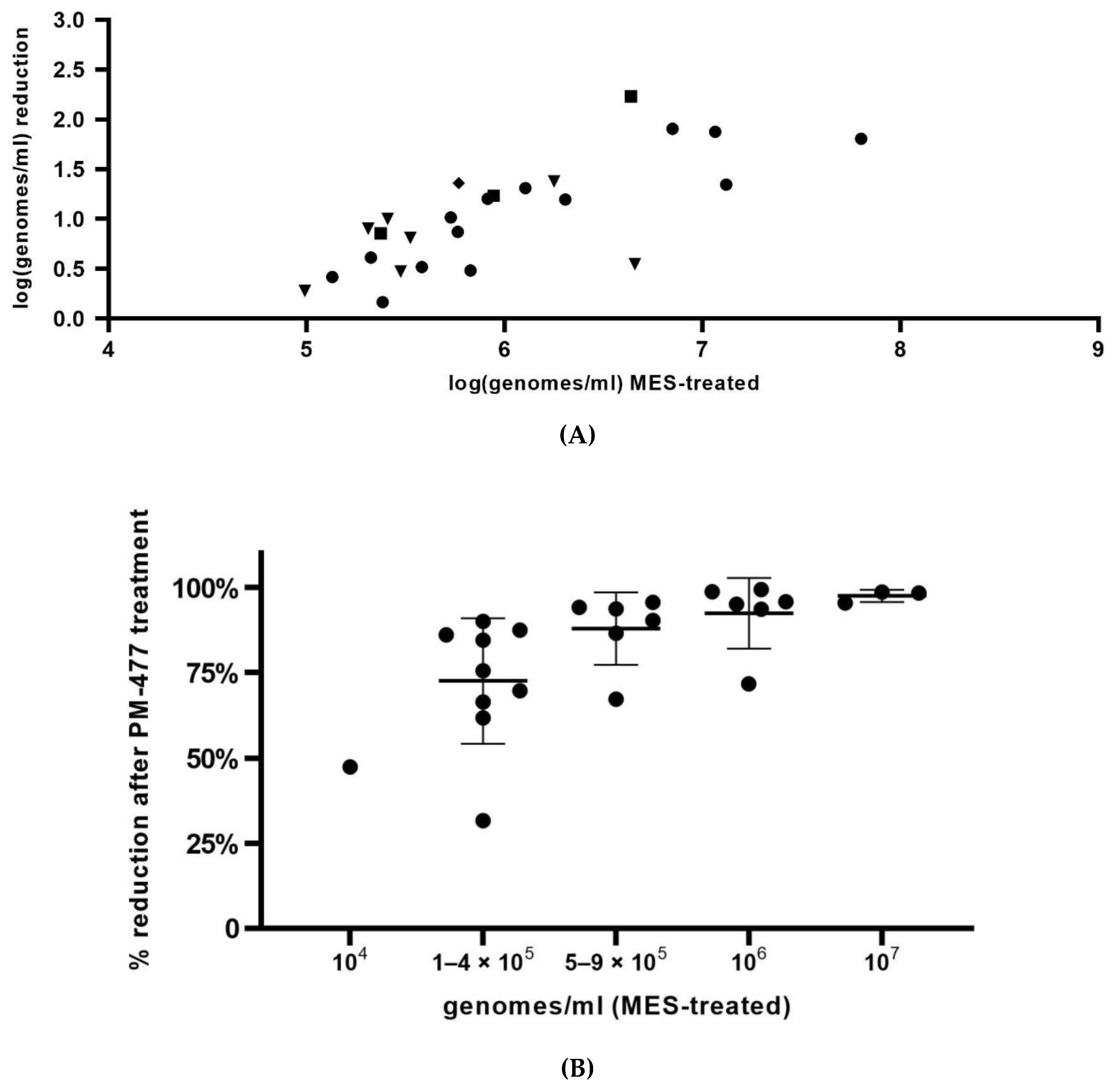
In summary, we optimized conditions of viability-qPCR for quantification of killing of planktonic cells of four different Gardnerella species by means of the PM-477 endolysin. We further validated this protocol in the clinical environment of vaginal samples from women with bacterial vaginosis. We were able to document up to log2.2 (99.4% reduction) reduction of Gardnerella cells after treatment with PM-477, despite the complex compositions of the samples. For the samples with the highest initial loads (log7 genomes/mL), we could achieve 95.5–98.7% (97.5% average) reduction in viable cells. Given that, as deduced above, the method of viability-qPCR may be reliable only when the live cells constitute between 1 and 100% of the sample; these values indicate that treatment with PM-477 reduced live Gardnerella to or below our limit of detection. The high Cq values (i.e., low number of live Gardnerella) measured in PM-477 treated samples cannot discriminate between remaining live cells and DNA from dead cells that has not been labelled by PMA.
Viability-qPCR remains a potential method for differentiation between live and dead bacterial cells in clinical samples, including clinical samples with biofilms [
9,
42], but seems to require optimization specific for different bacterial species and different sample types. Although it is not an absolute quantification method, documentation of full eradication, as is possible for the culture of planktonic cells, is difficult with viability-qPCR, which has higher quantification power than FISH. We previously found the FISH method to be only semi-quantitative whereby the true degree of killing of
Gardnerella cells was underestimated, possibly due to the staining of dead cells that still stick to the vaginal epithelial cells [
20].
In the present study, the bactericidal effect of endolysin PM-477, recently clearly documented to enable full eradication of planktonic
Gardnerella cells [
20], was confirmed in vaginal samples. We established viability-qPCR, which can detect reductions in live planktonic
Gardnerella cells of up to log4.0. In vaginal samples from women with BV, we measured strongly significant reductions of up to 99.4% (log2.2) of
Gardnerella cells. Reductions of
Gardnerella beyond this value were not measured. This might mean that the endolysin failed to eradicate the last cells from the vaginal biofilms. However, we deem it more likely that a higher log reduction was achieved but could not be documented as a consequence of technical limitations of the principle of viability-qPCR. At present, it is not possible to decide which is the case, because there are no valuable alternatives to confirm full eradication of (
Gardnerella) cells from (vaginal) biofilms in clinical samples.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}