Abstract
One of the biggest threats we face globally is the emergence of antimicrobial-resistant (AMR) bacteria, which runs in parallel with the lack in the development of new antimicrobials. Among these AMR bacteria pathogens belonging to the ESKAPE group can be highlighted (Enterococcus spp., Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacter spp.) due to their profile of drug resistance and virulence. Therefore, innovative lines of treatment must be developed for these bacteria. In this review, we summarize the different strategies for the treatment and study of molecular mechanisms of AMR in the ESKAPE pathogens based on the clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated (Cas) proteins’ technologies: loss of plasmid or cellular viability, random mutation or gene deletion as well directed mutations that lead to a gene’s loss of function.
1. CRISPR-Cas: An Adaptive Prokaryotic Immune System
Clustered regularly interspaced palindromic repeats (CRISPR) and the subsequent CRISPR-associated (Cas) proteins constitute an adaptive immune system in both bacteria and archaea. They were first identified in Escherichia coli in 1897 by Ishino and colleagues [1] and renamed as CRISPR by Jansen and colleagues [2]. However, it was Spanish microbiologist Francis Mojica who deepened their function and significance while studying the halophilic archaeon Haloferax mediterranei [3,4,5]. Finally, Doudna and Charpentier’s groups unraveled the process through which CRISPR-derived RNAs (crRNA) are processed, directed by the transactivating CRISPR RNAs (tracrRNA) [6,7,8,9]. Both of them were recently awarded with the Nobel Prize in Chemistry 2020 “for the development of a method for genome editing”.
The structure of a CRISPR-Cas system consists of an array of exogenous DNA sequences (known as spacers) flanked by similarly sized and identical direct and inverted repeats (known as palindromic repeats), as well as upstream cas genes. These spacers derive from foreign phages and/or plasmids entering the bacterial cell, and its chronologic insertion into the CRISPR array implies the acquisition of a “memory fragment” of those invaders. If the exogenous DNA enters the cell in the future, the bacterium specifically recognizes the DNA sequences matching its spacers and cleaves them through the Cas proteins’ nuclease activity, serving as a sequence-specific bacterial defense [10].
To avoid autoimmunity caused by the bacterium targeting its own DNA, protospacer adjacent motifs (PAMs) are 2–6 base pair (bp) sequences located within the invader DNA, near the Cas protein’s target. Without a PAM sequence, the CRISPR-Cas system is unable to bind to the target sequence and induce the strand separation required for the nuclease domain to act [9,11,12].
CRISPR-Cas systems can be classified into two main classes, each one divided into different types. Class I encompasses types I, III and IV, in which the identification of the target sequence and its cleavage is handled by a set of different proteins (Cas5, Cas7, SS, etc.). For class II, both identification and cleavage of the target sequence is performed by a single enzyme: Cas9 for type II, Cas12 for type V and Cas13 for type VI (Figure 1).
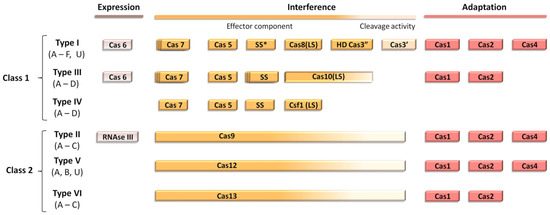
Figure 1.
Modular organization of the different classes of CRISPR-Cas systems. Scheme adapted from Ishino et al. SS* indicates that the putative small subunit (SS) might be fused to the large subunit in several type I subtypes [13].
CRISPR-Cas system’s activity is usually classified into three different steps, as reviewed by Strich and Chertow in 2018: adaptation, crRNA maturation and interference [11]. Firstly, PAM sequences within the exogenous DNA are recognized and cleaved into small fragments by Cas1, Cas2 and Cas4 enzymes. This process, known as adaptation, consists of the sequential integration of the resulting DNA fragments into the CRISPR array, next to the AT-rich motif. This requires the intervention of further enzymes, such as the integrated host factor (IHF), Cas9, Csn2 or tracrRNA, and their participation depends on the CRISPR-Cas system’s class. Then, crRNA maturation occurs when the CRISPR array is transcribed, and the resulting pre-crRNA is processed into individual crRNAs by the Cas proteins, which specifically recognize repeats. The secondary structure of the transcript given by its palindromic repeats, which form a loop, is essential for spacer identification and cleavage by Cas proteins in systems I-E, V-A and VI-A. For type III-A, a Cas6 dimer is responsible for crRNA maturation, and no loop formation is needed, whereas in type type II-A, RNase III cleaves the CRISPR transcript upon formation of a complex between the spacer, a tracrRNA and a Cas9 protein. Finally, interference takes place when the crRNA:Cas protein complex (crRNA:tracrRNA:Cas protein complex in type II-A) is directed to the invading DNA, specifically recognizes the target sequence and cleaves it, causing a fatal double-strand break [11].
2. CRISPR-Cas: A New Concept of Antimicrobials
During the last decades, the slow rate of new antimicrobial development when compared to the rapidity through which microorganisms (including parasites, virus and, specially, fungi and bacteria) acquire resistance to them has been a hot topic in microbiology. Costs derived from drug research and development, together with the limited duration of the antimicrobial treatment until the resolution of the infection, dramatically reduce the benefits of this industry. In addition, the risk of an eventual loss of efficacy due to the emergence of AMR makes the antimicrobial drug industry unappealing to pharmaceutical companies, which tend to focus on more profitable topics such as chronic diseases [10,14].
In this context, new approaches against pathogenic bacteria have emerged, with different mechanisms of action: antimicrobial peptides, metal nanoparticles, bacteriophages and gene edition tools [15]. The latter are of great interest due to their ability to target and cleave precise sequences within the bacterial genome in a species-specific manner, resulting in antimicrobials with the narrowest possible spectrum. These gene edition tools are zinc fingers [16,17], transcription activation-like effector nucleases (TALENs) [18], peptide nucleic acids [19], RNA interference (RNAi) [20] and CRISPR-Cas systems [10,21]. In the first three, specificity is given by protein–DNA interactions, which require protein engineering for its development. This makes it challenging, expensive and time-consuming to reshape the effector proteins in order to adapt them to new targets. However, CRISPR-Cas specificity is achieved through RNA-DNA interactions, with RNA engineering being much more affordable and thus a perfect candidate for a new concept of antibiotics based on gene edition [11].
CRISPR-Cas can be used following three general strategies: (i) it can be directed to cleave species-specific genes to treat acute infections, resulting in a deployment of the bacteria of interest while maintaining the host’s microbiome unaltered [22]; (ii) it can be directed to cleave drug-resistance genes, eliminating bacteria harboring them while maintaining the viability of the wild-type susceptible clones and thus decolonizing patients [23]; or (iii) it can be directed to modify or silence resistance genes, introducing mutations that cause resistance genes’ loss of function while maintaining bacterial viability in a process known as resensitization [24] (Figure 2).
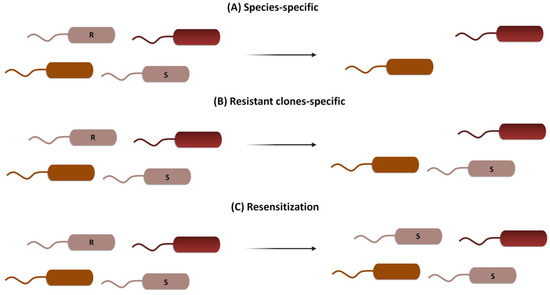
Figure 2.
Three different CRISPR-Cas strategies to combat AMR. (A) Species-specific targeting kills both susceptible and resistant clones of the same species, leaving the rest of the microbial population unaltered. (B) Resistant clones-specific targeting kills only bacteria harboring genes for AMR, leaving susceptible clones and the rest of the population unaffected. (C) Resensitization turns resistant clones into susceptible ones by specifically targeting resistance genes, without an effect on the rest of the microbial population.
Within all Cas proteins, the ones which were used the most to address AMR were the following: (a) Cas9, which specifically recognizes its target and cleaves it, causing a double-strand break [25]; (b) dCas9, a defective Cas9 protein lacking the double-strand nuclease activity which specifically recognizes its target and stays attached to that region, hampering the binding of the RNA polymerase and thus the formation of the transcription preinitiation complex [26]; (c) nSpCas9:rAPOBEC1, a Cas9 protein without nuclease activity fused to a deaminase, which causes the conversion of cytidine bases into thymine ones, thus creating a stop codon [24]; and (d) Cas13a, an RNA-specific endonuclease which indiscriminately cleaves RNA fragments upon activation by the recognition of its specific DNA sequence [27] (Figure 3).
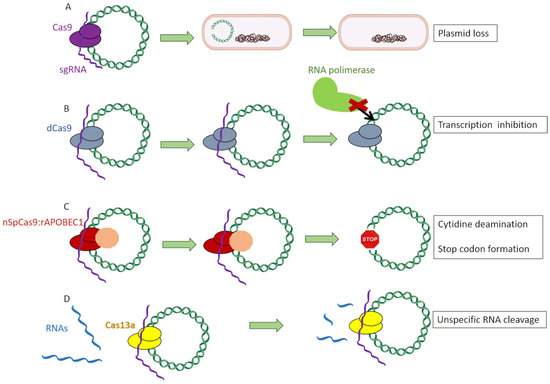
Figure 3.
Different Cas proteins used to target antimicrobial genes: (A) Cas9, which specifically recognizes its target and induces a double-strand break. (B) dCas9, a defective Cas9 protein lacking the double-strand nuclease activity which specifically recognizes its target and stays attached to that region, hampering the binding of the RNA polymerase and thus the formation of the transcription preinitiation complex. (C) nSpCas9:rAPOBEC1, a Cas9 protein without nuclease activity fused to a deaminase, which causes the conversion of cytidine bases into thymine ones, thus creating a stop codon. (D) Cas13a protein, an RNA-specific endonuclease which indiscriminately cleaves RNA fragments upon activation by the recognition of its specific DNA sequence.
Among all CRISPR-Cas types, the most broadly used for genetic engineering is type II due to its simplicity and the fact that it employs a single nuclease with two catalytic domains for interference (Cas9), in contrast to the protein complex required for the types among class 1 [11,28]. The general scheme through which type II CRISPR-Cas system can be used to target bacterial genes is as follows: firstly, a chimeric sgRNA (an artificial RNA construct which fuses crRNA and tracrRNA) is designed to be complementary to the target sequence in the bacterial gene of interest. The chosen sequence has to be unique within the bacterial genome and mobilome so that it only attaches to the desired gene, as well as possessing a PAM. Then, sgRNA:Cas9 complexes are recruited and cleave the target sequence within the bacterial genome, producing blunt ends [29]. This chromosomal double break poses a serious risk for the bacterial cell’s integrity, and it can be mended through nonhomologous end-joining repair (NHEJ) [30], which is error prone and often leads to loss of protein function by inserting aleatory nucleotides or even the loss of the cell’s viability [10]. The cytotoxicity of targeting chromosomal self-genes has been previously shown by Vercoe and colleagues in Pectobacterium artrosepticum strains [31]. In this study, the bacteria’s own CRISPR machinery was exploited by introducing expression vectors which coded for specific crRNAs targeting three nonessential chromosomal genes. As a result, a reduction of a hundred-thousand-fold in viable bacterial counts was observed, as well as the filamentation of surviving cells. In another study, Hullahali and colleagues demonstrated that the growth of Enterococcus faecalis strains is impaired when its own CRISPR machinery is modified to target self-genes [32]. However, this cytotoxicity can be prevented by inserting into the cell an artificial DNA fragment with a copy of the target gene, serving as template for homology-directed repair (HDR) [33] instead of NHEJ. If this copy is engineered with desired mutations, the targeted bacteria would be able to acquire them through a recombination process, resulting in a knockdown of the gene of interest while maintaining the cell’s viability (Figure 4).
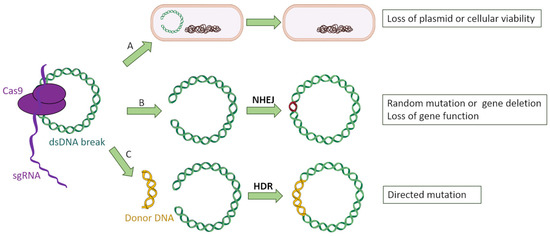
Figure 4.
Three possible events after CRISPR-Cas9 targeting of bacterial genes: (A) The double-strand break affecting a plasmid leads to its loss, whereas a double break into the bacterial chromosome is lethal for the microorganism. (B) The bacterium attempts to fix the double-strand break by nonhomologous end joining (NHEJ), introducing random mutations into the targeted gene which causes a loss of function. (C) The bacterium uses a donor DNA fragment designed with the desired mutations to repair the double-strand break, incorporating those mutations.
3. CRISPR-Cas: A Species-Specific Treatment for ESKAPE Infections
Morbidity and mortality associated with bacterial resistance is restlessly increasing. The European Society for Clinical Microbiology and Infectious Diseases (ESCMID) has delimited three different categories to classify resistant bacteria: multidrug-resistant (MDR) bacteria, when there is in vitro resistance to at least one agent in three or more antimicrobial categories; extensively drug-resistant (XDR) bacteria, when there is resistance to at least one agent in all but two or fewer categories; and pandrug-resistant (PDR) bacteria, where there is resistance to all antimicrobials [34].
According to the Centers for Disease Control and Prevention (CDC), in the year 2019, more than 2.8 million infections occurred in the US with an antibiotic-resistant causative agent, of which 35,000 resulted in the patient’s death [35]. Additionally, the estimated cost to treat MDR infections in the US was calculated to be more than 4.6 billion dollars in 2017 [36]. Regarding Europe, MDR infections are responsible for about 33,000 deaths annually, with an estimated cost of 1.1 billion euros (1.3 billion dollars) [37].
In this context, in 2008, Rice set a list of six main pathogens for which the development of new antibiotics was (and still is) crucial: the ESKAPE bacteria. These are Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacter cloacae [38]. Organizations such as the Infectious Disease Society of America (IDSA) have emphasized the need of joining efforts to tackle these bacteria due to their virulence, prevalence in nosocomial environments and drug resistance [39,40].
In this review, we focus on research directed to address AMR in the ESKAPE group by the edition of drug resistance or virulence genes with the different CRISPR-Cas technology available. Moreover, we also review the literature focused on the use of the CRISPR-Cas technology to study the molecular mechanisms of AMR (Table 1). For those purposes, a profound search in the Pubmed database was performed between 15 December 2020 and 31 March 2021. The included words were: CRISPR, treatment, ESKAPE, Enterococcus spp., Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacter spp.
3.1. Enterococcus faecium and E. faecalis
Enterococcus spp. is a genus of Gram-positive cocci arranged in pairs or short chains. Despite being part of the gastrointestinal microbiota, two species are often found to cause infection: E. faecalis and E. faecium [41]. Their ability to survive on inert surfaces for long periods of time has made them an important issue in hospital-acquired infections, and mutations and/or overproduction of a penicillin binding protein (PBP) of the class B, known as PBP5, confer them intrinsic resistance to the majority of β-lactams. The exception is ampicillin, which is effective in the majority of E. faecalis strains [42]. Enterococci are also intrinsically resistant to aminoglucosides, thus reducing the possible therapeutic options. In addition to that, in the last years the continuous increase in vancomycin-resistant enterococci (VRE) has been of special concern, with the number of infections in Europe almost doubling from 2007 to 2015, according to the ECDC annual report [43].
To date, we only have knowledge of a single gene edition study in E. faecium using CRISPR technology. In 2020, de Maat and colleagues harnessed the high recombination rates in E. faecium to insert two copies of the green fluorescent protein (GFP) into the macrolide resistance gene msrC [44]. This was performed by firstly transform the vancomycin-resistant E. faecium E745 clinical strain with a pVLP3004 plasmid encoding the Cas9 protein and a tracrRNA. Afterward, a second plasmid, named pVDM1001, was used, encoding the specific crRNA and a donor DNA to serve as a template for HDR. This dual plasmid strategy is an adaptation from Oh and Van Pijkeren’s work with Lactobacillus reuteri [45]. Successful edition was assessed by fluorescence measuring after plasmid curing; however, researchers did not study macrolide MICs after GFP insertion; therefore, unfortunately, resensitization could not be tested.
Furthermore, several studies have been published regarding another enterococcal species: E. faecalis. An interesting approach to treat enterococcal infections consists of harnessing the bacterium’s own CRISPR machinery. This was studied by Dr. Palmer’s group, who observed that Type II CRISPR2 orphan locus, which lacks the cas genes, could be reactivated to target pheromone-responsive plasmids (PRP) in the presence of Cas9 enzymes [46]. PRPs are E. faecalis specific plasmids in which high-frequency conjugation is enhanced by the constant production of small signaling peptides by recipient cells and often harbor resistance and virulence factor genes [47]. In their study, Price and colleagues activated the CRISPR2 locus, which lacks the cas genes, against the PRP pCF10 by introducing into the MDR E. faecalis T11 strain a CRISPR1-derived Cas9 enzyme [46]. In addition to that, they analyzed the effect of both restriction-modification and CRISPR-Cas3 systems against the pAM714 PRP. A year later, this group reactivated the same orphan type II CRISPR2 system in the vancomycin-resistant E. faecalis V583 strain to target mobile genetic elements. The authors noted that, despite what was previously published, in E. faecalis, a spacer and its corresponding target could temporarily coexist. In the presence of selective pressure (exposure to 15 µg/mL chloramfenicol and 50 µg/mL erythromycin), the toxic type II CRISPR2 spacers were gradually lost, whereas in the absence of antimicrobials, it was the CRISPR targets that were eliminated [32].
Finally, in 2019, Rodrigues and colleagues integrated a CRISPR system targeting tetM and ermB genes—which confer resistance to tetracycline and erythromycin, respectively—into a PRP, named as pKH88[sp-tetM] and pKH88[sp-ermB] [48]. These plasmids were transmitted by conjugation using E. faecalis CK135 as a donor strain and E. faecalis OG1SSp as a recipient strain, successfully removing antibiotic resistance in vitro. Afterward, using an in vivo C57BL6/J mouse model, it was seen that despite the low recombination rate obtained, transconjugants who successfully acquired the PRP became unable to gain erythromycin-resistance genes. This led the authors to suggest the possibility of using probiotic E. faecalis strains harboring these PRPs to hamper patient’s colonization by resistant E. faecalis strains.
3.2. Staphylococcus aureus
Staphylococcus aureus is a species of Gram-positive, coagulase and catalase positive cocci arranged in clusters and one of the main bacterial pathogens. In the last decades, the spread of methicillin-resistant S. aureus (MRSA), which harbors the mecA gene located in the SCCmec (staphylococcal cassette chromosome), has raised special concern [49]. The mecA gene is responsible for the production of PBP2a, which has low affinity for β-lactam antibiotics and results in resistance to penicillins, cephalosporins—except for the novel antibiotics ceftaroline and ceftobiprole—and carbapenems [50].
In 2014, Bikard and colleagues studied the ability of CRISPR-Cas systems to eliminate specific strains of S. aureus from mixed cultures without affecting other strains of the same species through phagemids [23]. Phagemids are genetic-engineering constructs which fuse a plasmid’s replication origin with filamentous phage’s coat proteins and related genes. For those purposes, a phagemid (pDB121) was designed to target the mecA gene in a 50/50 mix with a clinical MRSA strain (USA300φ) and a RN4220 strain lysogenized—and thus phage-immunized—with φNM1, named RNφ. The MRSA proportion dramatically decreased from the original 50% to a 0.4%. However, the authors remarked two main disadvantages to this study: the inability of phagemids to produce copies of themselves, making it necessary to inoculate a greater number of phagemids than the number of target cells, and its unknown display in a more complex environment, such as diverse microbial populations within living organisms [23].
In 2017, Guan and colleagues transformed S. aureus AH1 strains with engineered plasmids (pLI-158 and pLI-252), encoding a CRISPR-Cas system with spacers targeting the mecA gene [51]. Upon transformation, 95% of the bacterial population was eliminated, thus proving the cytotoxicity caused by targeting the host’s own chromosome. Interestingly, the surviving 5% of transformants had overcome CRISPR’s fitness cost through three different ways: target deletion (87.5%), mutations in the cas genes (10.2%) and spacer deletion (2.3%). As expected, those strains in which the mecA gene was eliminated were resensitized to oxacillin, whereas the other two remained resistant.
On the same year, Liu and colleagues inserted the ermR gene in the S. aureus RN4220 strain into the mecA cassette [52]. Researchers engineered a plasmid, pLQ-KI-ermR, encoding an ermR targeting sgRNA, the cas9 gene and a donor DNA, which served as a template for HDR. The achieved gene-edition efficiency was 43–96%, claimed by the authors to be superior to the allelic replacement’s efficiency (17–80%). Finally, plasmid curation was achieved by introducing a temperature-sensitive replicon into the plasmid and afterward incubating transformants at 42 °C, reaching an efficiency of 99.5%. Despite the successful gene edition, researchers did not study MICs to erythromycin or oxacillin of the edited strains.
Simultaneously, Park and colleagues removed any virulence factors from the temperate phage φSaBov and engineered it to carry a CRISPR-Cas system targeting the nuc gene, an S. aureus species-specific gene which encodes a thermostable nuclease [25]. The resulting phage was produced in RF122 S. aureus strains and termed φSaBov-Cas9-nuc. In their in vitro studies, the authors achieved an almost total decolonization from CTH96 S. aureus strains, recovering no viable cells after treatment with 100 multiplicities of infection (MOI, the ratio between the infecting particles and the host cells). By contrast, the same phage lacking the CRISPR-Cas system had an insignificant effect, which was attributed by the authors to the phage’s own lytic cycle. In their in vivo studies, the authors used a skin infection model in C57BL/6 mice and found more than two orders of magnitude in CFU reduction when applying φSaBov-Cas9-nuc embedded into a hydrogel. This was not observed when the phage was applied directly into the dry skin, due to the latency of bacterial metabolic activity in inert surfaces, which is needed for the CRISPR-Cas system’s functionality. Finally, in order to increase the host’s specificity, φSaBov-Cas9-nuc’s tail fiber protein was complemented with that from the broader-spectrum phage φ11, resulting in a specificity extended to the human pathogenic clones ST1, ST5, ST8 and ST36 [25].
In 2019, the same group studied the effects of their engineered phage φSaBov-Cas9-nuc on biofilms, both in vitro and in vivo [53]. For tracking purposes, biofilm forming ATCC 6538 S. aureus strain was modified by homologous recombination to incorporate a green fluorescent protein (GFP) into its chromosome. In their experiments, Cobb and colleagues compared the in vitro efficacy of vancomycin, fosfomycin and the CRISPR-Cas system harbored in the phage φSaBov-Cas9-nuc. Whereas vancomycin showed no effect on biofilm even at high concentrations (1024 µg/mL), fosfomycin (64 µg/mL) and φSaBov-Cas9-nuc (1 × 108 pfu/mL) successfully cleared it. Furthermore, the phage therapy was shown to be superior to fosfomycin by fluorescent tracking of the GFP. A rat osteomyelitis model was afterward developed by inoculating the ATCC 6538-GFP S. aureus strain in a screw which was then placed into the rat’s femur. After 7 days of the procedure, the infection site was treated with an alginate gel containing 3 g of fosfomycin, 3 × 107 pfu/mL of phage or both fosfomycin and phage. Whereas in the surrounding soft tissue all three treatments were shown to yield lower bacterial counts than the control (alginate alone), only fosfomycin was effective for the femur. This is believed to be due to the lower phage concentration achieved in the site of infection, unable to reach the quantity of 1 × 108 pfu/mL previously described to be effective. This was due to the small volume of alginate that could fit into the bone’s incision and the higher density of the phage-containing gel, resulting in lower phage concentrations [53].
In 2019, Wu and colleagues resensitized S. aureus ATCC 6538 strains to lysostaphin [26], a multiple-catalytic activity enzyme which specifically cleaves S. aureus cell wall’s interpeptide crossbridges [54]. Resistance to this bacteriolytic enzyme is developed by modifying the negative charge of the cell wall teichoic acids, thus hampering lysostaphin adhesion to the bacterial surface. In this study, researchers constructed a plasmid encoding a nuclease-defective Cas9 protein (dCas9) from S. pyogenes and several sgRNAs targeting the tarO, tarG and tarH genes, responsible for the teichoic acids’ synthesis. These genes are regulated in a cascade process, ultimately controlled by the tarO gene [54]. Approximately, a 38% reduction in transcription of these genes was observed by RT-PCR, resulting in ≈1 log kill of cells when incubated with 3 µg/mL lysostaphin and in an extent comparable to the wild-type susceptible strains [26]. However, tarO deletion was shown to cause a greater increase in S. aureus lysostaphin susceptibility, suggesting that dCas9 gene silencing was not complete. To solve this issue, the authors propose to enhance the repressing efficiency by increasing the dCas9 copy number in the transforming plasmid, targeting different DNA strands depending on the promoter’s position and constructing chimeric dCas9-transcriptional repressors. This strategy of employing a dCas9 enzyme has been also studied by Chen and colleagues to edit several genes in S. aureus not related to drug resistance (cmyR, agrA, cntA, murR, agrA and sasE) [55].
Moreover, in 2017, Kang and colleagues for the first time studied Cas9 proteins covalently linked to branched polyethyleneimine (bPEI), a cationic polymer, to enhance CRISPR-Cas uptake by the target cells [56]. This nonviral delivery method was proven to be more effective at targeting the mecA gene in S. aureus CCARM 3798, 3803 and 3877 strains than the CRISPR-Cas system alone or carried by lipofectamine, an artificial transfection reagent used for mammalian cells. Furthermore, the amount of bPEI required to pack the CRISPR-Cas constructs (SpCas9-bPEI) was considerably lower than that for lipid formulations, claimed by the authors to be crucial to reduce toxicity and allow the delivery of higher concentrations.
Furthermore, in 2020, Kiga and colleagues studied the effect of a different kind of Cas enzyme, Cas13a, packaged in a bacteriophage capsid [27]. This protein’s peculiarity resides in its ability to indiscriminately cleave single-stranded RNAs whenever the enzyme recognizes a viral transcript, thus limiting the phage’s replication by stopping the bacterial metabolism and growth. Hence, the activation of the Cas13a protein can be lethal for the targeted bacteria. For this experiment, a CRISPR-Cas13a construct targeting the mecA gene (pKLC-SP_mecA) was inserted into the S. aureus 80α phage’s capsid. This was studied in different S. aureus strains with resistance (MRSA USA300 strain) or susceptibility to oxacillin (USA300-∆mecA and RN4220 strains), finding out that only the oxacillin-resistant had a significantly impaired growth.
Finally, regarding the study of AMR, in 2018, Penewit and colleagues used the CRISPR-Cas technology not to edit bacterial genes but to eliminate the unedited ones in a process known as CRISPR counterselection, leading to high-throughput recombination rates [57]. A plasmid was introduced into S. aureus ATCC 29213 strains encoding a recombinase from E. faecalis (EF2132), and cells were transformed with a recombineering oligonucleotide containing a copy of the rpoB gene with a single-nucleotide mutation (H481Y), which confers rifampin resistance and a silent mutation that disrupts a PAM motif present in the wild-type gene. Afterward, a plasmid encoding Cas9 proteins and sgRNAs targeting the wild-type rpoB gene was introduced, causing double-strand breaks in those cells in which recombination had not occurred. However, in those with successful recombination, the PAM motif was eliminated, and rpoB mutation was acquired, meaning that cells gained both CRISPR-Cas immunity and rifampin resistance. Thanks to the temperature-sensitive replicon in which plasmids were engineered, cells could be cured from those external elements after overnight incubation at 43 °C, and selection markers could be lost [57].
3.3. Klebsiella pneumoniae
Klebsiella pneumoniae is a facultative anaerobe, capsulated, Gram-negative rod belonging to the Enterobacterales order and commonly found in the human gastrointestinal tract [58]. The wild-type strains only manifest intrinsic resistance to aminopenicillins (ampicillin, amoxicillin) and carboxipenicillins (ticarcillin and piperacillin) due to the chromosomal β-Lactamase SHV-1 [59]. However, the ability of K. pneumoniae to acquire resistance to virtually all approved antimicrobials through a combination of plasmid-borne carbapenemases and other resistance mechanisms is of special concern [60,61,62,63].
In 2018, Wang and colleagues developed two different genetic engineering methods based on CRISPR technology to reverse antibiotic resistance in K. pneumoniae [24]. Firstly, they designed a plasmid (pCasKP) harboring the S. pyogenes cas9 gene and a sgRNA targeting the fosA gene, which encodes a glutathione transferase responsible for fosfomycin resistance [64]. Due to the lack of a NHEJ pathway in K. pneumoniae and the low yield of HDR observed when a donor DNA was offered as template, another plasmid (pSGKP) with the λ Red recombination system was used. This high-efficiency bacteriophage recombination machinery for double-strand breaks has been studied and well characterized in the last 50 years [65]. Thanks to the combination of both plasmids (pCasKP-pSGKP), the authors resensitized the K. pneumoniae clinical strain KP 5573 to fosfomycin with an editing efficiency of 100% (10 out of 10 randomly picked colonies) [24].
Afterward, they designed a second genetic engineering tool (pBECKP) by fusing the Cas9 nickase nSpCas9 with the murine cytidine deaminase rAPOBEC1, creating a chimeric protein capable of recognizing specific sequences within the bacterial genome, inducing single-stranded breaks and converting cytidine bases into thymine ones. This tool was used to create a premature stop codon in the fosA gene of K. pneumoniae KP5573 strains, leading to the production of a truncated protein and reversing fosfomycin resistance with a 100% efficiency (8 out of 8 randomly picked colonies) [24].
Finally, researchers exploited these two tools to resensitize the hypermucoviscous K. pneumoniae clinical strain KPCRE23 to carbapenems by both deleting (through pCasKP-pSGKP) or truncating (pBECKP) the blaKPC-2 carbapenemase gene and the two blaSHV and blaCTX-M-65 ESBL genes. In the first case, chromosomal blaSHV deletion yielded an efficiency of 4/12, whereas for plasmid-borne blaSHV and blaCTX-M-65 genes, no PCR product was observed upon transformation, nor for the wild-type plasmid and nor for the ESBL-deleted plasmid. The authors suggested a plasmid loss due to the critical double-strand break caused by Cas9, opening the possibility for the removal of drug-resistance plasmids by CRISPR-Cas constructs. Lastly, the pBECKP tool was used to generate a stop codon into the blaKPC-2 gene, with an efficiency of 100% (8 out of 8 randomly picked colonies) and a reduction in imipenem MICs from 64 to 1 µg/mL, resulting in KPCRE23 resensitization to carbapenems [24].
Finally, in 2020, Hao and colleagues introduced CRISPR-Cas encoding plasmids (pCasCure) into carbapenem-resistant strains of K. pneumoniae to resensitize them to imipenem and meropenem in a very efficient manner. First of all, the KPC-2 encoding plasmid IncFIIK-pKpQIL from K. pneumoniae 13001 (ST258) harboring the blaKPC-2 gene was cured with an efficiency ranging from 98.6 ± 2.4% to 100%. Then, the KPC-2 encoding plasmid IcnN from K. pneumoniae Kp97_58 (ST111) harboring the gene blaKPC-2 was cured with an efficiency ranging from 96.5 ± 2.4% to 97.9 ± 2.1%. Afterward, the OXA-48-like encoding plasmid p72_X3_OXA181 from K. pneumoniae 5193 (ST307) harboring the gene blaOXA-48-like was cured with an efficiency of 98.6 ± 1.2%. Finally, although the OXA-48 encoding plasmid IncL-pOXA48 from K. pneumoniae 49210 (ST23) harboring the gene blaOXA-48 could not be removed due to IS1R-mediated recombination events, the target gene was found to be truncated, and sensitivity to carbapenems was restored. In this last strain, an additional sgRNA targeting the IncL replicon was inserted into the pCasCure CRISPR-plasmid, resulting in a plasmid curing efficiency ranging from 99.3 ± 1.2% to 100 ± 0%. In all of the strains, MICs to carbapenems decreased from >16 mg/L to lower than 0.25 mg/mL, proving a successful resensitization [66].
Finally, regarding the use of CRISPR-Cas to study the molecular mechanisms of AMR, in 2019, Sun and colleagues engineered carbapenem-resistant strains of K. pneumoniae to study colistin and tigecycline resistance by using the abovementioned dual-plasmid genome-editing system (pCasKP-pSGKP) [24]. These two antibiotics are considered to be last-resort options in carbapenem-resistant Enterobacterales’ infections. First of all, researchers used the Y4 strain of K. pneumoniae (susceptible to colistin, with a MIC of 0.25 mg/L) to target and delete the mgrB gene via CRISPR-Cas [67]. This gene is responsible for the production of a transmembrane regulatory protein which downregulates the PhoPQ-PmrAB pathway, a system which modifies de phosphate groups within the lypopolysaccharide’s lipid A by inserting amino-arabinose residues and thus changing the negative charges for positive ones. The elimination of mgrB gene results in an accumulation of positive-charged LPS within the bacterial cell wall and thus in colistin resistance due to like charges repulsion [68,69]. Upon mgrB deletion through pCasKP-pSGKP, Y4 K. pneumoniae strains’ MIC to colistin increased from 0.25 to 16 mg/L, classified as resistant by both antimicrobial susceptibility testing agencies, CLSI (Clinical and Laboratory Standards Institute) [70] and EUCAST (European Committee on Antimicrobial Susceptibility Testing) [71].
On the other hand, the Y17 K. pneumoniae strain (resistant to tigecycline, with a MIC of 8 mg/L) was used to analyze the effects of mutations in tetA and ramR genes. Whereas the tetA gene encodes for a tetracycline-specific efflux pump, the ramR gene downregulates the AcrAB efflux system via the ramA gene [72,73]. In their research work, Sun and colleagues used the pBECKP plasmid to create a stop codon into the tetA gene, resulting in a reduction of tigecycline from 8 to 2 mg/L, although it was insufficient to resensitize the strain (EUCAST sets susceptibility below 0.5 mg/L). In addition, ramA gene was deleted by using the dual-plasmid pCasKP-pSGKP editing system, increasing tigecycline MICs in K. pneumoniae Y17 from 8 to 64 mg/L, thus proving the implications of both genes in tigecycline resistance [67].
3.4. Acinetobacter baumannii
Acinectobacter baumannii is a strictly aerobic, nonfermentative, Gram-negative cocobacilli of special interest in hospital environments. Their ability to survive on inert surfaces and to adhere to materials such as latex in gloves has made them one of the main etiologic agents in hospital-acquired infections. In addition, a combination of efflux pumps and the impermeability of its outer membrane, together with its ability to acquire mobile genetic elements, confers extremely high drug-resistance rates to these bacteria [74,75].
In A. baumannii, little has been studied regarding the use of CRISPR-Cas systems as antimicrobial treatment. In 2018, Karlapudi and colleagues analyzed the abaI gene from A. baumannii through different bioinformatic tools to design suitable sgRNAs for its silencing [76]. AbaI is responsible for the synthesis of acylhomoserine-lactones, well-studied quorum-sensing autoinducers that regulate biofilm synthesis in A. baumannii [77,78].
Between 2019 and 2020, Wang and colleagues adapted the previously cited double-plasmid CRISPR tool in K. pneumoniae [24] to be used in A. baumannii gene editing. Given the intrinsic HDR activity of this species, a single plasmid harboring a CRISPR-Cas system was engineered (pCasAb). However, this recombination activity was found to be insufficient for repairing double-strand breaks per se; therefore, a recombination machinery was engineered in another plasmid as well (pSGAb). This plasmid carried the RecA recombination system, which was found to be more efficient for A. baumannii than the λ-Red (previously used in K. pneumoniae) and Rac-RecET systems [79]. However, to avoid plasmid loss risk when targeting extrachromosomal genes related to plasmid double-strand breaks, another CRISPR-Cas system was designed. In this case, the Cas9 nuclease was replaced by a chimeric nickase nSpCas9 fused with the murine cytidine deaminase rAPOBEC1 (pBECAb-apr) in an analogous way as it was performed with K. pneumoniae. This way, artificial stop codons could be made at desired locations by substituting Cs for Ts, silencing genes without hampering the DNA’s integrity. With this system, three β-lactamase encoding genes in A. baumannii XH386 were targeted (blaOXA-23, blaTEM-1D and blaADC-25), and susceptibility of the resulting mutants to imipenem and sulbactam was tested. Deletion of TEM-1D (labeled as ∆TEM) yielded no difference in imipenem susceptibility when compared to the WT, whereas deletion of ADC-25 (labeled as ∆ADC) resulted in a 2-fold increase and deletion of OXA-23 (labeled as ∆OXA) in an 8-fold increase. When simultaneously targeting more than one β-lactamase, the greatest effect was obtained in the ∆TEM∆ADC∆OXA and ∆ADC∆OXA mutants, suggesting no significant role of TEM-1D in imipenem’s susceptibility. On the other hand, ∆ADC, ∆OXA and ∆ADC∆OXA mutants did not show any reduction in susceptibility of A. baumannii to sulbactam, whereas ∆TEM mutants displayed a 2-fold increase. The greatest change was observed in the ∆TEM∆ADC∆OXA mutant, with a 4-fold increase in susceptibility to sulbactam. These results showed that TEM-1D was the main β-lactamase responsible for sulbactam resistance, suggesting the CRISPR-based plasmid pBECAb-apr to be a useful tool to elucidate the contribution of the different β-lactamases to drug resistance [80,81].
3.5. Pseudomonas aeruginosa
Pseudomonas aeruginosa is a strictly aerobic and nonfermenting Gram-negative rod commonly found as an opportunistic human pathogen. As in A. baumannii, its high drug-resistance rates, mainly acquired by a combination of low permeability of the outer membrane and active drug expelling out of the cell, are of special concern. The acquisition of mobile genetic elements encoding drug-resistance genes such as β-lactamases also plays an important role in antibiotic resistance. In addition, its ability to colonize patients with chronic diseases (such as cystic fibrosis or bronchiectasis) and form biofilm hampers antibiotic activity and the eradication of the pathogen [82].
In 2019, Xu and colleagues exploited the native CRISPR-Cas system found in the MDR P. aeruginosa PA154197 strain, classified as type I-F [83,84]. This strategy has been already used in species in which gene edition is usually inefficient due to poor genetic homeostasis and intrinsic cytotoxicity of heterologous Cas9 proteins, such as clostridia [85,86]. To avoid this issue, it is possible to harness the bacterium’s own CRISPR-Cas machinery by introducing into the cell sgRNA encoding plasmids, whose transcripts direct native Cas proteins to the desired targets.
After confirming the functionality of the native CRISPR-Cas system in the P. aeruginosa PA154197 strain, Xu and colleagues designed the plasmid pAY5233, which encoded sgRNAs targeting the mexB gene. This gene is responsible for the production of a component of the MexAB-OprM efflux pump in P. aeruginosa, which expels molecules such as antimicrobial drugs out of the cell, conferring resistance to them [87]. Targeting the mexB gene yielded no transformants, suggesting the potential toxicity of chromosomal double-strand breaks. Afterward, a new strategy was used, targeting this same gene but at the same time introducing into the pAY5233 plasmid, renamed as pAY5235, a donor DNA template for HDR. With this approach, it was possible to achieve deletion of mexB and regulatory genes mexF and mexH with a success rate above 90%, and changes in antimicrobial drug susceptibility were the analyzed. For ∆mexB mutants, MICs to antipseudomonal β-lactams were reduced by more than 128-fold (carbencillin), 64-fold (aztreonam, piperacillin/tazobactam), 32-fold (meropenem) and 8-fold (ceftazidime). Quinolone susceptibility was slightly increased, with a 2-fold reduction in MICs to levofloxacin and ciprofloxacin [84].
In addition, in order to study quinolone resistance, another CRISPR-Cas-based gene edition strategy was used. Due to the roll of gyrA as an essential gene involved in DNA uncoiling [88], its knocking down would compromise cell’s viability. Because of this, gyrA gene from the P. aeruginosa PA154197 strain was substituted by gyrA gene from the PAO1 strain, which exhibits MICs to both ciprofloxacin and levofloxacin of 0.25 µg/mL. The same approach was used with regulatory genes mexR and mexT, reverting mutations found in the PA154197 strain, which are absent in the quinolone-susceptible PAO1 strains. Results showed that a combination of the three gene substitutions (gyrAPAO1, mexRPAO1 and mexTPAO1 strains) through the CRISPR-harboring plasmid pAY5235 yielded a 128-fold reduction in both ciprofloxacin and levofloxacin MICs. This was much higher than the gyrAPAO1 substitution alone, proving a synergistic effect of the three genes in quinolone resistance in the PA154197 strains [84].
Another approach when facing MDR infections is antivirulence therapy, which focuses on inhibiting the mechanisms through which bacteria communicate with each other (quorum sensing, QS), synthesize biofilm and toxins or arrange their functional membrane microdomains, rather than directly addressing antibiotic resistance [89]. Following this idea, in 2018, Chen and colleagues designed a dual-plasmid strategy (pCasPA/pACRISPR) which combined CRISPR-Cas technology with the λ-Red recombination system and a donor DNA template to edit different virulence regulatory genes [90]. These were the acyl-homoserine lactone receptor encoding genes rhlR and lasR (which are components of the QS systems in P. aeruginosa), the nalD gene (which encodes a repressor of the MexAB-OprM efflux pump), the pigment and QS regulators rsaL and algR (affecting, among others, pyoverdine and pyocyanin production) and the rhamnolipid synthesis regulator rhlB (involved in motility and biofilm disruption) [91,92,93,94,95,96,97].
Furthermore, given the toxicity of the heterologous Cas protein and the large size of the transforming plasmids, these authors developed an additional gene edition tool to increase transformation efficacy and broaden its applicability to other species within the Pseudomonas genus. This was achieved by engineering the pnCasPA-BEC plasmid, encoding for a Cas protein fused with a murine cytidine deaminase and several sgRNAs, which direct the Cas protein to the target sequence. As a result, rhlR and rhlB genes were successfully edited with an efficiency of 11/12 in both of them for the P. aeruginosa PAO1 strain and 14/15 and 17/17, respectively, for the P. aeruginosa PAK strain [90].
Finally, in 2020, Xiang and colleagues reduced the expression of the prtR regulatory gene in the P. aeruginosa PAO1 and PAK strains by transformation with the pHERD20T-dCas9-prtR plasmid [98]. This was achieved by engineering an inducible vector encoding for a dCas protein and the corresponding sgRNA and directed by an arabinose dependent promoter. As a result, transcriptome analysis in the P. aeruginosa PAO1 strain showed that in those strains in which prtR expression was inhibited, 902 genes were downregulated and 587 upregulated. These included the downregulation of genes related to alginate biosynthesis, iron acquisition, proteases and rhamnolipid synthesis, and the upregulation of genes associated with pyocin synthesis and other virulence factors such as the type 6 secretion system. This is consistent with the known targets of the prtR repressor [99].
3.6. Enterobacter spp.
Enterobacter spp. is a genus of Gram-negative bacilli which belongs to the normal human microbiota. However, this bacterium can act as an opportunistic pathogen in a variety of infections, especially those of nosocomial origin (sepsis, urinary tract infections, pneumonia or postsurgical peritonitis) [100]. This genus is characterized by the presence of a chromosomal AmpC β-lactamase, which depending on the degree of de-repression, can confer resistance to third generation cephalosporins, penicillins and their combinations with inhibitors and even ertapenem. In addition, these bacteria can acquire extended spectrum β-lactamases and carbapenemases through mobile genetic elements, considerably reducing the available therapeutic options.
In 2020, Hao and colleagues electroporated the engineered plasmid pCasCure (previously detailed for K. pneumoniae) to resensitize carbapenem-resistant Enterobacter species to these antibiotic family [66]. By targeting their blaKPC-3-harboring plasmids, the E. hormaechei 34978 and the E. xiangfangensis 34399 strains were cured with efficiencies up to 95.8 ± 2.1% and 95.1 ± 2.4%, respectively.
In the same year, Tagliaferri and colleagues targeted the plasmid-borne blaTEM-1 gene in the E. hormaechei 4962 clinical isolate [101]. This plasmid, coding for the Cas9 protein and a specific sgRNA targeting the blaTEM-1 gene, was successfully electroporated in the E. coli 189A clinical isolate, as seen by qPCR and phenotypic testing, with a re-sensitization to ampicillin, cefazolin, cefuroxime, ceftriaxone and cefotaxime. However, plasmid curing efficiencies in the E. hormaechei 4962 strain were considerably lower, with plasmid maintenance despite a substantial reduction in copies per cell. Furthermore, the concomitant presence of AmpC, CTX-M-9 and OXA-9 β-Lactamases within the Enterobacter species hampered phenotypic verification of the curing of plasmids.
4. Discussion
To date, only a few studies have been performed to analyze the ability of the CRISPR-Cas technology to modify genes related to antibiotic resistance or virulence factors in the ESKAPE group. These studies are mainly focused on in vitro experiments, and the goal of the experiment is often to edit bacterial genes independently of their function, just for the sake of the edition, rather than addressing AMR. In other experiments, the goal was to analyze the relationship between a specific mutation or gene with the acquisition of resistance to a particular antibiotic. The fact that CRISPR was not being seen by most researchers as a potential treatment but more as a genetic study tool explains why in some experiments MICs were not performed after CRISPR-directed gene edition and re-sensitization could not be studied.
Furthermore, some pathogens of the ESKAPE group such as S. aureus or K. pneumoniae have been studied more profoundly than others such as A. baumannii, P. aeruginosa or Enterobacter sp. (Table 1). It should also be noted that a wide range of studies focusing on E. coli [102,103,104,105,106] or other species of Pseudomonas [107,108] have not been included in this review as they are not members of the ESKAPE group. In other cases, studies were not analyzed because, although the studied bacteria belonged to the ESKAPE group, the edited genes were related to metabolic pathways rather than antibiotic resistance. Regarding E. faecalis, the studies performed by Dr. Palmer’s group were included in this review for their transcendence and extent, despite the fact that this species is not strictly included into the ESKAPE group [32,46,48,109].

Table 1.
Summary of the CRISPR-Cas strategies used in the treatment and study of the molecular mechanisms of AMR in bacteria belonging to the ESKAPE group.
One of the main reasons why CRISPR-Cas is still not considered as a potential antimicrobial treatment is the delivery issue. While in vitro plasmid electroporation is the method of choice to introduce the CRISPR-Cas system into the bacterial cells in the vast majority of the studies we present in this review, that would not always be possible to perform in vivo. In those cases, some other strategies are to be considered, such as phage-delivery and phagemids [110], conjugative plasmids [105] or polymeric nanoparticles [56] (Figure 5).
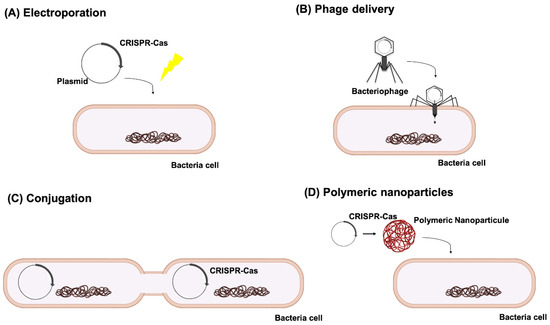
Figure 5.
Different means of CRISPR-Cas delivery into the target cells: (A) plasmid electroporation, (B) phage delivery, (C) conjugation and (D) polymeric nanoparticles.
In the in vivo experiments we reviewed, conjugative plasmids were used for E. faecalis [48] and phage-delivery for S. aureus [25,53]. The obtained efficacies were sensibly lower than in the in vitro experiments, which is explained by the authors by the complexity of the environment in an in vivo model, with external factors affecting plasmid conjugation and the limited amount of phage which could be delivered into the infection site, respectively. Moreover, Rodrigues and colleagues proposed a novel strategy to treat patients colonized by MDR E. faecalis strains [48]. After editing these strains in vitro, researchers found them unable to regain resistance to erythromycin, opening the possibilities to probiotic treatments with CRISPR-Cas edited strains to gradually modify the patient’s microbiome. This approach resembles the one proposed by Ruotsalainen and colleagues [105], who designed “midbiotics” (plasmid-probiotics) targeting ESBL encoding genes. These authors highlighted the advantages of conjugative plasmids over phages, such as a broader host range and protection against the bacterium’s own restriction-modification system.
Another issue of concern when applying the CRISPR-Cas technology into the therapeutics field is the possible side effects of potential off-target modifications in the host’s genome, despite the specificity given by the PAM motive. In the first place, analyzing the host’s genome for potential similarities with the designed sgRNA should be an important anticipation step. On the other hand, using bacteriophages or phagemids as delivery constructs may serve to narrow the CRISPR-Cas system’s specificity thanks to phage tropism, thus avoiding its entry into the host’s cells. This could also be achieved by conjugative plasmids, which require both a donor and a recipient bacterium to be mobilized. In addition, immunity against the CRISPR-Cas system has also been seen as a potential risk. In their studies, Simhadri and colleagues analyzed 200 human serum samples and discovered the presence of antiCas9 antibodies, 10% against Cas9 proteins from S. aureus and 2.5% of S. pyogenes origin [111]. This could be a greater problem than delivery itself, not only due to the potential loss of efficacy upon Cas9 opsonization but also because of the immune response which could be triggered with the treatment. To answer these questions, further in vivo studies focusing on the safety of CRISPR-Cas antimicrobials and their interactions with the host’s immune system should be made.
To conclude, further studies should be performed to deepen the promising applications of CRISPR-Cas as an antimicrobial treatment, specially focusing in the in vivo experiments. The ability of CRISPR-Cas to target single bacterial clones, leaving the rest of the microbiome unaltered, or even to resensitize MDR bacteria without affecting their viability, contrasts with the collateral damage caused to the patient by broad-spectrum antibiotics. With further in vivo studies focusing on CRISPR’s efficacy in complex environments such as the gastrointestinal tract, potential off-target mutations, or the host’s immune response, CRISPR-Cas antimicrobials could be proven to be effective, pathogen-specific and secure.
Author Contributions
Writing-original draft preparation, M.G.d.A.; writing-review and editing, M.G.-B., L.B., O.P., I.B., L.F.-G.; visualization M.F.-Q., M.L., G.B.; writing-review and editing, supervision and funding, M.T. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by grants PI16/01163 and PI19/00878 awarded to M. Tomás within the State Plan for R+D+I 2013–2016 (National Plan for Scientific Research, Technological Development and Innovation 2008–2011) and cofinanced by the ISCIII-Deputy General Directorate for Evaluation and Promotion of Research-European Regional Development Fund “A way of Making Europe” and Instituto de Salud Carlos III FEDER, Spanish Network for the Research in Infectious Diseases (REIPI, RD16/0016/0006) and by the Study Group on Mechanisms of Action and Resistance to Antimicrobials, GEMARA (SEIMC, http://www.seimc.org/, Accessed on 21 June 2021). I. Bleriot was financially supported by pFIS program (ISCIII, FI20/00302). O. Pacios and M. López was financially supported by a grant IN606A-2020/035 and IN606B-2018/008, respectively, (GAIN, Xunta de Galicia), and M. Gonzalez-Bardanca was financially supported by the Rio Hortega program (ISCIII, CM20/00198).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakatura, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isoenzyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, R.; Van Embden, J.D.A.; Gaastra, W.; Schouls, L.M. Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 2002, 43, 1565–1575. [Google Scholar] [CrossRef]
- Mojica, F.J.M.; Juez, G.; Rodriguez-Valera, F. Transcription at different salinities of Haloferax mediterranei sequences adjacent to partially modified PstI sites. Mol. Microbiol. 1993, 9, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Mojica, F.J.M.; Ferrer, C.; Juez, G.; Rodríguez-Valera, F. Long stretches of short tandem repeats are present in the largest replicons of the Archaea Haloferax mediterranei and Haloferax volcanii and could be involved in replicon partitioning. Mol. Microbiol. 1995, 17, 85–93. [Google Scholar] [CrossRef]
- Mojica, F.J.M.; Díez-Villaseñor, C.; García-Martínez, J.; Soria, E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 2005, 60, 174–182. [Google Scholar] [CrossRef]
- O’Connell, M.R.; Oakes, B.L.; Sternberg, S.H.; East-Seletsky, A.; Kaplan, M.; Doudna, J.A. Programmable RNA recognition and cleavage by CRISPR/Cas9. Nature 2014, 516, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Haurwitz, R.E.; Jinek, M.; Wiedenheft, B.; Zhou, K.; Doudna, J.A. Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science 2010, 329, 1355–1358. [Google Scholar] [CrossRef] [Green Version]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Gholizadeh, P.; Köse, Ş.; Dao, S.; Ganbarov, K.; Tanomand, A.; Dal, T.; Aghazadeh, M.; Ghotaslou, R.; Rezaee, M.A.; Yousefi, B.; et al. How CRISPR-Cas system could be used to combat antimicrobial resistance. Infect. Drug Resist. 2020, 13, 1111–1121. [Google Scholar] [CrossRef] [Green Version]
- Strich, J.R.; Chertow, D.S. CRISPR-cas biology and its application to infectious diseases. J. Clin. Microbiol. 2018, 57. [Google Scholar] [CrossRef] [Green Version]
- Gleditzsch, D.; Pausch, P.; Müller-Esparza, H.; Özcan, A.; Guo, X.; Bange, G.; Randau, L. PAM identification by CRISPR-Cas effector complexes: Diversified mechanisms and structures. RNA Biol. 2019, 16, 504–517. [Google Scholar] [CrossRef]
- Ishino, Y.; Krupovic, M.; Forterre, P. History of CRISPR-Cas from encounter with a mysterious repeated sequence to genome editing technology. J. Bacteriol. 2018, 200. [Google Scholar] [CrossRef] [Green Version]
- Gajdács, M. The Concept of an Ideal Antibiotic: Implications for Drug Design. Molecules 2019, 24, 892. [Google Scholar] [CrossRef] [Green Version]
- Pacios, O.; Blasco, L.; Bleriot, I.; Fernandez-Garcia, L.; Bardanca, M.G.; Ambroa, A.; López, M.; Bou, G.; Tomás, M. Strategies to combat multidrug-resistant and persistent infectious diseases. Antibiotics 2020, 9, 65. [Google Scholar] [CrossRef] [Green Version]
- Shahbazi Dastjerdeh, M.; Kouhpayeh, S.; Sabzehei, F.; Khanahmad, H.; Salehi, M.; Mohammadi, Z.; Shariati, L.; Hejazi, Z.; Rabiei, P.; Manian, M. Zinc finger nuclease: A new approach to overcome beta-lactam antibiotic resistance. Jundishapur J. Microbiol. 2016, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseini, N.; Khanahmad, H.; Esfahani, B.; Bandehpour, M.; Shariati, L.; Zahedi, N.; Kazemi, B. Targeting of cholera toxin A (ctxA) gene by zinc finger nuclease: Pitfalls of using gene editing tools in prokaryotes. Res. Pharm. Sci. 2020, 15, 182–190. [Google Scholar] [CrossRef]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goltermann, L.; Nielsen, P.E. PNA Antisense Targeting in Bacteria: Determination of Antibacterial Activity (MIC) of PNA-Peptide Conjugates. In Methods in Molecular Biology; Humana Press Inc.: New York, NY, USA, 2020; Volume 2105, pp. 231–239. [Google Scholar] [CrossRef]
- Edson, J.A.; Kwon, Y.J. RNAi for silencing drug resistance in microbes toward development of nanoantibiotics. J. Control. Release 2014, 189, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Sünderhauf, D.; Pursey, E.; Klümper, U.; Westra, E.; Gaze, W.; van Houte, S. AMR gene removal by conjugative delivery of CRISPR-Cas9. Access Microbiol. 2019, 1, 213. [Google Scholar] [CrossRef]
- Gomaa, A.A.; Klumpe, H.E.; Luo, M.L.; Selle, K.; Barrangou, R.; Beisel, C.L. Programmable removal of bacterial strains by use of genome- targeting CRISPR-cas systems. mBio 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Bikard, D.; Euler, C.W.; Jiang, W.; Nussenzweig, P.M.; Goldberg, G.W.; Duportet, X.; Fischetti, V.A.; Marraffini, L.A. Exploiting CRISPR-cas nucleases to produce sequence-specific antimicrobials. Nat. Biotechnol. 2014, 32, 1146–1150. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, S.; Chen, W.; Song, L.; Zhang, Y.; Shen, Z.; Yu, F.; Li, M.; Ji, Q. CRISPRCas9 and CRISPR-assisted cytidine deaminase enable precise and efficient genome editing in Klebsiella pneumoniae. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef] [Green Version]
- Park, J.Y.; Moon, B.Y.; Park, J.W.; Thornton, J.A.; Park, Y.H.; Seo, K.S. Genetic engineering of a temperate phage-based delivery system for CRISPR/Cas9 antimicrobials against Staphylococcus aureus. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Zha, J.; Koffas, M.A.G.; Dordick, J.S. Reducing Staphylococcus aureus resistance to lysostaphin using CRISPR-dCas9. Biotechnol. Bioeng. 2019, 116, 3149–3159. [Google Scholar] [CrossRef] [PubMed]
- Kiga, K.; Tan, X.E.; Ibarra-Chávez, R.; Watanabe, S.; Aiba, Y.; Sato’o, Y.; Li, F.Y.; Sasahara, T.; Cui, B.; Kawauchi, M.; et al. Development of CRISPR-Cas13a-based antimicrobials capable of sequence-specific killing of target bacteria. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Van Der Oost, J.; Westra, E.R.; Jackson, R.N.; Wiedenheft, B. Unravelling the structural and mechanistic basis of CRISPR-Cas systems. Nat. Rev. Microbiol. 2014, 12, 479–492. [Google Scholar] [CrossRef]
- Loureiro, A.; Da Silva, G.J. Crispr-cas: Converting a bacterial defence mechanism into a state-of-the-art genetic manipulation tool. Antibiotics 2019, 8, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Vercoe, R.B.; Chang, J.T.; Dy, R.L.; Taylor, C.; Gristwood, T.; Clulow, J.S.; Richter, C.; Przybilski, R.; Pitman, A.R.; Fineran, P.C. Cytotoxic Chromosomal Targeting by CRISPR/Cas Systems Can Reshape Bacterial Genomes and Expel or Remodel Pathogenicity Islands. PLoS Genet. 2013, 9, e1003454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hullahalli, K.; Rodrigues, M.; Elife, K.P. Exploiting CRISPR-Cas to manipulate Enterococcus faecalis populations. eLife 2017. [Google Scholar] [CrossRef]
- Haber, J.E. DNA Repair: The Search for Homology. BioEssays 2018, 40, e1700229. [Google Scholar] [CrossRef]
- Magiorakos, A.P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef] [Green Version]
- U.S. Department of Health and Human Services. Antibiotic Resistance Threats in the United States. Cent. Dis. Control. Prev. 2019, 1–113. [Google Scholar] [CrossRef] [Green Version]
- Nelson, R.E.; Hatfield, K.M.; Wolford, H.; Samore, M.H.; Scott, R.D.; Reddy, S.C.; Olubajo, B.; Paul, P.; Jernigan, J.A.; Baggs, J. National Estimates of Healthcare Costs Associated with Multidrug-Resistant Bacterial Infections among Hospitalized Patients in the United States. Clin. Infect. Dis. 2021, 72, S17–S26. [Google Scholar] [CrossRef]
- ECDC Antimicrobial Resistance Tackling the Burden in the European Union. Eur. Cent. Dis. Prev. Control. 2019, pp. 1–20. Available online: https://www.oecd.org/health/health-systems/AMR-Tackling-the-Burden-in-the-EU-OECD-ECDC-Briefing-Note-2019.pdf (accessed on 21 June 2021).
- Rice, L.B. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: No ESKAPE. J. Infect. Dis. 2008, 197, 1079–1081. [Google Scholar] [CrossRef] [PubMed]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad bugs, no drugs: No ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boucher, H.W.; Talbot, G.H.; Benjamin, D.K.; Bradley, J.; Guidos, R.J.; Jones, R.N.; Murray, B.E.; Bonomo, R.A.; Gilbert, D. 10 × ’20 Progress--Development of New Drugs Active against Gram-Negative Bacilli: An Update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2013, 56, 1685–1694. [Google Scholar] [CrossRef] [Green Version]
- Arias, C.A.; Murray, B.E. The rise of the Enterococcus: Beyond vancomycin resistance. Nat. Rev. Microbiol. 2012, 10, 266–278. [Google Scholar] [CrossRef] [Green Version]
- Miller, W.R.; Munita, J.M.; Arias, C.A. Mechanisms of antibiotic resistance in enterococci. Expert Rev. Anti. Infect. Ther. 2014, 12, 1221–1236. [Google Scholar] [CrossRef]
- EARS-net Antimicrobial resistance in the EU/EEA (EARS-Net), Annual Epidemiological Report for 2019. Surveill. Rep. 2020. Available online: https://www.ecdc.europa.eu/en/publications-data/surveillance-antimicrobial-resistance-europe-2019#no-link (accessed on 21 June 2021).
- De Maat, V.; Stege, P.B.; Dedden, M.; Hamer, M.; Van Pijkeren, J.P.; Willems, R.J.L.; Van Schaik, W. CRISPR-Cas9-mediated genome editing in vancomycin-resistant Enterococcus faecium. FEMS Microbiol. Lett. 2020, 366. [Google Scholar] [CrossRef]
- Oh, J.H.; Van Pijkeren, J.P. CRISPR-Cas9-assisted recombineering in Lactobacillus reuteri. Nucleic Acids Res. 2014, 42. [Google Scholar] [CrossRef]
- Price, V.J.; Huo, W.; Sharifi, A.; Palmer, K.L. CRISPR-Cas and Restriction-Modification Act Additively against Conjugative Antibiotic Resistance Plasmid Transfer in Enterococcus faecalis. Am. Soc. Microbiol. 2016. [Google Scholar] [CrossRef] [Green Version]
- Licht, T.R.; Laugesen, D.; Jensen, L.B.; Jacobsen, B.L. Transfer of the pheromone-inducible plasmid pCF10 among Enterococcus faecalis microorganisms colonizing the intestine of mini-pigs. Appl. Environ. Microbiol. 2002, 68, 187–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, M.; Mcbride, S.W.; Hullahalli, K.; Palmer, K.L.; Duerkop, B.A. Conjugative Delivery of CRISPR-Cas9 for the Selective Depletion of Antibiotic-Resistant Enterococci. Am. Soc. Microbiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S.; De Lencastre, H.; Garau, J.; Kluytmans, J.; Malhotra-Kumar, S.; Peschel, A.; Harbarth, S. Methicillin-resistant Staphylococcus aureus. Nat. Rev. Dis. Prim. 2018, 4. [Google Scholar] [CrossRef]
- Peacock, S.J.; Paterson, G.K. Mechanisms of methicillin resistance in Staphylococcus aureus. Annu. Rev. Biochem. 2015, 84, 577–601. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Wang, W.; Sun, B. Chromosomal Targeting by the Type III-A CRISPR-Cas System Can Reshape Genomes in Staphylococcus aureus. mSphere 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Jiang, Y.; Shao, L.; Yang, P.; Sun, B.; Yang, S.; Chen, D. CRISPR/Cas9-based efficient genome editing in Staphylococcus aureus. Acta Biochim. Biophys. Sin. 2017, 49, 764–770. [Google Scholar] [CrossRef] [Green Version]
- Cobb, L.H.; Park, J.Y.; Swanson, E.A.; Beard, M.C.; McCabe, E.M.; Rourke, A.S.; Seo, K.S.; Olivier, A.K.; Priddy, L.B. CRISPR-Cas9 modified bacteriophage for treatment of Staphylococcus aureus induced osteomyelitis and soft tissue infection. PLoS ONE 2019, 14, e0220421. [Google Scholar] [CrossRef] [Green Version]
- Kumar, J.K. Lysostaphin: An antistaphylococcal agent. Appl. Microbiol. Biotechnol. 2008, 80, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhang, Y.; Yeo, W.S.; Bae, T.; Ji, Q. Rapid and Efficient Genome Editing in Staphylococcus aureus by Using an Engineered CRISPR/Cas9 System. J. Am. Chem. Soc. 2017, 139, 3790–3795. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.K.; Kwon, K.; Ryu, J.S.; Lee, H.N.; Park, C.; Chung, H.J. Nonviral Genome Editing Based on a Polymer-Derivatized CRISPR Nanocomplex for Targeting Bacterial Pathogens and Antibiotic Resistance. Bioconjug. Chem. 2017, 28, 957–967. [Google Scholar] [CrossRef]
- Penewit, K.; Holmes, E.A.; McLean, K.; Ren, M.; Waalkes, A.; Salipante, S.J. Efficient and scalable precision genome editing in Staphylococcus aureus through conditional recombineering and CRISPR/Cas9-mediated counterselection. mBio 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Sun, X.; Ma, X. Systematic review and meta-analysis of mortality of patients infected with carbapenem-resistant Klebsiella pneumoniae. Ann. Clin. Microbiol. Antimicrob. 2017, 16, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaves, J.; Ladona, M.G.; Segura, C.; Coira, A.; Reig, R.; Ampurdanés, C. SHV-1 β-lactamase is mainly a chromosomally encoded species-specific enzyme in Klebsiella pneumoniae. Antimicrob. Agents Chemother. 2001, 45, 2856–2861. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Zhao, Z.; Ge, Y.; He, F. Rapid emergence of a pandrug-resistant Klebsiella pneumoniae ST11 isolate in an inpatient in a teaching hospital in China after treatment with multiple broad-spectrum antibiotics. Infect. Drug Resist. 2020, 13, 799–804. [Google Scholar] [CrossRef] [Green Version]
- Longo, L.G.A.; de Sousa, V.S.; Kraychete, G.B.; Justo-da-Silva, L.H.; Rocha, J.A.; Superti, S.V.; Bonelli, R.R.; Martins, I.S.; Moreira, B.M. Colistin resistance emerges in pandrug-resistant Klebsiella pneumoniae epidemic clones in Rio de Janeiro, Brazil. Int. J. Antimicrob. Agents 2019, 54, 579–586. [Google Scholar] [CrossRef]
- Jia, H.; Chen, H.; Ruan, Z. Unravelling the genome sequence of a pandrug-resistant Klebsiella pneumoniae isolate with sequence type 11 and capsular serotype KL64 from China. J. Glob. Antimicrob. Resist. 2019, 19, 40–42. [Google Scholar] [CrossRef]
- Jordt, H.; Stalder, T.; Kosterlitz, O.; Ponciano, J.M.; Top, E.M.; Kerr, B. Coevolution of host–plasmid pairs facilitates the emergence of novel multidrug resistance. Nat. Ecol. Evol. 2020, 4, 863–869. [Google Scholar] [CrossRef]
- Ito, R.; Mustapha, M.M.; Tomich, A.D.; Callaghan, J.D.; McElheny, C.L.; Mettus, R.T.; Shanks, R.M.Q.; Sluis-Cremer, N.; Doi, Y. Widespread fosfomycin resistance in gram-negative bacteria attributable to the chromosomal fosA gene. mBio 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Murphy, K.C. λ Recombination and Recombineering. EcoSal Plus 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, M.; He, Y.; Zhang, H.; Liao, X.P.; Liu, Y.H.; Sun, J.; Du, H.; Kreiswirth, B.N.; Chen, L. CRISPR-Cas9-mediated carbapenemase gene and plasmid curing in carbapenem-resistant enterobacteriaceae. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Wang, Y.; Dong, N.; Shen, L.; Zhou, H.; Hu, Y.; Gu, D.; Chen, S.; Zhang, R.; Ji, Q. Application of CRISPR/Cas9-based genome editing in studying the mechanism of pandrug resistance in Klebsiella pneumoniae. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [Green Version]
- Poirel, L.; Jayol, A.; Bontron, S.; Villegas, M.V.; Ozdamar, M.; Türkoglu, S.; Nordmann, P. The mgrB gene as a key target for acquired resistance to colistin in Klebsiella pneumoniae. J. Antimicrob. Chemother. 2015, 70, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Cannatelli, A.; D’Andrea, M.M.; Giani, T.; Di Pilato, V.; Arena, F.; Ambretti, S.; Gaibani, P.; Rossolini, G.M. In vivo emergence of colistin resistance in Klebsiella pneumoniae producing KPC-type carbapenemases mediated by insertional inactivation of the PhoQ/PhoP mgrB regulator. Antimicrob. Agents Chemother. 2013, 57, 5521–5526. [Google Scholar] [CrossRef] [Green Version]
- Humphries, R.M.; Abbott, A.N.; Hindler, J.A. Understanding and addressing CLSI breakpoint revisions: A primer for clinical laboratories. J. Clin. Microbiol. 2019, 57, e00203-19. [Google Scholar] [CrossRef] [Green Version]
- Leclercq, R.; Cantón, R.; Brown, D.F.J.; Giske, C.G.; Heisig, P.; Macgowan, A.P.; Mouton, J.W.; Nordmann, P.; Rodloff, A.C.; Rossolini, G.M.; et al. EUCAST expert rules in antimicrobial susceptibility testing. Clin. Microbiol. Infect. 2013, 19, 141–160. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Cai, Y.; Liu, X.; Bai, N.; Liang, B.; Wang, R. The emergence of clinical resistance to tigecycline. Int. J. Antimicrob. Agents 2013, 41, 110–116. [Google Scholar] [CrossRef]
- Chiu, S.K.; Huang, L.Y.; Chen, H.; Tsai, Y.K.; Liou, C.H.; Lin, J.C.; Siu, L.K.; Chang, F.Y.; Yeh, K.M. Roles of ramR and tet(A) mutations in conferring tigecycline resistance in carbapenem-resistant Klebsiella pneumoniae clinical isolates. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [Green Version]
- Vila, J.; Pachón, J. Therapeutic options for Acinetobacter baumannii infections. Expert Opin. Pharmacother. 2008, 9, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Tafaj, S.; Kostyanev, T.; Xavier, B.B.; Fluit, A.C.; Rodrigues, C.F.; Lammens, C.; Osmalli, D.; Raka, L.; Goossens, H.; Malhotra-Kumar, S.; et al. Clonal transmission of multidrug-resistant Acinetobacter baumannii harbouring blaOXA-24-like and blaOXA-23-like genes in a tertiary hospital in Albania. J. Glob. Antimicrob. Resist. 2020, 23, 79–81. [Google Scholar] [CrossRef]
- Karlapudi, A.P.; Venkateswarulu, T.C.; Tammineedi, J.; Srirama, K.; Kanumuri, L.; Kodali, V.P. In silico sgRNA tool design for CRISPR control of quorum sensing in Acinetobacter species. Genes Dis. 2018, 5, 123–129. [Google Scholar] [CrossRef]
- Mayer, C.; Muras, A.; Romero, M.; López, M.; Tomás, M.; Otero, A. Multiple quorum quenching enzymes are active in the nosocomial pathogen Acinetobacter baumannii ATCC17978. Front. Cell. Infect. Microbiol. 2018, 8. [Google Scholar] [CrossRef]
- Fernandez-Garcia, L.; Ambroa, A.; Blasco, L.; Bleriot, I.; López, M.; Alvarez-Marin, R.; Fernández-Cuenca, F.; Martinez-Martinez, L.; Vila, J.; Rodríguez-Baño, J.; et al. Relationship between the Quorum Network (Sensing/Quenching) and Clinical Features of Pneumonia and Bacteraemia Caused by A. baumannii. Front. Microbiol. 2018, 9, 3105. [Google Scholar] [CrossRef]
- Fu, J.; Bian, X.; Hu, S.; Wang, H.; Huang, F.; Seibert, P.M.; Plaza, A.; Xia, L.; Müller, R.; Stewart, A.F.; et al. Full-length RecE enhances linear-linear homologous recombination and facilitates direct cloning for bioprospecting. Nat. Biotechnol. 2012, 30, 440–446. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Z.; Ji, Q. CRISPR-Cas9-Based Genome Editing and Cytidine Base Editing in Acinetobacter baumannii. STAR Protoc. 2020, 1, 100025. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Z.; Chen, Y.; Hua, X.; Yu, Y.; Ji, Q. A Highly Efficient CRISPR-Cas9-Based Genome Engineering Platform in Acinetobacter baumannii to Understand the H2O2-Sensing Mechanism of OxyR. Cell Chem. Biol. 2019, 26, 1732–1742.e5. [Google Scholar] [CrossRef] [Green Version]
- Pang, Z.; Raudonis, R.; Glick, B.R.; Lin, T.J.; Cheng, Z. Antibiotic resistance in Pseudomonas aeruginosa: Mechanisms and alternative therapeutic strategies. Biotechnol. Adv. 2019, 37, 177–192. [Google Scholar] [CrossRef]
- Xu, Z.; Li, Y.; Yan, A. Repurposing the Native Type I-F CRISPR-Cas System in Pseudomonas aeruginosa for Genome Editing. STAR Protoc. 2020, 1, 100039. [Google Scholar] [CrossRef]
- Xu, Z.; Li, M.; Li, Y.; Cao, H.; Miao, L.; Xu, Z.; Higuchi, Y.; Yamasaki, S.; Nishino, K.; Woo, P.C.Y.; et al. Native CRISPR-Cas-Mediated Genome Editing Enables Dissecting and Sensitizing Clinical Multidrug-Resistant P. aeruginosa. Cell Rep. 2019, 29, 1707–1717. [Google Scholar] [CrossRef] [PubMed]
- Pyne, M.E.; Bruder, M.R.; Moo-Young, M.; Chung, D.A.; Chou, C.P. Harnessing heterologous and endogenous CRISPR-Cas machineries for efficient markerless genome editing in Clostridium. Sci. Rep. 2016, 6, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zong, W.; Hong, W.; Zhang, Z.T.; Wang, Y. Exploiting endogenous CRISPR-Cas system for multiplex genome editing in Clostridium tyrobutyricum and engineer the strain for high-level butanol production. Metab. Eng. 2018, 47, 49–59. [Google Scholar] [CrossRef]
- Tsutsumi, K.; Yonehara, R.; Ishizaka-Ikeda, E.; Miyazaki, N.; Maeda, S.; Iwasaki, K.; Nakagawa, A.; Yamashita, E. Structures of the wild-type MexAB–OprM tripartite pump reveal its complex formation and drug efflux mechanism. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Jalal, S.; Ciofu, O.; Høiby, N.; Gotoh, N.; Wretlind, B. Molecular mechanisms of fluoroquinolone resistance in Pseudomonas aeruginosa isolates from cystic fibrosis patients. Antimicrob. Agents Chemother. 2000, 44, 710–712. [Google Scholar] [CrossRef] [Green Version]
- Martínez, O.F.; Cardoso, M.H.; Ribeiro, S.M.; Franco, O.L. Recent advances in anti-virulence therapeutic strategies with a focus on dismantling bacterial membrane microdomains, toxin neutralization, quorum-sensing interference and biofilm inhibition. Front. Cell. Infect. Microbiol. 2019, 9, 74. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhang, Y.; Zhang, Y.; Pi, Y.; Gu, T.; Song, L.; Wang, Y.; Ji, Q. CRISPR/Cas9-based Genome Editing in Pseudomonas aeruginosa and Cytidine Deaminase-Mediated Base Editing in Pseudomonas Species. iScience 2018, 6, 222–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, S.; Moustafa, D.A.; Stergioula, V.; Smith, C.D.; Goldberg, J.B.; Bassler, B.L. The PqsE and RhlR proteins are an autoinducer synthase–receptor pair that control virulence and biofilm development in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2018, 115, E9411–E9418. [Google Scholar] [CrossRef] [Green Version]
- Kostylev, M.; Kim, D.Y.; Smalley, N.E.; Salukhe, I.; Peter Greenberg, E.; Dandekar, A.A. Evolution of the Pseudomonas aeruginosa quorum-sensing hierarchy. Proc. Natl. Acad. Sci. USA 2019, 116, 7027–7032. [Google Scholar] [CrossRef] [Green Version]
- Morita, Y.; Cao, L.; Gould, V.C.; Avison, M.B.; Poole, K. nalD encodes a second repressor of the mexAB-oprM multidrug efflux operon of Pseudomonas aeruginosa. J. Bacteriol. 2006, 188, 8649–8654. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Xu, C.; Pan, X.; Dong, Y.; Ren, H.; Jin, Y.; Bai, F.; Cheng, Z.; Jin, S.; Wu, W. Mechanisms of RsaL mediated tolerance to ciprofloxacin and carbenicillin in Pseudomonas aeruginosa. Curr. Genet. 2019, 65, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Okkotsu, Y.; Tieku, P.; Fitzsimmons, L.F.; Churchill, M.E.; Schurr, M.J. Pseudomonas aeruginosa AlgR phosphorylation modulates rhamnolipid production and motility. J. Bacteriol. 2013, 195, 5499–5515. [Google Scholar] [CrossRef] [Green Version]
- Little, A.S.; Okkotsu, Y.; Reinhart, A.A.; Damron, F.H.; Barbier, M.; Barrett, B.; Oglesby-Sherrouse, A.G.; Goldberg, J.B.; Cody, W.L.; Schurr, M.J.; et al. Pseudomonas aeruginosa algr phosphorylation status differentially regulates pyocyanin and pyoverdine production. mBio 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Wood, T.L.; Gong, T.; Zhu, L.; Miller, J.; Miller, D.S.; Yin, B.; Wood, T.K. Rhamnolipids from Pseudomonas aeruginosa disperse the biofilms of sulfate-reducing bacteria. NPJ Biofilms Microbiomes 2018, 4. [Google Scholar] [CrossRef] [Green Version]
- Xiang, L.; Qi, F.; Jiang, L.; Tan, J.; Deng, C.; Wei, Z.; Jin, S.; Huang, G. CRISPR-dCas9-mediated knockdown of prtR, an essential gene in Pseudomonas aeruginosa. Lett. Appl. Microbiol. 2020, 71, 386–393. [Google Scholar] [CrossRef]
- Sun, Z.; Shi, J.; Liu, C.; Jin, Y.; Li, K.; Chen, R.; Jin, S.; Wu, W. PrtR homeostasis contributes to Pseudomonas aeruginosa pathogenesis and resistance against Ciprofloxacin. Infect. Immun. 2014, 82, 1638–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezzatesta, M.L.; Gona, F.; Stefani, S. Enterobacter cloacae complex: Clinical impact and emerging antibiotic resistance. Future Microbiol. 2012, 7, 887–902. [Google Scholar] [CrossRef] [PubMed]
- Tagliaferri, T.L.; Guimarães, N.R.; de Paula Martins Pereira, M.; Vilela, L.F.F.; Horz, H.P.; dos Santos, S.G.; Mendes, T.A.D.O. Exploring the Potential of CRISPR-Cas9 Under Challenging Conditions: Facing High-Copy Plasmids and Counteracting Beta-Lactam Resistance in Clinical Strains of Enterobacteriaceae. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef]
- Kim, J.S.; Cho, D.H.; Park, M.; Chung, W.J.; Shin, D.; Ko, K.S.; Kweon, D.H. Crispr/cas9-mediated re-sensitization of antibiotic-resistant Escherichia coli harboring extended-spectrum β-lactamases. J. Microbiol. Biotechnol. 2015, 26, 394–401. [Google Scholar] [CrossRef]
- Sun, L.; He, T.; Zhang, L.; Pang, M.; Zhang, Q.; Zhou, Y.; Bao, H.; Wang, R. Generation of newly discovered resistance gene mcr-1 knockout in Escherichia coli using the CRISPR/Cas9 system. J. Microbiol. Biotechnol. 2017, 27, 1276–1280. [Google Scholar] [CrossRef] [Green Version]
- Qiu, H.; Gong, J.; Butaye, P.; Lu, G.; Huang, K.; Zhu, G.; Zhang, J.; Hathcock, T.; Cheng, D.; Wang, C. CRISPR/Cas9/sgRNA-mediated targeted gene modification confirms the cause-effect relationship between gyrA mutation and quinolone resistance in Escherichia coli. FEMS Microbiol. Lett. 2018, 365. [Google Scholar] [CrossRef] [PubMed]
- Ruotsalainen, P.; Penttinen, R.; Mattila, S.; Jalasvuori, M. Midbiotics: Conjugative plasmids for genetic engineering of natural gut flora. Gut Microbes 2019, 10, 643–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, P.; Cui, S.; Ma, Z.; Chen, L.; Li, X.; Zhao, R.; Xiong, W.; Zeng, Z. Reversal of mcr-1-mediated colistin resistance in Escherichia coli by CRISPR-Cas9 system. Infect. Drug Resist. 2020, 13, 1171–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Lu, L.B.; Liang, T.X.; Yang, L.R.; Wu, J.P. CRISPR-Assisted Multiplex Base Editing System in Pseudomonas putida KT2440. Front. Bioeng. Biotechnol. 2020, 8. [Google Scholar] [CrossRef]
- Wu, Z.; Chen, Z.; Gao, X.; Li, J.; Shang, G. Combination of ssDNA recombineering and CRISPR-Cas9 for Pseudomonas putida KT2440 genome editing. Appl. Microbiol. Biotechnol. 2019, 103, 2783–2795. [Google Scholar] [CrossRef] [PubMed]
- Hullahalli, K.; Rodrigues, M.; Nguyen, U.T.; Palmer, K. An attenuated CRISPR-cas system in Enterococcus faecalis permits DNA acquisition. mBio 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Fage, C.; Lemire, N.; Moineau, S. Delivery of CRISPR-Cas systems using phage-based vectors. Curr. Opin. Biotechnol. 2021, 68, 174–180. [Google Scholar] [CrossRef]
- Simhadri, V.L.; McGill, J.; McMahon, S.; Wang, J.; Jiang, H.; Sauna, Z.E. Prevalence of Pre-existing Antibodies to CRISPR-Associated Nuclease Cas9 in the USA Population. Mol. Ther. Methods Clin. Dev. 2018, 10, 105–112. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).