
Interplay between Non-Coding RNA Transcription, Stringent/Relaxed Phenotype and Antibiotic Production in Streptomyces ambofaciens


 ,
,  , , ,
, , ,  ,
,
Abstract
1. Introduction
2. Results
2.1. S. ambofaciens Transcriptome: Structure and Organization
2.2. Novel sRNAs and Putative Targets
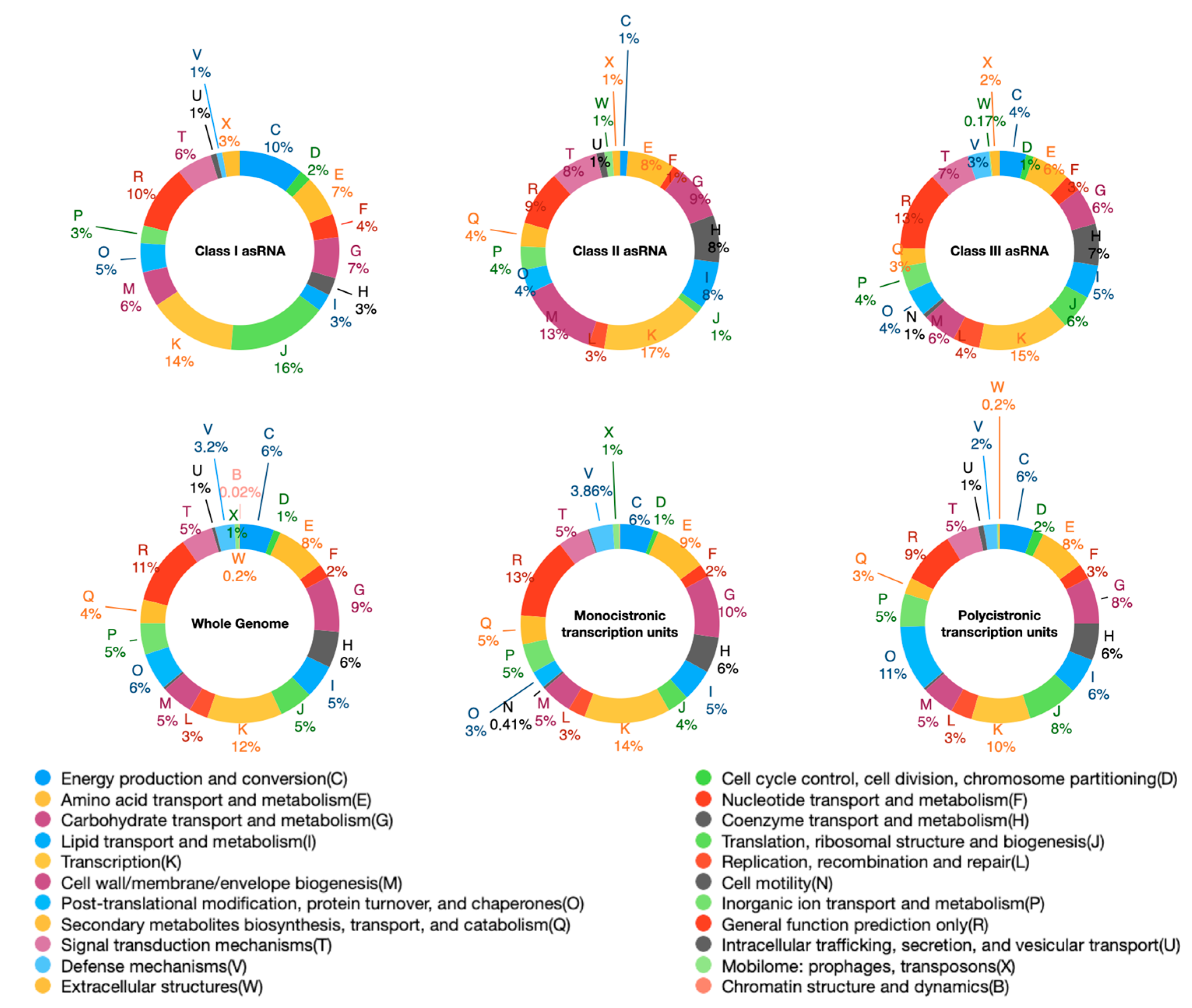
2.3. Antisense Transcription in S. ambofaciens
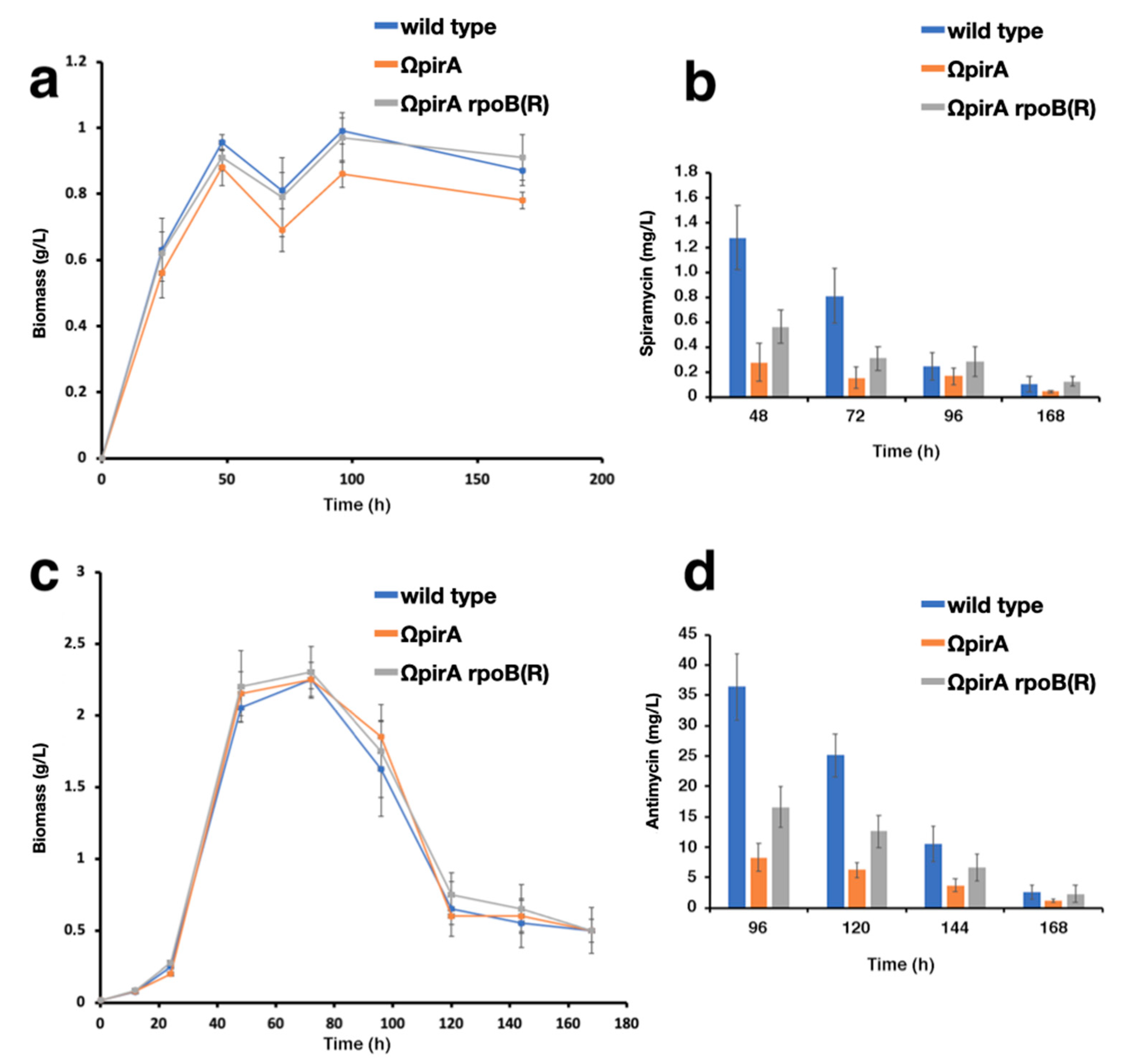
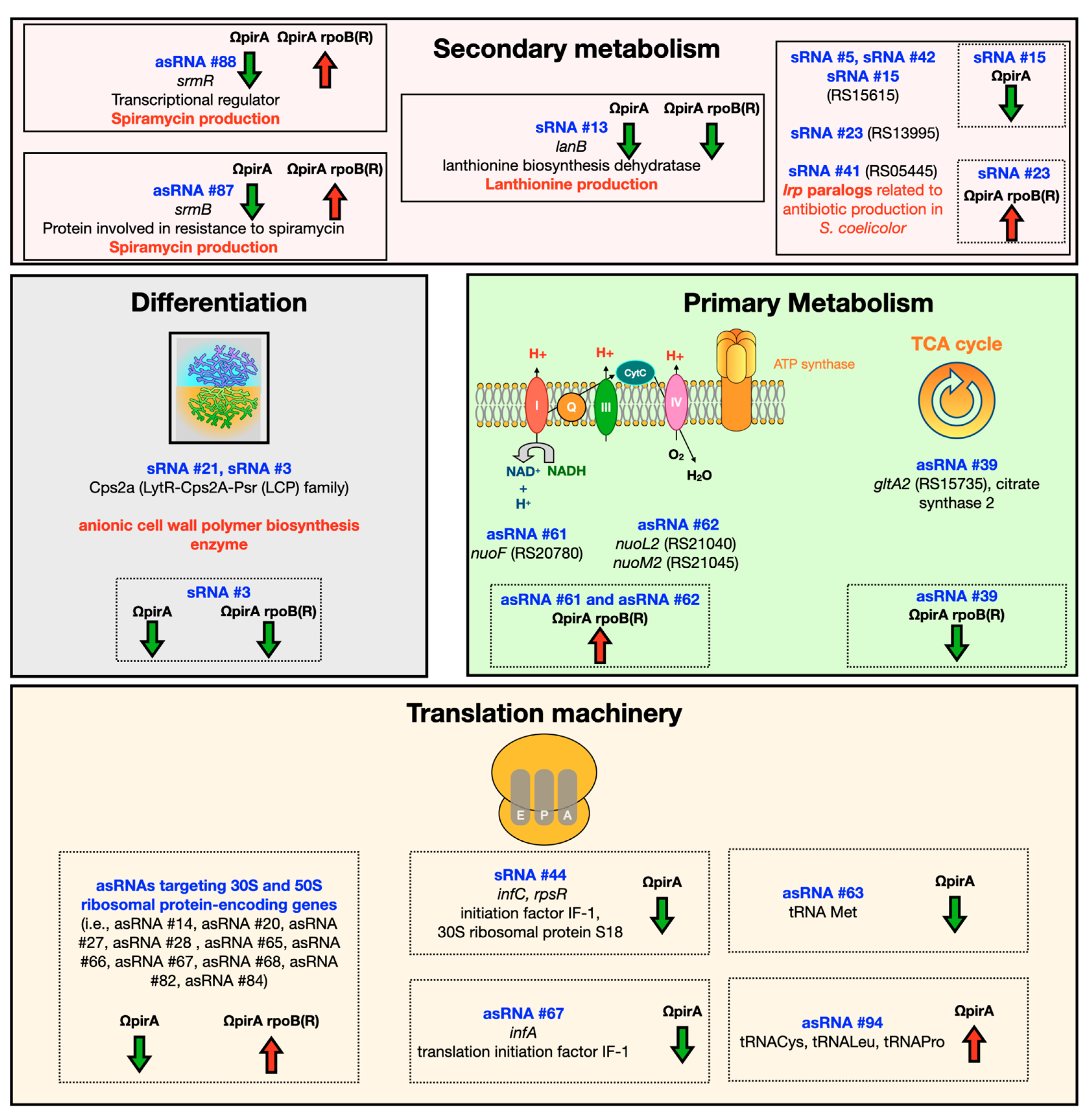
2.4. Expression of ncRNAs in S. ambofaciens ATCC 23877, and Derivative ΩpirA and ΩpirA rpoB(R) Mutants
3. Discussion and Conclusions
4. Material and Methods
4.1. Bacterial Strains, Media and Growth Conditions
4.2. Spiramycin Production Assay
4.3. Antimycin Production Assay
4.4. RNA Extraction and RNAseq Experiments
4.5. S. ambofaciens Transcriptome Re-definition, ncRNA Prediction and Antisense Transcripts Definition on the Main Chromosome
4.6. RNAseq Expression Analysis
4.7. ncRNAs Annotation and Functional Predictions
4.8. Code Availability
4.8.1. Gene Expression Analysis
4.8.2. Transcriptome Re-definition
4.8.3. ncRNAs Differential Expression
featureCounts -a <ncRNA.saf> -o <ncRNA_counts> -F SAF -t gene -s 1 -T 15 -p -B -C -J -G <GCF_001267885.1_ASM126788v1_genomic.fna> --fracOverlap 0.5 <BAM file list>
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brosse, A.; Guillier, M. Bacterial Small RNAs in Mixed Regulatory Networks. Microbiol. Spectr. 2018, 6. [Google Scholar] [CrossRef]
- Dutcher, H.A.; Raghavan, R. Origin, Evolution, and Loss of Bacterial Small RNAs. Microbiol. Spectr. 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, S.; Storz, G. Bacterial small RNA regulators: Versatile roles and rapidly evolving variations. Cold Spring Harb. Perspect. Biol. 2011, 3, a003798. [Google Scholar] [CrossRef]
- Waters, L.S.; Storz, G. Regulatory RNAs in bacteria. Cell 2009, 20, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Brophy, J.A.; Voigt, C.A. Antisense transcription as a tool to tune gene expression. Mol. Syst. Biol. 2016, 14, 854. [Google Scholar] [CrossRef] [PubMed]
- Saberi, F.; Kamali, M.; Najafi, A.; Yazdanparast, A.; Moghaddam, M.M. Natural antisense RNAs as mRNA regulatory elements in bacteria: A review on function and applications. Cell Mol. Biol. Lett. 2016, 28, 6. [Google Scholar] [CrossRef]
- Thomason, M.K.; Storz, G. Bacterial antisense RNAs: How many are there, and what are they doing? Annu. Rev. Genet. 2010, 44, 167–188. [Google Scholar] [CrossRef] [PubMed]
- Costa, F.F. Non-coding RNAs: Lost in translation? Gene 2007, 15, 1–10. [Google Scholar] [CrossRef]
- Hüttenhofer, A.; Schattner, P.; Polacek, N. Non-coding RNAs: Hope or hype? Trends Genet. 2005, 21, 289–297. [Google Scholar] [CrossRef]
- Lybecker, M.; Bilusic, I.; Raghavan, R. Pervasive transcription: Detecting functional RNAs in bacteria. Transcription 2014, 5, e944039. [Google Scholar] [CrossRef]
- Raghavan, R.; Sloan, D.B.; Ochman, H. Antisense transcription is pervasive but rarely conserved in enteric bacteria. mBio 2012, 7, e00156-12. [Google Scholar] [CrossRef] [PubMed]
- Wade, J.T.; Grainger, D.C. Pervasive transcription: Illuminating the dark matter of bacterial transcriptomes. Nat. Rev. Microbiol. 2014, 12, 647–653. [Google Scholar] [CrossRef]
- Lloréns-Rico, V.; Cano, J.; Kamminga, T.; Gil, R.; Latorre, A.; Chen, W.H.; Bork, P.; Glass, J.I.; Serrano, L.; Lluch-Senar, M. Bacterial antisense RNAs are mainly the product of transcriptional noise. Sci. Adv. 2016, 2, e1501363. [Google Scholar] [CrossRef] [PubMed]
- Georg, J.; Hess, W.R. cis-antisense RNA, another level of gene regulation in bacteria. Microbiol. Mol. Biol. Rev. 2011, 75, 286–300. [Google Scholar] [CrossRef]
- Gottesman, S. The small RNA regulators of Escherichia coli: Roles and mechanisms. Annu. Rev. Microbiol. 2004, 58, 303–328. [Google Scholar] [CrossRef]
- Gottesman, S. Micros for microbes: Non-coding regulatory RNAs in bacteria. Trends Genet. 2005, 21, 399–404. [Google Scholar] [CrossRef]
- Li, L.; Huang, D.; Cheung, M.K.; Nong, W.; Huang, Q.; Kwan, H.S. BSRD: A repository for bacterial small regulatory RNA. Nucleic Acids Res. 2013, 41, D233–D238. [Google Scholar] [CrossRef] [PubMed]
- Bejerano-Sagie, M.; Xavier, K.B. The role of small RNAs in quorum sensing. Curr. Opin. Microbiol. 2007, 10, 189–198. [Google Scholar] [CrossRef]
- Toledo-Arana, A.; Repoila, F.; Cossart, P. Small noncoding RNAs controlling pathogenesis. Curr. Opin. Microbiol. 2007, 10, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Moody, M.J.; Young, R.A.; Jones, S.E.; Elliot, M.A. Comparative analysis of non-coding RNAs in the antibiotic-producing Streptomyces bacteria. BMC Genom. 2013, 14, 558. [Google Scholar] [CrossRef]
- Pánek, J.; Bobek, J.; Mikulík, K.; Basler, M.; Vohradský, J. Biocomputational prediction of small non-coding RNAs in Streptomyces. BMC Genom. 2008, 9, 217. [Google Scholar] [CrossRef]
- Swiercz, J.P.; Bobek, J.; Haiser, H.J.; Berardo, C.D.; Tjaden, B.; Elliot, M.A. Small non-coding RNAs in Streptomyces coelicolor. Nucleic Acids Res. 2008, 36, 7240–7251. [Google Scholar] [CrossRef][Green Version]
- Vockenhuber, M.P.; Sharma, C.M.; Statt, M.G.; Schmidt, D.; Xu, Z.; Dietrich, S.; Liesegang, H.; Mathews, D.H.; Suess, B. Deep sequencing-based identification of small non-coding RNAs in Streptomyces coelicolor. RNA Biol. 2011, 8, 468–477. [Google Scholar] [CrossRef][Green Version]
- Champness, W.C.; Chater, K.F. The regulation and integration of antibiotic production and morphological differentiation. In Regulation of Bacterial Differentiation in Streptomyces spp.; Piggot, P., Moran, C.P., Youngman, P., Eds.; American Society for Microbiology: Washington, DC, USA, 1994; pp. 61–94. [Google Scholar]
- D’Alia, D.; Nieselt, K.; Steigele, S.; Muller, J.; Verburg, I.; Takano, E. Noncoding RNA of glutamine synthetase I modulates antibiotic production in Streptomyces coelicolor A3(2). J. Bacteriol. 2010, 192, 1160–1164. [Google Scholar] [CrossRef]
- Mikulik, K.; Bobek, J.; Zidkova, J.; Felsberg, J. 6S RNA modulates growth and antibiotic production in Streptomyces coelicolor. Appl. Microbiol. Biotechnol. 2014, 98, 7185–7197. [Google Scholar] [CrossRef]
- Palecková, P.; Felsberg, J.; Bobek, J.; Mikulík, K. tmRNA abundance in Streptomyces aureofaciens, S. griseus and S. collinus under stress-inducing conditions. Folia Microbiol. (Praha) 2007, 52, 463–470. [Google Scholar] [CrossRef]
- Šetinová, D.; Šmídová, K.; Pohl, P.; Musić, I.; Bobek, J. RNase III-Binding-mRNAs Revealed Novel Complementary Transcripts in Streptomyces. Front. Microbiol. 2018, 8, 2693. [Google Scholar] [CrossRef]
- Leistra, A.N.; Curtis, N.C.; Contreras, L.M. Regulatory non-coding sRNAs in bacterial metabolic pathway engineering. Metab. Eng. 2018, 52, 190–214. [Google Scholar] [CrossRef]
- Chew, W.K.; Segarra, I.; Ambu, S.; Mak, J.W. Significant reduction of brain cysts caused by Toxoplasma gondii after treatment with spiramycin coadministered with metronidazole in a mouse model of chronic toxoplasmosis. Antimicrob. Agents Chemother. 2012, 56, 1762–1768. [Google Scholar] [CrossRef] [PubMed]
- Poulet, P.P.; Duffaut, D.; Barthet, P.; Brumpt, I. Concentrations and in vivo antibacterial activity of spiramycin and metronidazole in patients with periodontitis treated with high-dose metronidazole and the spiramycin/metronidazole combination. J. Antimicrob. Chemother. 2005, 55, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Cosar, C.; Ninet, L.; Pinnert-Sindico, S.; Preud’homme, J. Activité trypanocide d’un antibiotique produit par un streptomyces [Trypanocide action of an antibiotic produced by a Streptomyces]. Comptes Rendus Hebd. Séances L’académie Sci. 1952, 234, 1498–1499. [Google Scholar]
- Thibessard, A.; Haas, D.; Gerbaud, G.; Aigle, B.; Lautru, S.; Perdonet, J.L.; Leblond, P. Complete genome sequence of Streptomyces ambofaciens ATCC 23877, the spiramycin producer. J. Biotechnol. 2015, 214, 117–118. [Google Scholar] [CrossRef] [PubMed]
- Fondi, M.; Pinatel, E.; Talà, A.; Damiano, F.; Consolandi, C.; Mattorre, B.; Fico, D.; Testini, M.; De Benedetto, G.E.; Siculella, L.; et al. Time-Resolved Transcriptomics and Constraint-Based Modeling Identify System-Level Metabolic Features and Overexpression Targets to Increase Spiramycin Production in Streptomyces ambofaciens. Front. Microbiol. 2017, 8, 835. [Google Scholar] [CrossRef]
- Talà, A.; Damiano, F.; Gallo, G.; Pinatel, E.; Calcagnile, M.; Testini, M.; Fico, D.; Rizzo, D.; Sutera, A.; Renzone, G.; et al. Pirin: A novel redox-sensitive modulator of primary and secondary metabolism in Streptomyces. Metab. Eng. 2018, 48, 254–268. [Google Scholar] [CrossRef]
- Talà, A.; Wang, G.; Zemanova, M.; Okamoto, S.; Ochi, K.; Alifano, P. Activation of dormant bacterial genes by Nonomuraea sp. strain ATCC 39727 mutant-type RNA polymerase. J. Bacteriol. 2009, 191, 805–814. [Google Scholar] [CrossRef]
- D’Argenio, V.; Petrillo, M.; Pasanisi, D.; Pagliarulo, C.; Colicchio, R.; Talà, A.; de Biase, M.S.; Zanfardino, M.; Scolamiero, E.; Pagliuca, C.; et al. The complete 12 Mb genome and transcriptome of Nonomuraea gerenzanensis with new insights into its duplicated "magic" RNA polymerase. Sci. Rep. 2016, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- McClure, R.; Balasubramanian, D.; Sun, Y.; Bobrovskyy, M.; Sumby, P.; Genco, C.A.; Vanderpool, C.K.; Tjaden, B. Computational analysis of bacterial RNA-Seq data. Nucleic Acids Res. 2013, 41, e140. [Google Scholar] [CrossRef]
- Tjaden, B. De novo assembly of bacterial transcriptomes from RNA-seq data. Genome Biol. 2015, 16, 1. [Google Scholar] [CrossRef]
- Tjaden, B. A computational system for identifying operons based on RNA-seq data. Methods 2020, 1, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Mai, J.; Rao, C.; Watt, J.; Sun, X.; Lin, C.; Zhang, L.; Liu, J. Mycobacterium tuberculosis 6C sRNA binds multiple mRNA targets via C-rich loops independent of RNA chaperones. Nucleic Acids Res. 2019, 47, 4292–4307. [Google Scholar] [CrossRef]
- Bertram, R.; Schlicht, M.; Mahr, K.; Nothaft, H.; Saier, M.H., Jr.; Titgemeyer, F. In silico and transcriptional analysis of carbohydrate uptake systems of Streptomyces coelicolor A3(2). J. Bacteriol. 2004, 186, 1362–1373. [Google Scholar] [CrossRef]
- Weinberg, Z.; Lünse, C.E.; Corbino, K.A.; Ames, T.D.; Nelson, J.W.; Roth, A.; Perkins, K.R.; Sherlock, M.E.; Breaker, R.R. Detection of 224 candidate structured RNAs by comparative analysis of specific subsets of intergenic regions. Nucleic Acids Res. 2017, 45, 10811–10823. [Google Scholar] [CrossRef]
- Heueis, N.; Vockenhuber, M.P.; Suess, B. Small non-coding RNAs in streptomycetes. RNA Biol. 2014, 11, 464–469. [Google Scholar] [CrossRef][Green Version]
- Vockenhuber, M.P.; Suess, B. Streptomyces coelicolor sRNA scr5239 inhibits agarase expression by direct base pairing to the dagA coding region. Microbiology 2012, 158 Pt 2, 424–435. [Google Scholar] [CrossRef]
- Vockenhuber, M.P.; Heueis, N.; Suess, B. Identification of metE as a second target of the sRNA scr5239 in Streptomyces coelicolor. PLoS ONE 2015, 10, e0120147. [Google Scholar] [CrossRef]
- Moody, M.J.; Jones, S.E.; Elliot, M.A. Complex intra-operonic dynamics mediated by a small RNA in Streptomyces coelicolor. PLoS ONE 2014, 9, e85856. [Google Scholar] [CrossRef]
- Lott, S.C.; Schäfer, R.A.; Mann, M.; Backofen, R.; Hess, W.R.; Voß, B.; Georg, J. GLASSgo-Automated and reliable detection of sRNA homologs from a single input ssequence. Front. Genet. 2018, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Raden, M.; Ali, S.M.; Alkhnbashi, O.S.; Busch, A.; Costa, F.; Davis, J.A.; Eggenhofer, F.; Gelhausen, R.; Georg, J.; Heyne, S.; et al. Freiburg RNA tools: A central online resource for RNA-focused research and teaching. Nucleic Acids Res. 2018, 46, W25–W29. [Google Scholar] [CrossRef]
- Schäfer, R.A.; Lott, S.C.; Georg, J.; Grüning, B.A.; Hess, W.R.; Voß, B. GLASSGo in Galaxy: High-throughput, reproducible and easy-to-integrate prediction of sRNA homologs. Bioinformatics 2020, 36, 4357–4359. [Google Scholar] [CrossRef]
- Busch, A.; Richter, A.S.; Backofen, R. IntaRNA: Efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions. Bioinformatics 2008, 24, 2849–2856. [Google Scholar] [CrossRef]
- Mann, M.; Wright, P.R.; Backofen, R. IntaRNA 2.0: Enhanced and customizable prediction of RNA-RNA interactions. Nucleic Acids Res. 2017, 45, W435–W439. [Google Scholar] [CrossRef]
- Tschowri, N. Cyclic Dinucleotide-Controlled Regulatory Pathways in Streptomyces Species. J. Bacteriol 2016, 198, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Millan-Oropeza, A.; Henry, C.; Lejeune, C.; David, M.; Virolle, M.J. Expression of genes of the Pho regulon is altered in Streptomyces coelicolor. Sci. Rep. 2020, 10, 8492. [Google Scholar] [CrossRef]
- Aínsa, J.A.; Ryding, N.J.; Hartley, N.; Findlay, K.C.; Bruton, C.J.; Chater, K.F. WhiA, a protein of unknown function conserved among Gram- positive bacteria, is essential for sporulation in Streptomyces coelicolor A3(2). J. Bacteriol. 2000, 182, 5470–5478. [Google Scholar] [CrossRef]
- Bush, M.J.; Chandra, G.; Bibb, M.J.; Findlay, K.C.; Buttner, M.J. Genome-wide chromatin immunoprecipitation sequencing analysis shows that WhiB is a transcription factor that cocontrols its regulon with WhiA to initiate developmental cell division in Streptomyces. mBio 2016, 7, e00523-16. [Google Scholar] [CrossRef]
- Geistlich, M.; Losick, R.; Turner, J.R.; Rao, R.N. Characterization of a novel regulatory gene governing the expression of a polyketide synthase gene in Streptomyces ambofaciens. Mol. Microbiol. 1992, 6, 2019–2029. [Google Scholar] [CrossRef] [PubMed]
- Karray, F.; Darbon, E.; Nguyen, H.C.; Gagnat, J.; Pernodet, J.L. Regulation of the biosynthesis of the macrolide antibiotic spiramycin in Streptomyces ambofaciens. J. Bacteriol. 2010, 192, 5813–5821. [Google Scholar] [CrossRef] [PubMed]
- Vigliotta, G.; Tredici, S.M.; Damiano, F.; Montinaro, M.R.; Pulimeno, R.; Di Summa, R.; Massardo, D.R.; Gnoni, V.G.; Alifano, P. Natural merodiploidy involving duplicated rpoB alleles affects secondary metabolism in a producer actinomycete. Mol. Microbiol. 2005, 55, 396–412. [Google Scholar] [CrossRef]
- Richardson, M.A.; Kuhstoss, S.; Huber, M.L.; Ford, L.; Godfrey, O.; Turner, J.R.; Rao, R.N. Cloning of spiramycin biosynthetic genes and their use in constructing Streptomyces ambofaciens mutants defective in spiramycin biosynthesis. J. Bacteriol. 1990, 172, 3790–3798. [Google Scholar] [CrossRef]
- Su, W.; Kumar, V.; Ding, Y.; Ero, R.; Serra, A.; Lee, B.S.T.; Wong, A.S.W.; Shi, J.; Sze, S.K.; Yang, L.; et al. Ribosome protection by antibiotic resistance ATP-binding cassette protein. Proc. Natl. Acad. Sci. USA 2018, 115, 5157–5162. [Google Scholar] [CrossRef]
- Liu, J.; Li, J.; Dong, H.; Chen, Y.; Wang, Y.; Wu, H.; Li, C.; Weaver, D.T.; Zhang, L.; Zhang, B. Characterization of an Lrp/AsnC family regulator SCO3361, controlling actinorhodin production and morphological development in Streptomyces coelicolor. Appl. Microbiol. Biotechnol. 2017, 101, 5773–5783. [Google Scholar] [CrossRef]
- Lee, H.J.; Gottesman, S. sRNA roles in regulating transcriptional regulators: Lrp and SoxS regulation by sRNAs. Nucleic Acids Res. 2016, 44, 6907–6923. [Google Scholar] [CrossRef]
- Kang, S.M.; Kim, D.H.; Jin, C.; Ahn, H.C.; Lee, B.J. The crystal structure of AcrR from Mycobacterium tuberculosis reveals a one-component transcriptional regulation mechanism. FEBS Open Bio 2019, 9, 1713–1725. [Google Scholar] [CrossRef]
- Goerlich, O.; Foeckler, R.; Holler, E. Mechanism of synthesis of adenosine(5′) tetraphospho (5′)adenosine (AppppA) by aminoacyl-tRNA synthetases. Eur. J. Biochem. 1982, 126, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Kisselev, L.L.; Justesen, J.; Wolfson, A.D.; Frolova, L.Y. Diadenosine oligophosphates (Ap(n)A), a novel class of signalling molecules? FEBS Lett. 1998, 8, 157–163. [Google Scholar] [CrossRef]
- Guranowski, A. Specific and nonspecific enzyme involved in the catabolism of mononucleoside and dinuceoside polyphosphases. Pharmacol. Ther. 2000, 87, 117–139. [Google Scholar] [CrossRef]
- Battchikova, N.; Eisenhut, M.; Aro, E.-M. Cyanobacterial NDH-1 complexes: Novel insights and remaining puzzles. Biochim. Biophys. Acta 2011, 1807, 935–944. [Google Scholar] [CrossRef]
- Khayrullina, G.A.; Raabe, C.A.; Hoe, C.H.; Becker, K.; Reinhardt, R.; Tang, T.H.; Rozhdestvensky, T.S.; Kopylov, A.M. Transcription analysis and small non-protein coding RNAs associated with bacterial ribosomal protein operons. Curr. Med. Chem. 2012, 19, 5187–5198. [Google Scholar] [CrossRef]
- Asato, Y. Control of ribosome synthesis during the cell division cycles of E. coli and Synechococcus. Curr. Issues Mol. Biol. 2005, 7, 109–117. [Google Scholar]
- Fu, Y.; Deiorio-Haggar, K.; Anthony, J.; Meyer, M.M. Most RNAs regulating ribosomal protein biosynthesis in Escherichia coli are narrowly distributed to Gammaproteobacteria. Nucleic Acids Res. 2013, 41, 3491–3503. [Google Scholar] [CrossRef]
- Blanco, G.; Sánchez, C.; Rodicio, M.R.; Méndez, C.; Salas, J.A. Identification of a growth phase-dependent promoter in the rplJL operon of Streptomyces coelicolor A3(2). Biochim. Biophys. Acta 2001, 1517, 243–249. [Google Scholar] [CrossRef]
- Küster, C.; Piepersberg, W.; Distler, J. Cloning and transcriptional analysis of the rplKA-or f31-rplJL gene cluster of Streptomyces griseus. Mol. Gen. Genet. 1998, 257, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Chater, K.F.; Chandra, G. The use of the rare UUA codon to define “expression space” for genes involved in secondary metabolism, development and environmental adaptation in Streptomyces. J. Microbiol. 2008, 46, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bush, M.J.; Tschowri, N.; Schlimpert, S.; Flärdh, K.; Buttner, M.J. c-di-GMP signaling and the regulation of developmental transitions in Streptomyces. Nat. Rev. Microbiol. 2015, 13, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Martinez, S.E.; Beavo, J.A.; Hol, W.G. GAF domains: Two-billion-year-old molecular switches that bind cyclic nucleotides. Mol. Interv. 2002, 2, 317–323. [Google Scholar] [CrossRef]
- Sardiwal, S.; Kendall, S.L.; Movahedzadeh, F.; Rison, S.C.; Stoker, N.G.; Djordjevic, S. A GAF domain in the hypoxia/NO-inducible Mycobacterium tuberculosis DosS protein binds haem. J. Mol. Biol. 2005, 353, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Tucker, N.P.; D’autréaux, B.; Spiro, S.; Dixon, R. Mechanism of transcriptional regulation by the Escherichia coli nitric oxide sensor NorR. Biochem. Soc. Trans. 2006, 34, 191–194. [Google Scholar] [CrossRef]
- Chater, K.F. Recent advances in understanding Streptomyces. F1000Research 2016, 5, 2795. [Google Scholar] [CrossRef]
- Hesketh, A.; Chen, W.J.; Ryding, J.; Chang, S.; Bibb, M. The global role of ppGpp synthesis in morphological differentiation and antibiotic production in Streptomyces coelicolor A3(2). Genome Biol. 2007, 8, R161. [Google Scholar] [CrossRef]
- Ochi, K. Metabolic initiation of differentiation and secondary metabolism by Streptomyces griseus: Significance of the stringent response (ppGpp) and GTP content in relation to A factor. J. Bacteriol. 1987, 169, 3608–3616. [Google Scholar] [CrossRef]
- Kieser, T.; Bibb, M.J.; Buttner, M.J.; Chater, K.F.; Hopwood, D.A. Practical Streptomyces Genetics; The John Innes Foundation: Norwich, UK, 2000. [Google Scholar]
- Calcagnile, M.; Bettini, S.; Damiano, F.; Talà, A.; Tredici, S.M.; Pagano, R.; Di Salvo, M.; Siculella, L.; Fico, D.; De Benedetto, G.E.; et al. Stimulatory effects of methyl-β-cyclodextrin on spiramycin production, and physical chemical characterization of nonhost@guest complexes. ACS Omega 2018, 3, 2470–2478. [Google Scholar] [CrossRef] [PubMed]
- De Benedetto, G.E.; Fanigliulo, M. A new CE-ESI-MS method for the detection of stable hemoglobin acetaldehyde adducts, potential biomarkers of alcohol abuse. Electrophoresis 2009, 30, 1798–1807. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 15, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 4, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Pinatel, E.; Peano, C. RNA Sequencing and Analysis in Microorganisms for Metabolic Network Reconstruction. Methods Mol. Biol 2018, 1716, 239–265. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Kalvari, I.; Argasinska, J.; Quinones-Olvera, N.; Nawrocki, E.P.; Rivas, E.; Eddy, S.R.; Bateman, A.; Finn, R.D.; Petrov, A.I. Rfam 13.0: Shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res. 2018, 46, D335–D342. [Google Scholar] [CrossRef]
- Kalvari, I.; Nawrocki, E.P.; Argasinska, J.; Quinones-Olvera, N.; Finn, R.D.; Bateman, A.; Petrov, A.I. Non-Coding RNA Analysis Using the Rfam Database. Curr. Protoc. Bioinform. 2018, 62, e51. [Google Scholar] [CrossRef]
- Richter, A.S.; Backofen, R. Accessibility and conservation: General features of bacterial small RNA-mRNA interactions? RNA Biol. 2012, 9, 954–965. [Google Scholar] [CrossRef]
- Nakamura, Y.; Gojobori, T.; Ikemura, T. Codon usage tabulated from the international DNA sequence databases. Nucleic Acids Res. 1998, 26, 334. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Nueda, M.J.; Ferrer, A.; Talón, M. maSigPro: A method to identify significantly differential expression profiles in time-course microarray experiments. Bioinformatics 2006, 22, 1096–1102. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcriptional Units | Number | Length (Median) |
|---|---|---|
| Monocistronic | 4433 | 902 |
| Polycistronic | 1154 | 2177 |
| sRNA | 45 | 100 |
| asRNA I | 119 | 166 |
| Transcriptional Units | Number | Overlap Length (Median) |
| asRNA I | 119 | 150 |
| asRNA II | 83 | 113 |
| asRNA III (cutoRNA) | 507 | 167 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinatel, E.; Calcagnile, M.; Talà, A.; Damiano, F.; Siculella, L.; Peano, C.; De Benedetto, G.E.; Pennetta, A.; De Bellis, G.; Alifano, P. Interplay between Non-Coding RNA Transcription, Stringent/Relaxed Phenotype and Antibiotic Production in Streptomyces ambofaciens. Antibiotics 2021, 10, 947. https://doi.org/10.3390/antibiotics10080947
Pinatel E, Calcagnile M, Talà A, Damiano F, Siculella L, Peano C, De Benedetto GE, Pennetta A, De Bellis G, Alifano P. Interplay between Non-Coding RNA Transcription, Stringent/Relaxed Phenotype and Antibiotic Production in Streptomyces ambofaciens. Antibiotics. 2021; 10(8):947. https://doi.org/10.3390/antibiotics10080947
Chicago/Turabian StylePinatel, Eva, Matteo Calcagnile, Adelfia Talà, Fabrizio Damiano, Luisa Siculella, Clelia Peano, Giuseppe Egidio De Benedetto, Antonio Pennetta, Gianluca De Bellis, and Pietro Alifano. 2021. "Interplay between Non-Coding RNA Transcription, Stringent/Relaxed Phenotype and Antibiotic Production in Streptomyces ambofaciens" Antibiotics 10, no. 8: 947. https://doi.org/10.3390/antibiotics10080947
APA StylePinatel, E., Calcagnile, M., Talà, A., Damiano, F., Siculella, L., Peano, C., De Benedetto, G. E., Pennetta, A., De Bellis, G., & Alifano, P. (2021). Interplay between Non-Coding RNA Transcription, Stringent/Relaxed Phenotype and Antibiotic Production in Streptomyces ambofaciens. Antibiotics, 10(8), 947. https://doi.org/10.3390/antibiotics10080947